

Abstract
Asthma is thought to result from an abnormal expansion of CD4 T cells reactive with airborne allergens, and pathology is controlled by several cytokines of the T helper type 2 (Th2) family. The exact molecules which are involved in generating allergen-reactive T cells are not clear. Studies with blocking reagents or knockout animals have shown that the CD28/B7 interaction partially controls development of allergic asthma in mouse models, but may not be the sole molecule involved. In this report, we have investigated the role of the tumor necrosis factor receptor family member OX40 in allergic inflammation using OX40-deficient mice. OX40 has been shown to participate in regulating clonal expansion and memory development of CD4 T cells and may synergize with CD28. Our studies demonstrate that OX40−/− mice, primed with the model allergen ovalbumin and challenged through the airways with aerosolized antigen, are severely impaired in their ability to generate a Th2 response characterized by high levels of interleukin (IL)-5, IL-4, and immunoglobulin E. Moreover, OX40−/− mice exhibit diminished lung inflammation, including an 80–90% reduction in eosinophilia and mucus production, less goblet cell hyperplasia, and significantly attenuated airway hyperreactivity. These studies highlight the potential importance of OX40 in development of allergic asthma and suggest that targeting OX40 may prove useful therapeutically.
Keywords: asthma, OX40, costimulation, allergy, inflammation
Introduction
Asthma is a multifactorial disease characterized by chronic inflammation in the lungs. The most common form, extrinsic asthma, is believed to be driven by an immune response to airborne allergens. The signature of the disease is massive infiltration of the bronchial mucosa by several cell types including lymphocytes, eosinophils, and mast cells, and this is accompanied by a pronounced elevation in serum IgE. Desquamation of the lung epithelium occurs, along with goblet cell hyperplasia, thickening of the mucosal layer, and mucus production within the bronchioles, with the overall result being impeded airflow and airway hyperreactivity (AHR).
Studies over the past few years, particularly in mice, have established a critical role for the type 2 cytokines IL-4, IL-5, IL-13, and recently IL-9, in the asthmatic response. Studies of antibody blocking or in knockout mice have clearly shown that IL-4 and IL-5 control many, although not all, phases of asthma 1 2, and recent data with recombinant IL-13 have also demonstrated that this cytokine can reproduce much of the asthmatic reaction 3. IL-5 and IL-4 may control eosinophilia and IgE 1 2, and IL-13 and IL-9 may control mucus production and AHR 4 5.
Although mast cells and eosinophils can produce type 2 cytokines, and are therefore potential contributors to the inflammatory response in asthma, there is abundant clinical and experimental evidence that initiation of the asthmatic reaction is dependent on T cells, particularly those of the CD4 subset. For example, depletion of CD4 cells from mice can prevent AHR and eosinophilia 6, and Th2 clones or primary CD4 T cells secreting IL-4, IL-5, and IL-13 can induce asthmatic symptoms if adoptively transferred into unimmunized recipients 4.
One strategy to control asthma is therefore to directly block T cell activation by inhibiting recognition of antigen and signaling through the T cell receptor. The usefulness of anti-CD4 and pharmacological inhibitors that prevent T cell signaling are being tested. The latter include corticosteroids, cyclosporin A, FK506, and rapamycin. Another strategy is to target costimulatory interactions that are essential for full activation and effector function of T cells. Two major interactions that are involved in the initial stages of a T cell response are those between CD28/B7 and CD40 ligand (CD40L)/CD40. Both have recently been targeted in mouse models of asthma. The majority of studies have examined CD28/B7 and demonstrated that blockade of this pathway can reduce type 2 cytokines, IgE, AHR, and eosinophilia 7 8 9. Contrasting data have been published on the role of CD40L/CD40. In one study, CD40 knockout mice were relatively normal in their ability to mount an eosinophil response and to develop AHR, even though IgE levels were reduced 10, whereas in a study of mice deficient in CD40L, a reduction in eosinophils and IL-4 was observed, but not IL-5 11. Thus, ligation of CD28 and perhaps CD40L appear to be involved in development of the CD4 response that leads to asthma.
As well as molecules that regulate early phases of T cell responses, additional costimulatory interactions have been described between inducible molecules which may be crucial for the development of a long-lasting T cell response (for reviews, see references 12 and 13). In particular, signaling through the TNFR family member OX40 (CD134) has recently been shown to control clonal expansion, cytokine production, and memory development of CD4 T cells in several experimental situations involving either Th1 or Th2 cytokines 14 15 16 17 18 19. Whether OX40 is required for T cell responses in most disease situations is not known, nor whether OX40 plays an obligatory role in development of Th2 responses. Ligation of OX40 in vitro and in vivo can enhance development of T cells secreting Th2 cytokines 15 19, and OX40 and OX40L knockout mice have been shown to be defective in the weak Th2 responses that are induced by priming with protein antigens 18 19. In support of a role in Th2 responses in disease scenarios, blocking OX40 suppressed the inflammatory response to Leishmania major in BALB/c mice 20. However, in contrast to this, there was no apparent requirement for OX40 in the Th2 response to the parasite Nippostrongylus brasiliensis, suggesting that there may not be an obligatory role for OX40 in every situation where Th2 cytokines are produced 21.
In this report, we have investigated the role of OX40 in development of allergic inflammation in a murine model of asthma using mice deficient in OX40. The results clearly demonstrate that much of the asthmatic Th2 response is dependent on OX40/OX40L interactions including production of high levels of IL-5, recruitment of large numbers of eosinophils, and induction of IgE. Importantly, goblet cell hyperplasia, mucus production, and AHR are all suppressed in the absence of OX40, demonstrating that this molecule plays a major role in development of the asthmatic reaction.
Materials and Methods
Induction of Allergic Airway Inflammation.
OX40−/− mice 19 21, backcrossed at least six times onto a C57BL/6 background, were compared with wild-type (Wt) C57BL/6 mice obtained from The Jackson Laboratory or produced as OX40+/+ littermates from crossing heterozygous knockouts. Groups of mice were sensitized by intraperitoneal injection of 20 μg OVA protein (chicken egg albumin; Sigma-Aldrich) and 2 mg aluminum hydroxide in PBS. After 4 wk, challenge doses of OVA were given through the airways by subjecting mice for 30 min to 10 ml of a solution of 5 mg/ml OVA induced into aerosol form in a Plexiglas chamber with a nebulizer. The challenge was performed once a day for four consecutive days and then the mice were assessed for allergic inflammation of the lungs 1–3 h after the last aerosol exposure.
Measurement of AHR.
Responsiveness to β-methacholine was assessed in conscious, unrestrained mice by barometric plethysmography, using apparatus and software from Buxco Electronics. Measurements were performed by standard methods using this apparatus similarly to that described previously 22. Each mouse was placed in a chamber and exposed to an aerosol of PBS (baseline readings) and then increasing concentrations of aerosolized β-methacholine (2.5–20 mg/ml; Sigma-Aldrich) for 3 min each. AHR is assessed using the parameter Penh (enhanced pause), which is calculated automatically based on the mean pressure generated in plethysmograph chambers during inspiration and expiration, combined with the time of each phase. Penh readings were taken for 5 min after each aerosol exposure and averaged. Values are represented as the mean percent change in Penh at each methacholine concentration compared with baseline readings, calculated from groups of four mice.
Characterization of Lung Histology and Cellular Infiltrates.
After AHR measurement, mice were anesthetized and the trachea cannulated. Bronchoalveolar lavage (BAL) was performed with 1 ml 0.9% NaCl (Baxter) and the fluid centrifuged at 400 g to separate cells from liquid. Fluid was used to determine lung cytokine content. The total number of BAL cells was determined by trypan blue exclusion, and then differential cell counts for eosinophils, neutrophils, lymphocytes, and monocytes were assessed by staining cytospins with Hema 3 stain (Fisher Scientific), a modified Wright-Giemsa stain.
Lungs were removed from mice that were not subjected to the bronchial lavage procedure. Samples were formalin fixed overnight, stored in 70% ethanol, and sectioned to 5 μm. Sections were stained with periodic acid-Schiff (PAS) as a measure of mucus production.
Cytokine Assays.
BAL fluid was assessed for cytokine content by standard ELISA protocols as described previously 19 using commercially available antibodies or those produced in house. Antibodies 11B11 and biotin-BVD6 (BD PharMingen) were used for IL-4, TRFK5, and biotin-TRFK4 for IL-5, R46A-2, and biotin-XMG1.2 (BD PharMingen) for IFN-γ. Standard curves were constructed with purified IL-4, IL-5, and IFN-γ (supernatants from the respective X63.Ag cell lines). The sensitivity of each assay was similar, with levels of detection being 50–100 pg/ml.
IgE Assay.
Mice were bled at the time of killing, after measurement of AHR. Total IgE was quantitated by ELISA using rabbit anti-IgE, rat anti-IgE, and horseradish peroxidase–conjugated rat anti-IgE as described previously 23. OVA-specific IgE was determined in standard ELISAs by first coating plates with OVA, followed by the secondary anti-IgE antibody. Values were converted to arbitrary units using sera from immunized mice as the standard.
Results and Discussion
The role of OX40 in the allergic inflammatory response in the lung was determined in the murine model of asthma, which is induced by sensitization with the protein OVA. Wt and OX40 knockout (−/−) animals were primed for 4 wk and then challenged once a day for 4 d with aerosolized OVA. This protocol produces a classic asthmatic reaction characterized by high levels of IgE, Th2 cytokine production, eosinophil infiltration in the lungs, mucus production, and development of AHR.
After the last aerosol exposure, the lungs were lavaged and the BAL fluid assessed for the presence of cellular infiltrates by differential cell counting. Control mice that were not rechallenged with OVA had no inflammatory response including the absence of cell infiltrates (data not shown). Wt mice challenged with OVA had three to four times the number of total cells in the BAL fluid compared with OX40-deficient mice challenged with OVA (Fig. 1, left). The predominant infiltrate in Wt mice were eosinophils as demonstrated many times before in this model, with lower numbers of neutrophils and lymphocytes (Fig. 1, right). In striking contrast, the number of eosinophils in the BAL of OX40-deficient animals was dramatically lower, as was the number of lymphocytes, whereas fairly equivalent numbers of neutrophils and monocytes were detected.
Figure 1.

Reduced eosinophilia is associated with allergic inflammation in OX40-deficient mice. Groups of Wt mice (black bars) and OX40−/− mice (white bars) were immunized with 20 μg OVA given intraperitoneally in alum. After 4 wk, each mouse was subjected to an aerosol of 5 mg/ml OVA for 30 min for four consecutive days. Approximately 7 h after the last aerosol, BALs were performed with 1 ml of PBS. The resultant fluid was analyzed for total cell numbers (left) and for numbers of neutrophils (neut), eosinophils (eosin), monocytes (mono), and lymphocytes (lym) by differential cell counting (right). Results are the mean number of cells ± SEM from four separate experiments with four mice per group in each experiment.
There is abundant evidence from many studies that IL-4 plays a major role in development of Th2 cells and induction of the antibody IgE, but whether IL-4 is an important effector molecule in many disease scenarios is largely unknown. In the case of asthma, there is some evidence that IL-4 is involved in the influx of eosinophils 24 25 26, although this has not been reproducibly seen. In contrast, studies of IL-5–deficient animals and T cells have repeatedly shown a major role for this cytokine in eosinophilia 2. To determine the role of OX40 in cytokine production in the lungs, BAL fluid was assessed for the presence of IL-4 and IL-5. In both cases, reduced production was seen in OX40-deficient animals compared with Wt controls (Fig. 2 a), with the predominant effect being on IL-5, which was reduced by 60–70%, directly correlating with the reduction in eosinophil numbers. As an independent assessment of the role of OX40 in development of the Th2 response, serum levels of IgE were determined (Fig. 2 b). Although the role of IgE in the asthmatic response is controversial 27, there is nonetheless a direct correlation between Th2 responses, allergic inflammation, and the production of this antibody 23. As with the reduction in Th2 cytokines, OVA-specific IgE (and total IgE; not shown) in OVA-challenged mice lacking OX40 was <50% of that induced in Wt mice. Thus, OX40 plays a major role in the development of the Th2 response that is seen locally in the lung and also systemically.
Figure 2.
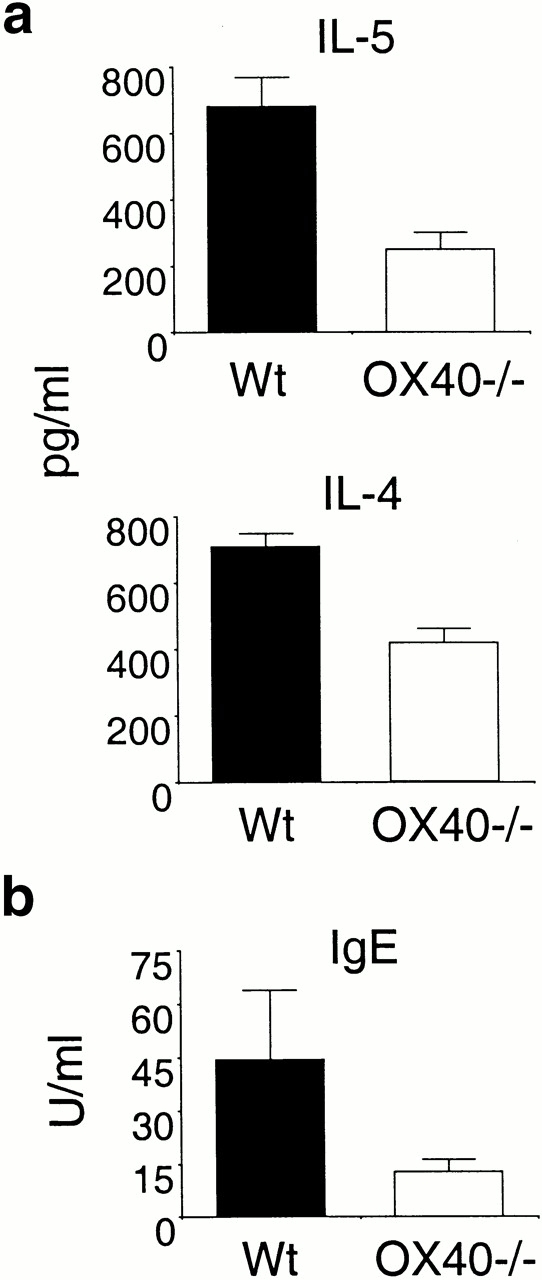
OX40-deficient mice are impaired in development of allergic Th2 responses. Wt and OX40−/− mice were immunized and challenged as in the legend to Fig. 1. (a) BAL fluid was assessed for IL-5 and IL-4 levels by ELISA. (b) Sera from the mice were analyzed for OVA-specific IgE levels by ELISA. Similar data were obtained when measuring total IgE levels (not shown). Results are the mean levels ± SEM from four separate experiments with four mice per group in each experiment.
The hallmark of allergic inflammation in the lungs is enhanced airway reactivity brought about by a combination of cellular infiltration, goblet cell hyperplasia in the bronchiole linings, and overproduction of mucus. To determine if the lack of OX40 resulted in suppressed AHR, mice were exposed to β-methacholine in aerosol form and subjected to analysis with a plethysmograph to determine Penh. Wt mice exhibited pronounced AHR increasing over a range of methacholine concentrations. In contrast, OX40-deficient animals were markedly less responsiveness at the low concentrations, although they were still capable of mounting an enhanced response when exposed to a high dose of methacholine (Fig. 3 a).
Figure 3.
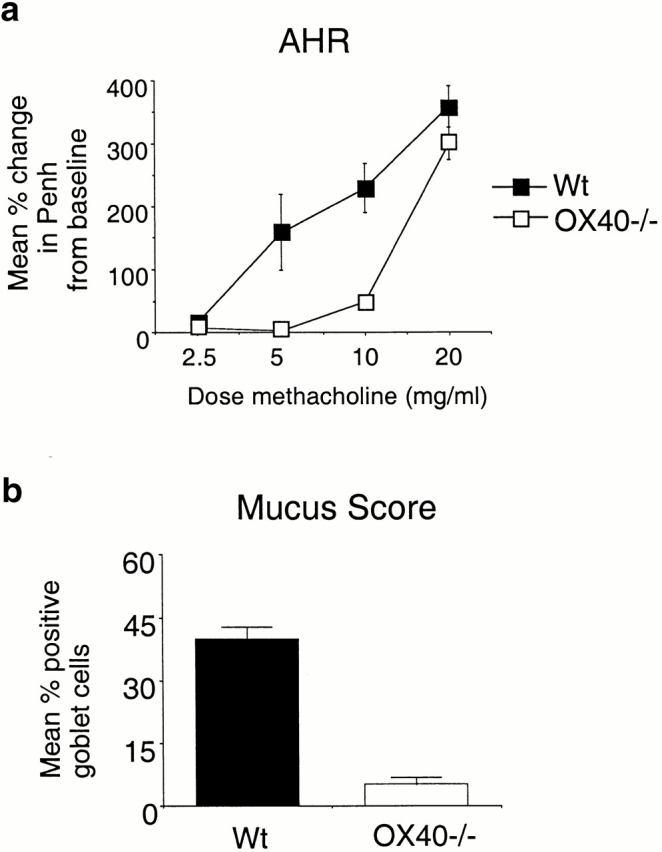
AHR and mucus production is reduced in the absence of OX40. Wt and OX40−/− mice were immunized and challenged as in Fig. 1. (a) 1–3 h after the last aerosol challenge, individual mice were assessed for AHR as described in Materials and Methods. Results are the mean percent change ± SEM in Penh levels above baseline, after exposure to graded doses of methacholine. Values are calculated from four experiments with four mice in each group per experiment. (b) 7 h after the last aerosol challenge, lungs were removed from mice that were not lavaged. Sections were fixed and stained with PAS. Mucus production was assessed by determining the percentage of goblet cells that stained strongly. Lung sections from four mice in each group were counted with at least 1,000 goblet cells analyzed per section, which comprised 70% or more of the total section. Results are the mean percentage of positive cells ± SEM from the four mice.
The lungs from sensitized and challenged mice were removed immediately after testing for AHR and examined histologically by staining for cellular architecture around the bronchioles and for mucus production with PAS base. As seen in Fig. 4, whereas Wt mice had prominent cellular infiltration around the bronchioles, there was substantially less in the OX40-deficient mice, correlating with the reduced numbers of cells, particularly eosinophils, seen in the BAL fluid. Additionally, the extent of hyperplasia of the goblet cells was significantly lower in the absence of OX40, although compared with a mouse that was not challenged with antigen (not shown), some inflammation was still evident. A characteristic feature of asthma pathophysiology is mucus overproduction in the goblet cells and the appearance of mucus plugs within the bronchioles 3. There was a dramatic difference in mucus production, with ∼40% of goblet cells staining strongly with PAS in Wt mice but only ∼5% staining strongly in OX40−/− animals (Fig. 3 b and 4). Additionally, mucus plugs within the bronchioles were common in lungs from Wt mice, whereas few were detectable in lungs from OX40 knockout mice (Fig. 4).
Figure 4.
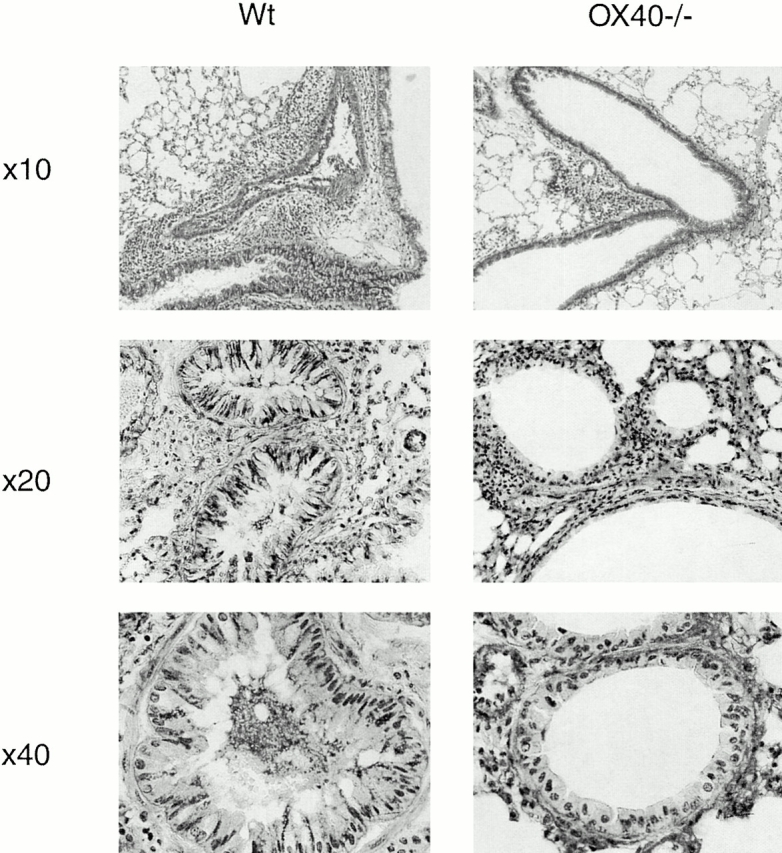
Lung inflammatory infiltrates, goblet cell hyperplasia, and mucus production are reduced in OX40-deficient animals. Wt and OX40−/− mice were immunized and challenged with OVA as in the legend to Fig. 1, and lungs sectioned from mice that did not undergo BAL. Sections were stained with PAS. Original magnifications: Top, ×10; middle, ×20; bottom, ×40. Note that dark areas that are not nuclei represent mucus. Prominent mucus plugs and mucus positive goblet cells are visible in Wt bronchioles in the middle and bottom pictures.
Collectively, these data demonstrate that OX40/OX40L interactions are integral to development of the asthmatic phenotype in the murine model used here and complement previous data that have shown a role for OX40 in generating T cell responses in several disease scenarios. Initial work on OX40 suggested that this molecule may preferentially regulate Th2 responses and suppress development of Th1 responses 15. The demonstration that L. major–induced inflammation in BALB/c mice was inhibited with a blocking reagent to OX40L supported this idea 20, particularly when coupled with studies that suggested that protective Th1 responses to Leishmania in BL/6 mice did not involve OX40 20 21. In contrast, other studies of the helminth parasite N. brasiliensis showed that OX40 was not absolutely necessary for development of a Th2 response 21, and subsequent studies from ourselves and others have shown that OX40 can be equally involved in priming for Th1 cytokines in many instances 16 17 19. In the study here, the lack of the asthmatic phenotype in OX40−/− mice could have been brought about by defective priming of Th2 cells, and/or by enhancement of Th1 cytokines that could directly antagonize Th2 associated pathology 28. Because we found no evidence for elevated levels of IFN-γ in BAL samples (data not shown), and both IL-5 and IL-4 production were reduced (Fig. 2), this argues against default development of Th1 cells when OX40 signals are absent.
In summary, the data in this paper conclusively show that OX40 ligation is integral to full development of allergic asthma in mice and adds to the growing list of responses that are controlled by OX40. Earlier reports that CD28/B7 interactions are also involved in allergic asthma 7 8 9 complement basic studies of T cell responses which have suggested that activation, expansion, and effector function are controlled by the synergistic action of multiple costimulatory receptors (for reviews, see references 12 and 13). We have proposed that OX40 functions to regulate the number of T cells that persist over time 19, with OX40 potentially acting both in synergy with CD28 and independently 14 29. In contrast, the predominant action of CD28 is to control the number of T cells that are initially generated 30, and because of its constitutive expression, CD28 obviously provides signals earlier than OX40, which has to be induced. Blocking one interaction in isolation has proved effective in suppressing development of the allergic inflammatory response, but significantly, none of the studies of CD28/B7 or our study here with OX40 have completely prevented all of the asthmatic symptoms. Therapies which use a combination approach to target CD28 and OX40 together may prove effective in completely preventing the development of allergen-specific T cells.
Acknowledgments
The authors would like to thank Nigel Killeen for originally making available the OX40-deficient mice.
This work was funded by a grant from the Sandler Program for Asthma Research awarded to Michael Croft. This is manuscript no. 407 from the La Jolla Institute for Allergy and Immunology.
References
- Coyle A.J., Le Gros G., Bertrand C., Tsuyuki S., Heusser C.H., Kopf M., Anderson G.P. Interleukin-4 is required for the induction of lung Th2 mucosal immunity. Am. J. Respir. Cell Mol. Biol. 1995;13:54–59. doi: 10.1165/ajrcmb.13.1.7598937. [DOI] [PubMed] [Google Scholar]
- Foster P.S., Hogan S.P., Ramsay A.J., Matthaei K.I., Young I.G. Interleukin 5 deficiency abolishes eosinophilia, airways hyperreactivity, and lung damage in a mouse asthma model. J. Exp. Med. 1996;183:195–201. doi: 10.1084/jem.183.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunig G., Warnock M., Wakil A.E., Venkayya R., Brombacher F., Rennick D.M., Sheppard D., Mohrs M., Donaldson D.D., Locksley R.M., Corry D.B. Requirement for IL-13 independently of IL-4 in experimental asthma. Science. 1998;282:2261–2263. doi: 10.1126/science.282.5397.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn L., Homer R.J., MacLeod H., Mohrs M., Brombacher F., Bottomly K. Th2-induced airway mucus production is dependent on IL-4Ralpha, but not on eosinophils. J. Immunol. 1999;162:6178–6183. [PubMed] [Google Scholar]
- Townsend M.J., Fallon P.G., Matthews D.J., Smith P., Jolin H.E., McKenzie A.N. IL-9-deficient mice establish fundamental roles for IL-9 in pulmonary mastocytosis and goblet cell hyperplasia but not T cell development. Immunity. 2000;13:573–583. doi: 10.1016/s1074-7613(00)00056-x. [DOI] [PubMed] [Google Scholar]
- Gavett S.H., Chen X., Finkelman F., Wills-Karp M. Depletion of murine CD4+ T lymphocytes prevents antigen-induced airway hyperreactivity and pulmonary eosinophilia. Am. J. Respir. Cell Mol. Biol. 1994;10:587–593. doi: 10.1165/ajrcmb.10.6.8003337. [DOI] [PubMed] [Google Scholar]
- Keane-Myers A., Gause W.C., Linsley P.S., Chen S.J., Wills-Karp M. B7-CD28/CTLA-4 costimulatory pathways are required for the development of T helper cell 2-mediated allergic airway responses to inhaled antigens. J. Immunol. 1997;158:2042–2049. [PubMed] [Google Scholar]
- Padrid P.A., Mathur M., Li X., Herrmann K., Qin Y., Cattamanchi A., Weinstock J., Elliott D., Sperling A.I., Bluestone J.A. CTLA4Ig inhibits airway eosinophilia and hyperresponsiveness by regulating the development of Th1/Th2 subsets in a murine model of asthma. Am. J. Respir. Cell Mol. Biol. 1998;18:453–462. doi: 10.1165/ajrcmb.18.4.3055. [DOI] [PubMed] [Google Scholar]
- Mark D.A., Donovan C.E., De Sanctis G.T., Krinzman S.J., Kobzik L., Linsley P.S., Sayegh M.H., Lederer J., Perkins D.L., Finn P.W. Both CD80 and CD86 co-stimulatory molecules regulate allergic pulmonary inflammation. Int. Immunol. 1998;10:1647–1655. doi: 10.1093/intimm/10.11.1647. [DOI] [PubMed] [Google Scholar]
- Hogan S.P., Mould A., Kikutani H., Ramsay A.J., Foster P.S. Aeroallergen-induced eosinophilic inflammation, lung damage, and airways hyperreactivity in mice can occur independently of IL-4 and allergen-specific immunoglobulins. J. Clin. Invest. 1997;99:1329–1339. doi: 10.1172/JCI119292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei X.F., Ohkawara Y., Stampfli M.R., Mastruzzo C., Marr R.A., Snider D., Xing Z., Jordana M. Disruption of antigen-induced inflammatory responses in CD40 ligand knockout mice. J. Clin. Invest. 1998;101:1342–1353. doi: 10.1172/JCI1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croft M., Dubey C. Accessory molecule and costimulation requirements for CD4 T cell response. Crit. Rev. Immunol. 1997;17:89–118. doi: 10.1615/critrevimmunol.v17.i1.40. [DOI] [PubMed] [Google Scholar]
- Watts T.H., DeBenedette M.A. T cell co-stimulatory molecules other than CD28. Curr. Opin. Immunol. 1999;11:286–293. doi: 10.1016/s0952-7915(99)80046-6. [DOI] [PubMed] [Google Scholar]
- Gramaglia I., Weinberg A.D., Lemon M., Croft M. OX40 liganda potent costimulatory molecule for sustaining primary CD4 T cell responses. J. Immunol. 1998;161:6510–6517. [PubMed] [Google Scholar]
- Flynn S., Toellner K.M., Raykundalia C., Goodall M., Lane P. CD4 T cell cytokine differentiationthe B cell activation molecule, OX40 ligand, instructs CD4 T cells to express interleukin 4 and upregulates expression of the chemokine receptor, blr-1. J. Exp. Med. 1998;188:297–304. doi: 10.1084/jem.188.2.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen A.I., McAdam A.J., Buhlmann J.E., Scott S., Lupher M.L., Jr., Greenfield E.A., Baum P.R., Fanslow W.C., Calderhead D.M., Freeman G.J., Sharpe A.H. Ox40-ligand has a critical costimulatory role in dendritic cell:T cell interactions. Immunity. 1999;11:689–698. doi: 10.1016/s1074-7613(00)80143-0. [DOI] [PubMed] [Google Scholar]
- Kopf M., Ruedl C., Schmitz N., Gallimore A., Lefrang K., Ecabert B., Odermatt B., Bachmann M.F. OX40-deficient mice are defective in Th cell proliferation but are competent in generating B cell and CTL responses after virus infection. Immunity. 1999;11:699–708. doi: 10.1016/s1074-7613(00)80144-2. [DOI] [PubMed] [Google Scholar]
- Murata K., Ishii N., Takano H., Miura S., Ndhlovu L.C., Nose M., Noda T., Sugamura K. Impairment of antigen-presenting cell function in mice lacking expression of OX40 ligand. J. Exp. Med. 2000;191:365–374. doi: 10.1084/jem.191.2.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gramaglia I., Jember A., Pippig S.D., Weinberg A.D., Killeen N., Croft M. The OX40 costimulatory receptor determines the development of CD4 memory by regulating primary clonal expansion. J. Immunol. 2000;165:3043–3050. doi: 10.4049/jimmunol.165.6.3043. [DOI] [PubMed] [Google Scholar]
- Akiba H., Miyahira Y., Atsuta M., Takeda K., Nohara C., Futagawa T., Matsuda H., Aoki T., Yagita H., Okumura K. Critical contribution of OX40 ligand to T helper cell type 2 differentiation in experimental leishmaniasis. J. Exp. Med. 2000;191:375–380. doi: 10.1084/jem.191.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pippig S.D., Pena-Rossi C., Long J., Godfrey W.R., Fowell D.J., Reiner S.L., Birkeland M.L., Locksley R.M., Barclay A.N., Killeen N. Robust B cell immunity but impaired T cell proliferation in the absence of CD134 (Ox40) J. Immunol. 1999;163:6520–6529. [PubMed] [Google Scholar]
- Hamelmann E., Schwarze J., Takeda K., Oshiba A., Larsen G.L., Irvin C.G., Gelfand E.W. Noninvasive measurement of airway responsiveness in allergic mice using barometric plethysmography. Am. J. Respir. Crit. Care Med. 1997;156:766–775. doi: 10.1164/ajrccm.156.3.9606031. [DOI] [PubMed] [Google Scholar]
- Zuberi R.I., Apgar J.R., Chen S.S., Liu F.T. Role for IgE in airway secretionsIgE immune complexes are more potent inducers than antigen alone of airway inflammation in a murine model. J. Immunol. 2000;164:2667–2673. doi: 10.4049/jimmunol.164.5.2667. [DOI] [PubMed] [Google Scholar]
- Brusselle G., Kips J., Joos G., Bluethmann H., Pauwels R. Allergen-induced airway inflammation and bronchial responsiveness in wild-type and interleukin-4-deficient mice. Am. J. Respir. Cell Mol. Biol. 1995;12:254–259. doi: 10.1165/ajrcmb.12.3.7873190. [DOI] [PubMed] [Google Scholar]
- Cohn L., Homer R.J., Marinov A., Rankin J., Bottomly K. Induction of airway mucus production by T helper 2 (Th2) cellsa critical role for interleukin 4 in cell recruitment but not mucus production. J. Exp. Med. 1997;186:1737–1747. doi: 10.1084/jem.186.10.1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn L., Tepper J.S., Bottomly K. IL-4-independent induction of airway hyperresponsiveness by Th2, but not Th1, cells. J. Immunol. 1998;161:3813–3816. [PubMed] [Google Scholar]
- Hamelmann E., Tadeda K., Oshiba A., Gelfand E.W. Role of IgE in the development of allergic airway inflammation and airway hyperresponsiveness—a murine model. Allergy. 1999;54:297–305. doi: 10.1034/j.1398-9995.1999.00085.x. [DOI] [PubMed] [Google Scholar]
- Lack G., Bradley K.L., Hamelmann E., Renz H., Loader J., Leung D.Y., Larsen G., Gelfand E.W. Nebulized IFN-gamma inhibits the development of secondary allergic responses in mice. J. Immunol. 1996;157:1432–1439. [PubMed] [Google Scholar]
- Akiba H., Oshima H., Takeda K., Atsuta M., Nakano H., Nakajima A., Nohara C., Yagita H., Okumura K. CD28-independent costimulation of T cells by OX40 ligand and CD70 on activated B cells. J. Immunol. 1999;162:7058–7066. [PubMed] [Google Scholar]
- June C.H., Bluestone J.A., Nadler L.M., Thompson C.B. The B7 and CD28 receptor families. Immunol. Today. 1994;15:321–331. doi: 10.1016/0167-5699(94)90080-9. [DOI] [PubMed] [Google Scholar]