

Abstract
Apoptotic and mitogenic stimuli activate c-Jun NH2-terminal kinases (JNKs) in T cells. Although T cells express both JNK1 and JNK2 isozymes, the absence of JNK2 alone can result in resistance to anti-CD3–induced thymocyte apoptosis and defective mature T cell proliferation. Similar defects in thymocyte apoptosis and mature T cell proliferation, the latter due to reduced interleukin 2 production, are also caused by JNK1 deficiency. Importantly, T cell function was compromised in Jnk1 +/−Jnk2 +/− double heterozygous mice, indicating that JNK1 and JNK2 play similar roles in regulating T cell function. The reduced JNK dose results in defective c-Jun NH2-terminal phosphorylation in thymocytes but not in peripheral T cells, in which nuclear factors of activated T cells (NK-ATs)–DNA binding activity is affected. Thus, JNK1 and JNK2 control similar functions during T cell maturation through differential targeting of distinct substrates.
Keywords: apoptosis, JNK1, JNK2, proliferation, T lymphocyte
Introduction
The c-Jun NH2-terminal kinase (JNK) signaling pathway is activated by a plethora of stimuli resulting in the potentiation of the activity of transcription factors such as c-Jun and activating transcription factor 2 (ATF2) in various cell types, which in turn regulates diverse biological processes. JNK signaling has also been implicated in multiple T cell functions. In general, T cell activation involves several steps, including the induction of immediate early genes, followed by the induction of IL-2, a growth factor whose secretion leads to autocrine and paracrine T cell proliferation 1. The JNKs are synergistically activated by costimulation of the TCR with antibodies to its CD3 component and the CD28 auxiliary receptor or by combined treatment with PMA and Ca2+ ionophore 2 3 4. This synergistic JNK activation response is unique to T and B cells 2 3 5 and was shown to correlate with IL-2 gene induction in the T cell tumor line Jurkat 2. Anergic T cells were shown to exhibit a defective JNK activation response 6 7.
The importance of JNK activity for T cell activation and IL-2 production is also suggested by the role of AP-1 dimers, composed of Jun and Fos subunits. Besides the binding to its own recognition sites, AP-1 can cooperate with the Ca2+-responsive, cyclosporin A–sensitive nuclear factor of activated T cells (NF-ATc), stabilizing the binding to the composite nuclear factors of activated T cells (NF-ATs) recognition element in the IL-2 promoter 8. In nonstimulated T cells, basal levels of AP-1 proteins are low but T cell activation results in rapid induction of jun and fos genes 2 9. This process is partially dependent on JNK activation 2 and consistent with the defect in JNK activation, anergic T cells exhibit a deficient AP-1 transcriptional response 10. In addition to stimulating preexisting transcription factors, such as c-Jun, ATF2, and Elk-1 involved in c-jun and c-fos induction, the JNKs phosphorylate newly synthesized Jun proteins and enhance their ability to activate transcription 11 12. Full induction of IL-2 expression also requires the stabilization of its mRNA, which in nonstimulated T cells has a very short half-life 13. Recent studies indicate that the JNKs are also involved in activation-induced IL-2 mRNA stabilization 14. Analysis of this process in Jurkat cells indicates that this function is unique to the JNKs and that neither extracellular signal–regulated kinases nor the p38s have such a role.
Although the loss of c-Jun, a classical JNK target, results in embryonic lethality impeding further immunological analysis 15, c-jun − /− lymphoid cells were generated by the recombination activating gene 2 (RAG2) complementation which showed incomplete restoration of thymocytes 16. Nonetheless, the importance of JNK-mediated phosphorylation of c-Jun in T cell functions has not been critically addressed. Besides c-Jun, the role of NF-ATc components (NF-ATc1–c4) in T cell functions has been extensively addressed. NF-ATc1−/− T cells show a decrease in activation-induced proliferation 17, whereas the absence of NF-ATc2 and NF-ATc3 results in the hyperactivation of peripheral T cells 18 19. Moreover, the overexpression of dominant negative (dn) NF-AT proteins in T cells results in markedly reduced IL-2 production 20, indicating that NF-ATc1 is a positive mediator of T cell activation. On the other hand, NF-ATc3 was shown to be a target of JNK that negatively regulates NF-ATc3 translocation to the nucleus in Jurkat cells 21.
JNK1 and JNK2 are ubiquitously expressed in adult mice, whereas JNK3 expression is mostly restricted to the brain 22. Although the predominant isoform of JNK2 exhibits higher affinity to c-Jun than the predominant JNK1 isoform in vitro, other isoforms of JNK1 exhibit high affinity to c-Jun while certain JNK2 isoforms generated by alternate splicing display low affinity towards c-Jun 11 23 24. To understand the physiological functions, knockout mice lacking individual Jnk genes and transgenic mice expressing a dn Jnk1 gene have been generated. The absence of JNK2 and JNK1 resulted in impaired T cell activation, although T and B lymphocyte development was unaltered 25 26 27. JNK2 was also found to be required for IFN-γ production by Th1 cells 26. Moreover, activation-induced thymocyte death was affected, as Jnk2 −/− double positive (DP) thymocytes were resistant to apoptosis induced by the pan-T cell–activating anti-CD3 antibody 25. These data suggested a role for JNK2 in T cell activation that is not fully compensated for by other JNK isoforms. JNK1 was shown to negatively regulate the production of Th2 cytokines, and this altered response of Th cells was attributed to increased nuclear accumulation of NF-ATs 27. In addition, Jnk1 −/− T cells were found to be hyperproliferative, whereas the expression of dn Jnk1 was shown to result in reduced T cell proliferation 27 28. Surprisingly, no increase in IL-2 production was found associated with increased proliferation of Jnk1 −/− T cells. We also generated mice lacking JNK1 to define the role of JNK1 in both T cell proliferation and apoptosis. No defects in T and B cell development were observed in mice lacking JNK1; however, these mice exhibited impaired activation of mature T cells, leading to reduced IL-2 production and proliferation which was rescued by exogenous IL-2. As shown for JNK2, immature thymocytes lacking JNK1 are resistant to anti-CD3–induced apoptosis, suggesting that JNK1 and JNK2 have similar functions in T cells. Importantly, Jnk1 +/−Jnk2 +/− double heterozygous mice exhibit essentially the same defects in T cell function as Jnk1 − /− or Jnk2 − /− single homozygous mice. In addition, we find that in mature T cells, JNK activity is required for the induction of NF-AT–DNA binding activity instead of being involved in c-Jun NH2-terminal phosphorylation.
Materials and Methods
Generation of Jnk1−/− Mice by Gene Targeting.
A targeting construct for the inactivation of the Jnk1 locus was made by subcloning a HindIII-XmnI fragment containing sequences encoding the first 73 amino acids of JNK1 in-frame with the lacZ gene in the pGNA vector. The diphtheria toxin A gene was used as a negative selection marker together with the MCI promoter in pGNA containing Jnk1 sequences 25. E14.1 embryonic stem (ES) cells were electroporated with Pme1-linearized Jnk1 targeting construct, selected in the presence of 300 μg/ml G418, and screened by Southern blotting for homologous recombination 25. DNA from ES cells was digested with HindIII and probed with a 1.8-kb Xba1 fragment (see Fig. 1 a). The probe detects a 7.5-kb fragment from the wild-type allele and a 9.5-kb fragment from the targeted allele. Targeted ES cell clones were injected into blastocysts of C57BL/6J mice, and the chimeras were transmitted efficiently to the germline. Heterozygous mice were interbred to obtain Jnk1 − /− mice, which were genotyped by PCR using the following primers: 5′-cacatacactcagtggatct-3′ and 5′-cactattgccttaagactcc-3′, which detect the wild-type allele, and 5′-cacatacactcagtggatct-3′ and 5′-ctgatcttccagataactgc-3′, which detect the mutant allele. For all experiments, sex-matched littermates of Jnk1 +/− intercrosses were used, and mice were handled according to the Austrian laws governing the use of animals for research.
Figure 1.

Targeted mutation of the murine Jnk1 gene in ES cells. (a) The structure of Jnk1 genomic DNA encoding a portion of the JNK1 protein kinase domain (top). The targeting vector (middle) replaced parts of exon 2 with a β-galactosidase gene fused in-frame with exon 2 and a Neor cassette driven by the Rous sarcoma virus promoter. The structure of the targeted allele after homologous recombination is depicted (bottom). DT-A, diphtheria toxin A gene used for negative selection. (b) Genotyping of offspring of mice heterozygous for the targeted Jnk1 mutation. Tail DNA was digested with HindIII and analyzed by Southern blot analysis using the probe indicated in panel a. (c) Western blot analysis of JNK protein expression in primary embryonic fibroblasts. Whole cell extracts (100 μg protein) from wild-type (+/+), heterozygous (+/−), and homozygous mutant (−/−) embryonic fibroblasts were separated by SDS-PAGE, transferred to a membrane, and probed with anti-JNK antibodies. The positions of the long (L) and short (S) isoforms of the JNK proteins are indicated.
Western Blot Analysis and JNK Assay.
Western blot analysis was performed using 100 μg of whole cell extracts of primary embryonic fibroblasts, splenic, lymph node, and thymic cells according to standard procedures. Cell extracts were prepared as described 23, separated on a 10% SDS-PAGE, transferred onto Immobilon-P membrane (Millipore), and probed with anti-JNK1 (333.8), anti-JNK (666.8; BD PharMingen), anti–c-Jun (Transduction Laboratories), and anti–phospho-c-Jun (S63; a gift from Drs. D. Lallemand and M. Yaniv, Pasteur Institute, Paris, France) antibodies. Kinase assays were performed using 60 μg of whole cell extracts and glutathione S-transferase (GST)–c-Jun (1-79) as a substrate and quantified on a Molecular Dynamics PhosphorImager. Relative JNK activity is represented by pixel values obtained by quantification of the intensity of the signal. Parallel samples were analyzed for protein expression by immunoblotting with the above antibodies as well as an antibody to actin (Sigma-Aldrich).
Flow Cytometric Analysis.
Single cell suspensions of thymi and spleens were prepared, and 106 cells were stained in PBS containing 1% FCS for 1 h at 4°C. Monoclonal antibodies used for staining were FITC-conjugated anti-CD8, anti-IgM, anti-CD43, anti–IL-2Rα, and PE-conjugated anti-B220, anti-CD4, and anti-CD3 (all from BD PharMingen). Analyses were performed on a FACScan™ flow cytometer (Becton Dickinson) using CELL Quest™ software.
Proliferation Assays and ELISA.
Total spleen and lymph node cells as well as T cells purified from spleens were used for the proliferation assays. Splenic T cells were purified by FACS® sorting using anti-CD4 and anti-CD8 antibodies. Cells were plated in 96-well plates, precoated with anti-CD3ε antibody for 2 h at 37°C, in the presence or absence of anti-CD28 antibody (BD PharMingen). Cells were also cultured in the absence or presence of 5 μg/ml concanavalin A. After 60 h, cultures were pulsed for 10–12 h with 1 μCi of [3H]thymidine/well, and cells were subsequently harvested and analyzed by standard procedures. T lymphocytes stimulated with anti-CD3ε antibody for 24 h were harvested, and the supernatants were used to measure IL-2 levels by ELISA (R&D Systems). 50 U/ml IL-2 was added to cells for proliferation assays when indicated. B cell proliferation was analyzed by the stimulation of B220+ spleen cells with 50 U/ml IL-4, 1 μg/ml soluble anti-CD40, or 10 μg/ml LPS. The magnitude of stimulated [3H]thymidine incorporation was used as an indicator of cell proliferation. The results are representative of at least three independent experiments, each using three pairs of mice. All experiments were performed in triplicates.
In Vivo Thymocyte Death Assay.
12-wk-old wild-type (Jnk1 +/+) and Jnk1 −/− mice were injected with either PBS or various doses of purified anti-CD3ε antibody (BD PharMingen) intraperitoneally. After 48 h, mice were killed and thymi were removed. Single cell suspensions were prepared and subjected to flow cytometric analysis after staining with the appropriate antibodies.
In Vitro Apoptosis Assays.
Freshly isolated thymocytes were cultured in DMEM containing 10% FCS, glutamine, pyruvate, penicillin, and streptomycin and were plated at 5 × 105 cells/ml in each well of a 24-well dish. For anti-CD3ε–induced cell death, wells were precoated with various concentrations of the antibody for 1 h at 37°C, washed three times in PBS, and thymocytes were subsequently cultured for the indicated periods in the presence of 1 μg/ml anti-CD28. Apoptosis was also induced by the addition of various concentrations of anti-Fas antibody (Jo-2 clone; BD PharMingen), dexamethasone (Fluka), or by UVC irradiation (40 or 80 J/m2) using a Stratagene cross-linker. After incubation for the indicated periods at 37°C in a 5% CO2 atmosphere, the cells were harvested and stained with FITC-conjugated annexin V (BD PharMingen) and propidium iodide and analyzed with a FACScan™ cytometer as described above. Thymocytes cultured in medium alone served as controls to measure spontaneous rate of cell death. Thymocyte viability is expressed as the percentage of viable thymocytes treated with apoptotic stimuli over viability of untreated thymocytes. The results are representative of four independent experiments, each using three pairs of mice. All experiments were performed in duplicates.
Nuclear Extracts and Electrophoretic Mobility Shift Assay.
Nuclear extracts were prepared from 107 spleen and lymph node cells as described 12. Binding reactions were conducted using the indicated amount of nuclear extracts and 3 × 104 cpm of 32P end-labeled double stranded oligonucleotides. The double stranded oligonucleotides used in the mobility shift assay were from the human osteocalcin AP-1 site (5′-GGGTGACTCACCGGGTGAA-3′) and from the distal NF-AT binding site of the human IL-2 promoter (5′-gatcggaggaaaaactgtttcatacagaaggcgt-3′) and human IL-4 promoter (5′-GTAATAAAATTTTCCAATGTAAA-3′). For supershift experiments, 2 μg of anti–c-Jun (SC-44; Santa Cruz Biotechnology, Inc.) and anti–NF-ATc1 and NF-ATc2 (a gift of Dr. G. Crabtree, Stanford Medical School, Stanford, CA) were added to the reactions.
Results
Generation of JNK1-deficient Mice.
The Jnk1 gene was inactivated by homologous recombination in ES cells using a targeting construct that lacks parts of the kinase domain (Fig. 1 a). Heterozygous mice were intercrossed to produce homozygous animals (Fig. 1 b), which were viable and exhibited no anomalies at birth. Western blot analysis confirmed the absence of both JNK1 long and short isoforms in primary embryonic fibroblasts (Fig. 1 c). Both JNK2 isoforms were present at similar levels in wild-type, Jnk1 +/−, and Jnk1 −/− cells (Fig. 1 c). These results indicate that the targeted mutation of the Jnk1 gene results in the complete absence of a functional gene product.
Lymphocyte Development Is Not Altered in the Absence of JNK1.
Although JNK signaling was suggested to be involved in the development of both T and B lymphocytes 29 30, neither was altered in the absence of JNK1 (data not shown). Thymi of Jnk1 −/− mice had normal numbers of CD4+CD8+, CD4+CD8−, CD4−CD8+, and CD4−CD8− cells (data not shown). Moreover, Jnk1 − /− splenic cells were comparable to wild-type cells as determined by staining with antibodies to CD4, CD8, CD3, heat stable antigen (HSA), TCR-α/β, TCR-γ/δ, Thy1.2, CD25, CD44, and B220 (data not shown). Cell numbers were also comparable between wild-type and mutant spleens, thymi, and lymph nodes.
Lack of JNK1 Results in Increased Resistance to Anti-CD3–induced Thymocyte Apoptosis In Vivo.
We next determined if T cell function is altered in the absence of JNK1. Both Jnk1 − /− and wild-type mice were injected intraperitoneally with various doses of the anti-CD3ε antibody, which is a potent activator of T cells, and analyzed for depletion of DP CD4+CD8+ thymocytes. Wild-type mice injected with 10 μg of anti-CD3 contained ∼55% DP cells compared with PBS-treated mice which had ∼89% DP cells, 48 h after injection (Table ). By contrast, Jnk1 − /− mice contained almost similar levels of DP cells (81%) compared with PBS-treated mice (87%; Table ). Injection of higher doses of the antibody resulted in increased thymocyte cell death both in wild-type and Jnk1 − /− mice. However, Jnk1 − /− mice were more tolerant to intermediate doses of the antibody, with ∼56% DP remaining after injection of 30 μg antibody, compared with only 38% survival in wild-type mice (Table ). At the highest dose of 60 μg antibody almost all the DP cells were dead (3% left) in wild-type mice, whereas ∼10% of the DP cells remained in Jnk1 − /− mice. Thus, absence of JNK1 confers partial resistance to anti-CD3 antibody–induced thymocyte apoptosis in vivo, as was found with Jnk2 − /− mice 25.
Table 1.
JNK1-deficient Thymocytes Are Resistant to Cell Death Induced by Anti-CD3 Antibody In Vivo
| Percent surviving CD4+CD8+ thymocytes | ||
|---|---|---|
| Treatment | JNK1+/+ | JNK1−/− |
| PBS | 89 ± 5 | 87 ± 6 |
| Anti-CD3 10 μg | 55 ± 7 | 81 ± 7 |
| Anti-CD3 30 μg | 38 ± 8 | 56 ± 5 |
| Anti-CD3 60 μg | 3 ± 3 | 10 ± 4 |
Lack of JNK1 results in the increased resistance of immature thymocytes to anti-CD3 antibody–induced cell death. 12-wk-old wild-type and Jnk1 −/− mice were injected with PBS or indicated amounts of anti-CD3 antibody and thymi were removed for analysis after 48 h. Single cell suspensions of thymocytes were stained with anti-CD4 and anti-CD8 fluorescent antibody conjugates and analyzed by flow cytometry. The results are representative of at least five mice per group.
Thymocyte Survival in Response to Various Apoptotic Stimuli In Vitro.
We investigated if the observed resistance of Jnk1 − /− thymocytes to anti-CD3 antibody–induced cell death in vivo is a cell-autonomous defect. Thymocytes from wild-type and Jnk1 −/− mice were incubated in vitro with anti-CD3 antibody, in the presence of anti-CD28 antibody that provides the auxiliary signal, and cell viability was determined. Wild-type thymocytes underwent cell death in a dose-dependent manner, when assayed at 24 h (Fig. 2 a). By comparison, Jnk1 −/− thymocytes were partially resistant to anti-CD3–induced cell death (Fig. 2 a). At the highest concentration of the antibody (100 μg/ml), 70% of wild-type thymocytes were viable compared with 85% of Jnk1 −/− thymocytes. A similar pattern was observed at lower doses of the anti-CD3 antibody: 77% of wild-type cells were alive compared with 92% live Jnk1 −/− cells at 10 μg/ml of anti-CD3; and 88% viability of wild-type cells compared with 98% viability of Jnk1 −/− cells at 1 μg/ml of antibody (Fig. 2 a). Jnk1 −/− thymocytes were still more resistant than their wild-type counterparts even after 48 h of incubation with anti-CD3 antibody. At this time point, ∼40% of the wild-type thymocytes incubated with 100 μg/ml of antibody were alive, compared with 62% of the Jnk1 −/− thymocytes (Fig. 2 b). This trend was also observed at a lower dose of antibody: 59% of wild-type thymocytes were viable compared with 76% viable Jnk1 −/− thymocytes (Fig. 2 b). These results were consistently and reproducibly obtained in many experiments. Nonetheless, the resistance to anti-CD3–induced death observed in vitro is not as extensive as the resistance observed in vivo, suggesting that the in vivo effect could be a result of both direct and indirect effects of the anti-CD3 treatment.
Figure 2.
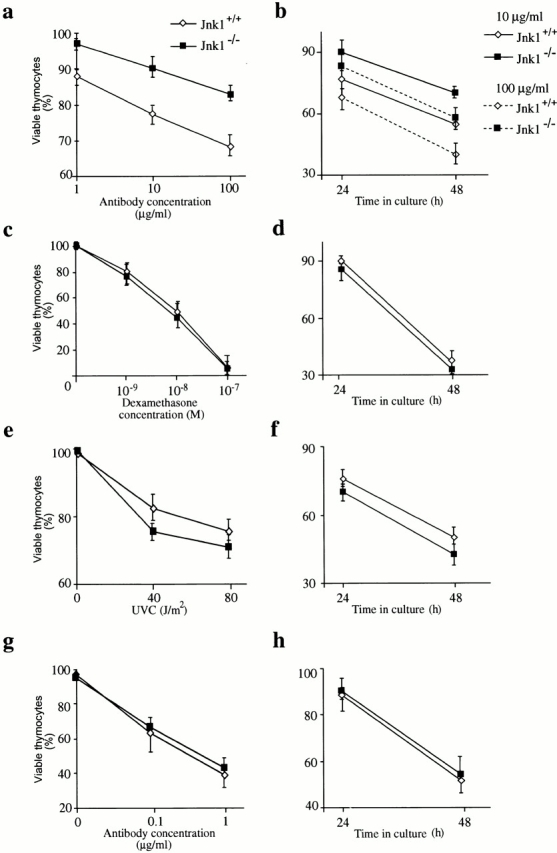
In vitro susceptibility of Jnk1 − /− thymocytes to death caused by direct incubation with anti-CD3 antibody and other apoptotic stimuli. (a–b) Thymocytes from wild-type (⋄) and Jnk1 − /− (▪) mice were cultured on 24-well plates coated with anti-CD3ε antibody for (a) 24 h or (b) the indicated time periods in the presence of the indicated concentrations of soluble anti-CD28 antibody, and cell viability was determined. There was no difference in spontaneous (i.e., basal) cell death rates between wild-type and Jnk1 − /− thymocytes. (c–h) Cell death was determined after treatment of thymocytes with: (c) various concentrations of dexamethasone; (e) various doses of UVC irradiation; (g) incubation with various concentrations of anti-Fas antibody for 24 h; (d) 10−9 M dexamethasone; (f) 80 J/m2 of UVC irradiation; or (h) 0.05 μg/ml of anti-Fas antibody for the indicated time periods. These results are representative of four independent experiments, each using three pairs of mice. All assays were conducted in triplicates. Standard deviations are indicated by vertical lines.
As JNK signaling was suggested to be generally involved in responses to apoptotic stimuli, we determined the response of wild-type and Jnk1 −/− thymocytes to other apoptosis-inducing agents. There was no appreciable difference in viability between wild-type and Jnk1 −/− thymocytes over various doses of dexamethasone, anti-Fas antibody, and UVC radiation regardless of the time point after treatment at which the cells were analyzed (Fig. 2c–h). Therefore, like JNK2, JNK1 is involved in transducing cell death signals in response to the anti-CD3 antibody but is not required in response to other proapoptotic stimuli.
Activation of Peripheral T Cells, but Not B Cells, Is Impaired in the Absence of JNK1.
It has been reported that cytokine production by activated peripheral T cells can induce the expression of cell death mediators or permissive factors that will result in DP thymocyte death in vivo 31. As the magnitude of anti-CD3–induced cell death in vitro was lower than what was observed in vivo, we examined if peripheral T cell activation was affected by the absence of JNK1. Total spleen cells, lymph node cells, and purified splenic T cells were stimulated with various doses of plate-bound anti-CD3 and soluble anti-CD28 antibodies or concanavalin A, and the proliferation rates were determined. Both wild-type and Jnk1 −/− total spleen cells proliferated vigorously and no significant differences were observed over a broad range of anti–CD3 antibody concentrations (Fig. 3 a). However, lymph node cells from Jnk1 −/− mice exhibited a proliferation defect that was apparent only at lower doses of the activating stimuli (Fig. 3 b). Purified splenic T cells from Jnk1 −/− mice also exhibited a proliferation defect which was present at all concentrations of stimuli (Fig. 3 c).
Figure 3.



Inefficient proliferation and IL-2 production by T cells lacking JNK1. (a–c) Cells from (a) spleen, (b) lymph nodes, or (c) purified splenic T cells, all from 4–8-wk-old Jnk1 +/+ or Jnk1−/− mice, were cultured in the presence of the indicated concentrations of plate-bound anti-CD3ε antibody in the absence or presence of the indicated amounts of anti-CD28 antibody. After 60 h, cells were pulsed for 12 h with 1 μCi [3H]thymidine/well and collected for measurement of [3H]thymidine incorporation into DNA. (d) IL-2 production was measured by ELISA on media collected 24 h after stimulation of purified splenic T cells as described above. (e) Proliferation rates of purified T cells cultured in the presence of 50 U/ml IL-2 plus anti-CD3 and/or anti-CD28 antibodies. Proliferation rates of splenic B cells cultured in medium alone (untreated), or in the presence of either 50 U/ml IL-4 plus 1 μg/ml soluble anti-CD40 or 10 μg/ml LPS. The results are representative of four independent experiments, each using three matched pairs of mice. All assays were conducted in triplicates. Standard deviations are shown as vertical lines.
The levels of IL-2 produced 24 h after T cell activation from purified Jnk1 −/− splenic T cells were significantly reduced at lower doses of the activating anti-CD3 and anti-CD28 antibodies (Fig. 3 d). However, the levels of IL-2 production by both wild-type and Jnk1 −/− T cells were comparable at the highest dose of the activating stimuli (Fig. 3 d). These data correlate with the proliferation data, suggesting that the proliferation defect is due to insufficient production of IL-2. Indeed, whereas the IL-2 receptor α chain was expressed at similar levels in both wild-type and Jnk1 − /− cells (data not shown), the addition of exogenous IL-2 completely suppressed the proliferation defect of Jnk1 −/− T cells (Fig. 3 e).
As JNK signaling was suggested to be involved in B cell activation 32, we determined if JNK1-deficient B cells were impaired in their response to activating stimuli. Proliferation rates of splenic B cells stimulated with various doses of LPS or anti-CD40 and IL-4 were comparable between wild-type and Jnk1 −/− mice (Fig. 3 f, and data not shown). Moreover, Jnk1 −/− B cells normally upregulated CD23 expression upon CD40 stimulation (data not shown). Thus, JNK1 deficiency does not appear to affect B cell activation.
Defective Apoptosis and Proliferation in Double Heterozygous Jnk1+/− Jnk2+/− Cells.
The apoptosis and proliferation defects displayed by Jnk1 − /− thymocytes and peripheral T cells are similar to those found in T cells lacking JNK2 25. This raised the possibility that JNK1 and JNK2 have similar and overlapping roles in T cell signal transduction. The phenotypes exhibited by either Jnk1 −/− or Jnk2 −/− mice could therefore be due to a reduction in the total amount of JNK activity. Unfortunately, Jnk1 −/−Jnk2 −/− double mutant fetuses die at E11.5 33 34, and the analysis of double mutant T cells could only be performed in chimaeras with mutant ES cells 35. To test whether gene dosage could account for the observed T cell defects, T cell function was analyzed in Jnk1 +/−Jnk2 +/− double heterozygotes. Thymocytes from wild-type, Jnk1 +/−, Jnk2 +/−, Jnk1 −/−, Jnk2 −/−, Jnk1 +/−Jnk2 +/−, Jnk1 −/−Jnk2 +/−, and Jnk1 +/− Jnk2 −/− mice were isolated and treated with anti-CD3 and anti-CD28 antibodies to determine the apoptotic response. Jnk1 +/−Jnk2 +/− double heterozygote thymocytes were as resistant as Jnk1 −/− or Jnk2 −/− single knockout thymocytes (Fig. 4), suggesting that the loss of a single allele of each Jnk gene is functionally equivalent to the loss of both alleles of a single gene. Single heterozygotes, Jnk1 +/− Jnk2 +/+ or Jnk1 +/+Jnk2 +/−, however, had no phenotype (Fig. 4). Moreover, loss of three Jnk alleles, namely Jnk1 −/− Jnk2 +/− or Jnk1 +/−Jnk2 −/−, did not enhance the defect in activation-induced cell death (Fig. 4).
Figure 4.

Jnk1 +/−Jnk2 +/− thymocytes are resistant to anti-CD3 activation–induced cell death. Thymocytes from mice of various genotypes were cultured with anti-CD3ε and anti-CD28 antibodies for 24 h and their viability was determined as described. The results are representative of three independent experiments, each using three matched pairs of mice. All assays were conducted in triplicates. Standard deviations are shown as vertical lines.
Peripheral T cells from the above mice were also examined for the proliferative response to anti-CD3 plus anti-CD28 stimulation. Total spleen cells activated with either anti-CD3 antibody alone or concanavalin A exhibited a vigorous proliferative response regardless of genotype (Fig. 5 a, and data not shown). However, lymph node T cells from all mutant mice displayed a marked proliferative defect when exposed to low doses of anti-CD3 antibody (Fig. 5 b). Both Jnk1 −/− and Jnk2 −/− T cells proliferated much less efficiently than wild-type cells when stimulated with 1 μg/ml of anti-CD3 antibody (Fig. 5 b). Most importantly, lymph node T cells from Jnk1 +/−Jnk2 +/−, as well as from Jnk1 −/−Jnk2 +/− or Jnk1 +/−Jnk2 −/− mice, exhibited a similar proliferative defect to the single knockout cells (Fig. 5 b). Nevertheless, T cells from single heterozygotes, Jnk1 +/− Jnk2 +/+ or Jnk1 +/+Jnk2 +/−, did not display any proliferation defect (Fig. 5 b).
Figure 5.
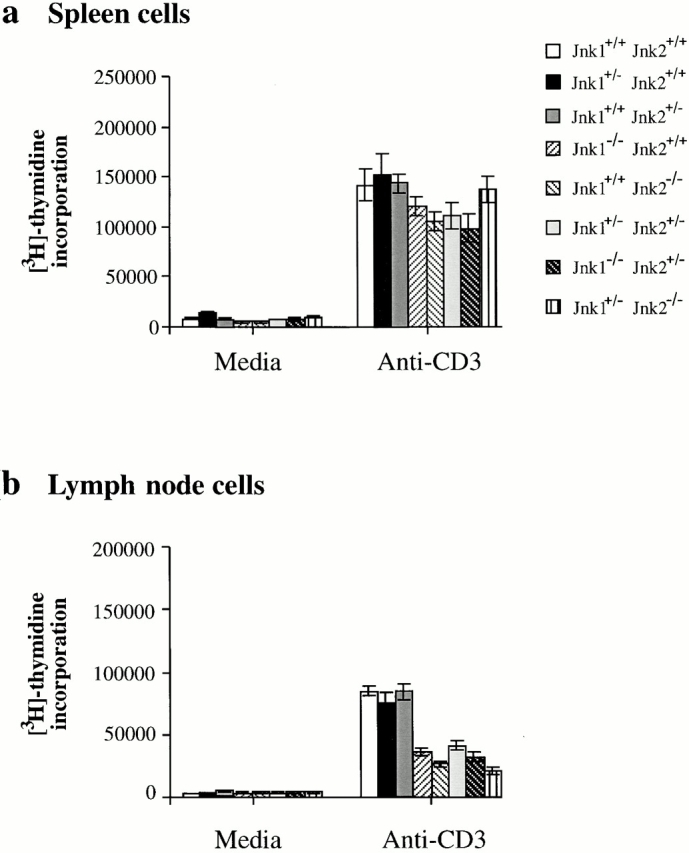
Impaired proliferation of Jnk1+ / −Jnk2+/− T cells. (a) Spleen cells and (b) lymph node cells were cultured with 1 μg/ml of anti-CD3 antibody and proliferation rates were determined as described above. The results are representative of three independent experiments, each using three matched pairs of mice. All assays were conducted in triplicates. Standard deviations are shown as vertical lines.
c-Jun Phosphorylation in the Absence of JNK1.
Activation of the JNK pathway leads to phosphorylation of various substrates, including c-Jun, and results in target gene induction 36. We investigated whether the defects observed in Jnk1 − /− thymocytes and mature T cells were due to defective c-Jun phosphorylation. Both wild-type and JNK1-deficient thymocytes were stimulated with anti-CD3 and anti-CD28, and cell lysates prepared at various time points after stimulation were used to determine total JNK activity, using GST–c-Jun (1-79) as a substrate. JNK activity was elevated 30 min after stimulation of wild-type thymocytes (Fig. 6 a). This increase persisted at 75 min after stimulation, but kinase activity returned to near basal levels by 240 min (Fig. 6 a). By contrast, JNK activity was only marginally elevated between 30 and 75 min after stimulation in Jnk1 − /− thymocytes and the entire response appeared to be delayed (Fig. 6 a). We also examined the NH2-terminal phosphorylation of endogenous c-Jun by Western blot analysis with anti–phospho-c-Jun (S63) antibodies. An increase in the amount of JNK-phosphorylated c-Jun was observed at ∼10 min and remained elevated for at least 240 min after stimulation in wild-type thymocytes (Fig. 6 a). After the appearance of phosphorylated c-Jun, the total amount of c-Jun protein also increased and peaked at 240 min after stimulation (Fig. 6 a). These results are consistent with the known autoregulation of c-jun transcription 37. However, in Jnk1 −/− thymocytes no c-Jun NH2-terminal phosphorylation could be detected, and the total amount of c-Jun was also very low and not induced upon stimulation (Fig. 6 a). These results indicate that c-Jun is an important target for JNK1 in anti-CD3–activated thymocytes.
Figure 6.
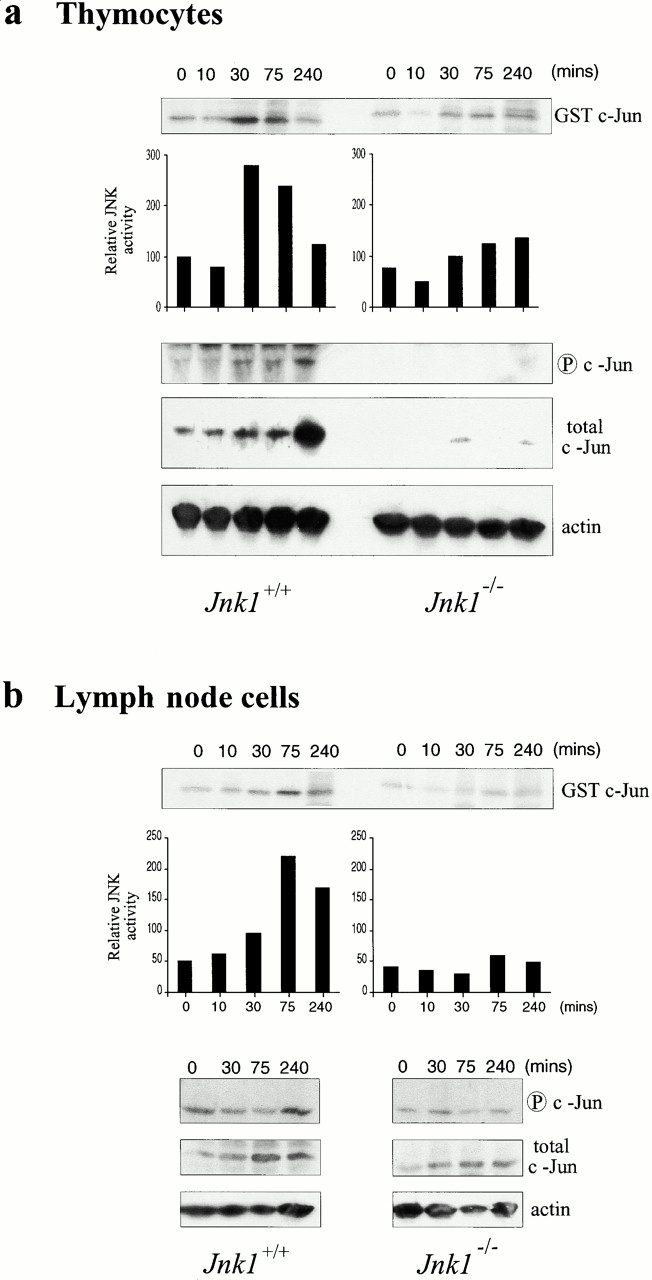
JNK activity and c-Jun status in Jnk1 − /− cells. (a) Thymocytes and (b) lymph node cells from wild-type and Jnk1 − /− mice were stimulated for the indicated times with 10 μg/ml anti-CD3 plus 0.1 μg/ml anti-CD28 antibodies, and JNK activity was determined in whole cell extracts, using solid-state kinase assay and GST–c-Jun (1-79) as a substrate. The same extracts were analyzed for their content of phospho-c-Jun (S63), total c-Jun, and actin by Western blot analysis.
Total JNK activity was also elevated in wild-type peripheral T cells activated with anti-CD3 and anti-CD28 antibodies, starting at 10 min after stimulation and peaking at ∼75 min after stimulation (Fig. 6 b). By contrast, no increase in total JNK activity was observed in Jnk1 − /− peripheral T cells (Fig. 6 b). Western blot analysis of endogenous c-Jun phosphorylation and expression revealed that although total c-Jun protein amount was modestly elevated after the stimulation of wild-type cells, only a marginal increase in c-Jun NH2-terminal phosphorylation could be detected (Fig. 6 b). These results suggest that by contrast to thymocytes, c-Jun might not be an important downstream target for JNK in peripheral T cells. Analysis of Jnk1 − /− peripheral T cells revealed only a slight increase in the total c-Jun expression, less than in activated wild-type T cells, and no significant increase in c-Jun NH2-terminal phosphorylation (Fig. 6 b).
NF-AT Binding Activity Is Reduced in JNK1 Mutant Peripheral T Cells.
To identify potential targets for JNK1 in peripheral T cells, we performed electrophoretic mobility shift assay using nuclear extracts from nonactivated and activated spleen and lymph node cells. The regulation of AP-1 binding activity was essentially identical in wild-type and Jnk1 −/− lymph node and spleen cells (Fig. 7 a, and data not shown). AP-1 binding activity was induced ∼3 h after stimulation and was still present at 16 h. The detected activity was sequence specific based on competition analysis, and all three Jun members were present at similar levels (data not shown). Therefore, we examined whether the activity of another critical T cell transcription factor, NF-AT, was altered in the absence of JNK1, as the absence of NF-ATc1 results in reduced T cell proliferation 17. Nuclear extracts from wild-type lymph node T cells activated with anti-CD3 and anti-CD28 antibodies exhibited enhanced NF-AT–DNA binding activity to the NF-AT binding site of the IL-2 promoter 3 h after stimulation. However, NF-AT activity was markedly reduced in Jnk1 −/− extracts (Fig. 7 b), although using five times higher amount of Jnk1 −/− extracts, NF-AT–DNA binding activity could be detected (Fig. 7 c). The presence of NF-ATc1 and NF-ATc2 in the specific complex was confirmed by supershift analysis with the respective antibodies (Fig. 7 c). As NF-AT–DNA binding on the IL-2 promoter is dependent on the AP-1 site, we also examined the NF-AT–DNA binding activity using the site from the IL-4 promoter, which is a high affinity binding site to which NF-AT binds in an AP-1–independent manner 8. Using increasing amount of extracts, we found NF-AT–DNA binding activity in the wild-type extracts (Fig. 7 d). This activity was reduced in the absence of JNK1, indicating that the defect is not due to any modification of the AP-1 component (Fig. 7 d). However, this defect was not absolute as we could detect NF-AT–DNA binding activity with higher doses of extracts (Fig. 7 d). The reduced NF-AT–DNA binding activity in Jnk1 − /− T cell extracts is not due to inefficient cytoplasmic exit or nuclear translocation of NF-ATc1, as we observed comparable amounts of NF-ATc1 in both wild-type and Jnk1 − /− nuclear extracts after stimulation (Fig. 7 e).
Figure 7.
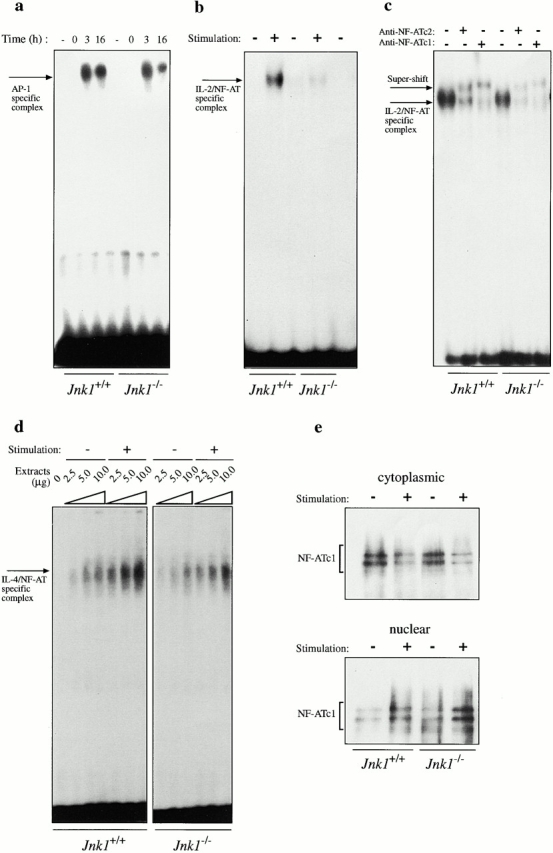
AP-1– and NF-AT–DNA-binding activity in JNK1-deficient T cells. (a) AP-1–DNA binding activity was measured using 2.5 μg nuclear extracts from untreated or 10 μg/ml anti-CD3 and 0.1 μg/ml anti-CD28–stimulated lymph node cells for the indicated period of time. The proteins were incubated with an AP-1 binding site oligonucleotide probe, and the specific protein–DNA complex is indicated. (b–d) NF-AT–DNA binding activity from untreated (−) or anti-CD3– and anti-CD28–stimulated (3 h; +) lymph node cells of wild-type and Jnk1 − /− mice was examined using oligonucleotide probes corresponding to either the low affinity NF-AT binding site from the IL-2 promoter (b and c) or the high affinity NF-AT binding site from the IL-4 promoter (d). 2.5 μg of nuclear extracts was used in b. In the supershift experiments, 2.5 μg of wild-type extracts and 12.5 μg of Jnk1 − /− extracts were used and the specific complex was supershifted by both anti–NF-ATc1 and anti–NF-ATc2 antibodies (c). Increasing amounts of extracts were used in d as indicated. The specific protein–DNA complex is indicated. (e) Cytoplasmic and nuclear extracts from unstimulated (−) and stimulated (+) wild-type and Jnk1 − /− lymph node T cells were prepared as described in b–d, and NF-ATc1 protein was detected by Western blot analysis.
Discussion
Mice lacking JNK1 were generated to evaluate the specific roles of JNK isozymes in T cell function. We found that both JNK1 and JNK2 have similar and stage-dependent roles in regulating T cell function. In thymocytes, JNK1 is necessary for efficient apoptosis induced by anti-CD3 antibody, whereas in mature T cells, it is necessary for efficient proliferation and IL-2 production. At both developmental stages, JNK1 uses different targets in regulating the two different processes. c-Jun phosphorylation is reduced in the absence of JNK1 in thymocytes, whereas it is not affected in mature T cells. In contrast, NF-AT–DNA binding activity is reduced in Jnk1 − /− mature T cells. These data underscore the importance of regulation of divergent targets by JNK1 in transmitting signals to the nucleus, which to our knowledge is the first demonstration of such distinct control of gene expression by a signaling cascade in different developmental stages of a cell type.
As found previously with Jnk2 − /− thymocytes 25, immature Jnk1 − /− thymocytes are resistant to anti-CD3 antibody–induced death, both in vitro and in vivo, yet are able to respond efficiently to other apoptotic stimuli. Thus, like JNK2, JNK1 is involved in the transduction of apoptotic signals generated by the massive activation of the TCR with the anti-CD3 antibody. This defect in activation-induced death in Jnk1 − /− mice was pronounced when the anti-CD3 antibody was administered in vivo rather than presented in a plate-bound form to isolated thymocytes. As peripheral mature T cells are also activated by anti-CD3 antibody–injection resulting in the release of cytokines, including IL-2 31, it could be envisaged that the major consequence of the JNK1 or JNK2 deficiencies is reduced responsiveness of mature T cells to activating stimuli. The activation defect could result in decreased production of death cytokines or cytokines that sensitize immature thymocytes to death signals. Indeed, peripheral Jnk1 − /− and Jnk2 − /− T cells exhibit a proliferation defect at limiting doses of activating stimuli, due to reduced IL-2 production. This proliferation defect was fully rescued by the addition of exogenous IL-2. Interestingly, thymocytes from mice that are deficient in the α and β subunits of the IL-2 receptor or IL-2 itself exhibit decreased sensitivity to apoptosis induced by in vitro incubation with anti-CD3 antibody 38 39 40. Thus, the resistance of Jnk1 − /− or Jnk2 − /− thymocytes to in vivo administration of anti-CD3 antibody is probably due to a combination of a thymocyte-autonomous defect and decreased production of death-promoting cytokines in the periphery. It should be noted, however, that JNK activity is not required for all forms of thymocyte apoptosis, as both Jnk1 − /− or Jnk2 − /− thymocytes were susceptible to apoptosis induced by anti-Fas antibody, dexamethasone, and UVC irradiation as wild-type cells.
These results strongly suggest that JNK1 and JNK2 have similar functions in T cell signal transduction, possibly through phosphorylation of a common set of substrates. This hypothesis was tested using double heterozygote Jnk1 +/−Jnk2 +/− mice, which lack half of the Jnk1 and Jnk2 gene doses. The single heterozygotes displayed no defect whatsoever in activation-induced cell death or proliferation of peripheral T cells. However, thymocytes and T cells derived from Jnk1+ / −Jnk2+/− double heterozygous mice had the same defects as either Jnk1 − /− or Jnk2 − /− T cells. Thus, both JNK1 and JNK2 make important but very similar contributions to the activation of peripheral T cells and induction of thymocyte apoptosis in response to TCR activation with anti-CD3. A requirement for JNK activity in anti-CD3–induced apoptosis and peripheral T cell activation has also been demonstrated by ectopic expression of catalytically inactive JNK1 in thymocytes 28.
The JNKs phosphorylate various substrates, among which c-Jun has been shown to be a critical and ubiquitous target in many cell types 36. Nonetheless, the absence of JNK1 or JNK2 leads to defective endogenous c-Jun phosphorylation upon stimulation with the anti-CD3 antibody only in immature thymocytes and not in mature T cells. Consistent with these findings, we found using mutant mice homozygous for a c-jun allele, in which the serines 63 and 73 were replaced with alanines, that c-Jun phosphorylation is essential for thymocyte apoptosis (unpublished observations). However, c-Jun does not seem to be a critical target for either JNK1 or JNK2 in activated mature peripheral T cells. Total c-Jun levels in peripheral T cells do not increase as dramatically in response to TCR activation as in activated thymocytes. Consistent with these findings, mature T cells from the phosphorylation-defective c-Jun mutant mice do not exhibit any proliferation defect (unpublished observations). These results strongly suggest that as T cells mature, JNK activation becomes linked to the phosphorylation of substrates other than c-Jun. Whereas c-Jun NH2-terminal phosphorylation is linked to activation-induced thymocyte apoptosis, the phosphorylation of these yet to be identified substrates is linked to T cell proliferation.
Reduced peripheral T cell proliferation in both Jnk1 − /− and Jnk2 − /− mutants correlates with reduced NF-AT–DNA binding activity. Although both AP-1 and NF-AT–DNA binding activities were induced upon activation of wild-type T cells, only AP-1 DNA binding activity was induced to normal levels in Jnk1 − /− or Jnk2 − /− mutants. Lack of NF-ATc1 has been shown to result in reduced T cell proliferation 17. Moreover, overexpression of a dn NF-AT in T cells markedly inhibits IL-2 production 20. It is therefore likely that the reduction in NF-AT activity may account for the proliferation defect of JNK-deficient T cells. It should be noted that the proliferation defect is not absolute, as higher levels of stimulation result in increased proliferation. Consistent with this, we found that NF-AT–DNA binding activity is not abrogated in the absence of JNK1, but only reduced. Thus, the reduction in NF-AT–DNA binding activity correlates with the proliferation defect in the absence of JNK1. It is not yet clear how JNK activation is linked to nduction of NF-AT activity. As the binding of NF-AT in the IL-4 promoter does not require cooperation from AP-1 8, it is possible that the primary target for JNK phosphorylation could be a nuclear partner, NF-ATn, whose phosphorylation could assist NF-AT proteins in binding DNA. Alternatively, NF-AT proteins themselves could be a direct target for JNK-mediated phosphorylation. In this regard, it was reported that NF-ATc3 is phosphorylated by the JNKs resulting in its exclusion from the nucleus 41. However, these results imply that JNKs negatively regulate NF-AT function, whereas our data indicate that the JNKs exert a positive effect on NF-AT function, at least in peripheral T cells. No differences in the levels of NF-ATc1 and NF-ATc2 or their subcellular distribution were detected between wild-type and Jnk1 − /− cells. Thus, defective nuclear translocation cannot explain the defect in induction of NF-AT–DNA binding activity. Although Dong et al. 27 recently reported that the lack of JNK1 results in nuclear accumulation of NF-ATc1 in Th cells, we have not been able to identify such a defect in mature T cells. Our results clearly indicate that JNK1 has a positive rather than a negative effect on NF-AT and T cell activation and that JNK1 and JNK2 play similar and overlapping roles in T cell function.
Recently, lack of JNK1 was reported to result in hyperproliferation of purified CD4+ Th peripheral T cells without an increase in IL-2 production 27, whereas the absence of JNK2 had no effect on mature T cell proliferation but affected IFN-γ production by Th1 cells 26. In neither case was a defect in IL-2 production observed, which generally reflects the proliferative capacity of T cells. Furthermore, lack of both JNK1 and JNK2 results in hyperproliferation and increased IL-2 production in CD4+ Th cells 35. However, transgenic mice expressing a catalytically inactive, dn Jnk1 gene showed reduced mature T cell proliferation 28. Most of the experiments performed with these JNK-deficient mice employed CD4+ Th1 and Th2 T cells generated by in vitro differentiation 26 27. In contrast, we examined peripheral T cells isolated from spleen and lymph nodes which, with the exception of mitogenic stimulation, were not subjected to any long-term in vitro manipulation. Our results clearly support the observations with the dn Jnk1 transgenic mice and indicate that JNK1 positively regulates proliferation. Moreover, thymocytes from the dn Jnk1 mice are also resistant to anti-CD3–induced apoptosis, similar to JNK1 mutant mice. These data strongly argue for an essential role for JNK1 in positively regulating mature T cell proliferation.
The observed proliferation defect correlates with and is probably caused by decreased IL-2 production, as it is completely suppressed by exogenous IL-2. In addition to JNK1 and JNK2, the JNK activating kinase JNKK1/SEK1 was also implicated in regulating T cell function 30. Chimeras generated using Sek1 − /− ES cells gave different results 30 42. Nishina et al. 30 found defective T cell activation, whereas Swat et al. 42 found normal activation of T cells lacking SEK1. In the latter, SEK1-deficient lymphocytes were found to be capable of JNK activation. Thus, it appears that the magnitude of the stimuli and the level of JNK activation are important factors determining the efficiency of T cell activation. Taken together, our results indicate that JNK1 has a positive rather than a negative effect on NF-AT and T cell activation and that JNK1 and JNK2 have similar and overlapping roles in T cell function. Although c-Jun is a relevant target of both JNKs in thymocytes, it does not seem to be an important target in peripheral T cells, where both JNK isozymes contribute to the induction of NF-AT rather than AP-1 activity.
Acknowledgments
We thank Christian Theussl for the blastocyst injections.
The Research Institute of Molecular Pathology is supported by Boehringer Ingelheim. Work in E.F. Wagner's lab was supported by the Austrian Federal Ministry of Science, Transport, and the Arts, and the Austrian Industrial Research Promotion Fund; work in M. Karin's lab was supported by grants from the National Institutes of Health and the State of California Cancer Research Program.
Footnotes
K. Sabapathy's present address is National Cancer Centre, Division of Cellular and Molecular Research, 11 Hospital Dr., Singapore 169610, Singapore. T. Kallunki's present address is Apoptosis Laboratory, Danish Cancer Society, Strandboulevarden 49, DK-2100, Copenhagen, Denmark.
Abbreviations used in this paper: dn, dominant negative; DP, double positive; ES, embryonic stem; GST, glutathione S-transferase; JNK, c-Jun NH2-terminal kinase; NF-ATc, cyclosporin A–sensitive NF-AT; NF-AT, nuclear factors of activated T cell.
References
- Crabtree G.R. Contingent genetic regulatory events in T lymphocyte activation. Science. 1989;243:355–361. doi: 10.1126/science.2783497. [DOI] [PubMed] [Google Scholar]
- Su B., Jacinto E., Hibi M., Kallunki T., Karin M., Ben-Neriah Y. JNK is involved in signal integration during costimulation of T lymphocytes. Cell. 1994;77:727–736. doi: 10.1016/0092-8674(94)90056-6. [DOI] [PubMed] [Google Scholar]
- Jacinto E., Werlen G., Karin M. Cooperation between Syk and Rac1 leads to synergistic JNK activation in T lymphocytes. Immunity. 1998;8:31–41. doi: 10.1016/s1074-7613(00)80456-2. [DOI] [PubMed] [Google Scholar]
- Werlen G., Jacinto E., Xia Y., Karin M. Calcineurin preferentially synergizes with PKC-theta to activate JNK and IL-2 promoter in T lymphocytes. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:3101–3111. doi: 10.1093/emboj/17.11.3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Healy J.I., Dolmetsch R.E., Timmerman L.A., Cyster J.G., Thomas M.L., Crabtree G.R., Lewis R.S., Goodnow C.C. Different nuclear signals are activated by the B cell receptor during positive versus negative signaling. Immunity. 1997;6:419–428. doi: 10.1016/s1074-7613(00)80285-x. [DOI] [PubMed] [Google Scholar]
- DeSilva D.R., Feeser W.S., Tancula E.J., Scherle P.A. Anergic T cells are defective in both jun NH2-terminal kinase and mitogen-activated protein kinase signaling pathways. J. Exp. Med. 1996;183:2017–2023. doi: 10.1084/jem.183.5.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Whaley C.D., Mondino A., Mueller D.L. Blocked signal transduction to the ERK and JNK protein kinases in anergic CD4+ T cells. Science. 1996;271:1272–1276. doi: 10.1126/science.271.5253.1272. [DOI] [PubMed] [Google Scholar]
- Jain J., McCaffrey P.G., Valge-Archer V.E., Rao A. Nuclear factor of activated T cells contain Fos and Jun. Nature. 1992;356:801–804. doi: 10.1038/356801a0. [DOI] [PubMed] [Google Scholar]
- Rincon M., Flavell R.A. AP-1 transcriptional activity requires both T-cell receptor-mediated and co-stimulatory signals in primary T lymphocytes. EMBO (Eur. Mol. Biol. Organ.) J. 1994;13:4370–4381. doi: 10.1002/j.1460-2075.1994.tb06757.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang S.-M., Bart B., Tran A.C., Brorson D., Schwartz R.H., Lenardo M.J. Transactivation by AP-1 is a molecular target of T cell clonal anergy. Science. 1992;257:1134–1138. doi: 10.1126/science.257.5073.1134. [DOI] [PubMed] [Google Scholar]
- Kallunki T., Deng T., Hibi M., Karin M. c-Jun can recruit JNK to phosphorylate dimerization partners via specific docking interactions. Cell. 1996;87:929–939. doi: 10.1016/s0092-8674(00)81999-6. [DOI] [PubMed] [Google Scholar]
- Li B., Tournier C., Davis R.J., Flavell R.A. Regulation of IL-4 expression by the transcription factor JunB during T helper cell differentiation. EMBO (Eur. Mol. Biol. Organ.) J. 1999;18:420–432. doi: 10.1093/emboj/18.2.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindstein T., June C.H., Ledbetter J.A., Stella G., Thompson C.B. Regulation of lymphokine messenger RNA stability by a surface mediated T cell activation pathway. Science. 1989;244:339–343. doi: 10.1126/science.2540528. [DOI] [PubMed] [Google Scholar]
- Chen C.-Y., Konczak F., Wu Z., Karin M. Stabilization of interleukin-2 mRNA by the c-Jun NH2-terminal kinase pathway through a 5′ response element. Science. 1998;280:1945–1949. doi: 10.1126/science.280.5371.1945. [DOI] [PubMed] [Google Scholar]
- Hilberg F., Aguzzi A., Howells N., Wagner E.F. c-jun is essential for normal mouse development and hepatogenesis. Nature. 1993;365:179–181. doi: 10.1038/365179a0. [DOI] [PubMed] [Google Scholar]
- Chen J., Stewart V., Spyrou G., Hilberg F., Wagner E.F., Alt F.W. Generation of normal T and B lymphocytes by c-jun deficient embryonic stem cells. Immunity. 1994;1:65–72. doi: 10.1016/1074-7613(94)90010-8. [DOI] [PubMed] [Google Scholar]
- Ranger A.M., Hodge M.R., Gravallese E.M., Oukka M., Davidson L., Alt F.W., de la Brousse F.C., Hoey T., Grusby M., Glimcher L.H. Delayed lymphoid repopulation with defects in IL-4-driven responses produced by inactivation of NF-ATc. Immunity. 1998;8:125–134. doi: 10.1016/s1074-7613(00)80465-3. [DOI] [PubMed] [Google Scholar]
- Hodge M.R., Ranger A.M., de la Brousse F.C., Hoey T., Grusby M.J., Glimcher L.H. Hyperproliferation and dysregulation of IL-4 expression in NF-ATp-deficient mice. Immunity. 1996;4:397–405. doi: 10.1016/s1074-7613(00)80253-8. [DOI] [PubMed] [Google Scholar]
- Oukka M., Ho I.C., de la Brousse F.C., Hoey T., Grusby M.J., Glimcher L.H. The transcription factor NFAT4 is involved in the generation and survival of T cells. Immunity. 1998;9:295–304. doi: 10.1016/s1074-7613(00)80612-3. [DOI] [PubMed] [Google Scholar]
- Chow C.W., Rincon M., Davis R.J. Requirement for transcription factor NF-AT in interleukin-2 expression. Mol. Cell. Biol. 1999;19:2300–2307. doi: 10.1128/mcb.19.3.2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow C.W., Rincon M., Cavenagh J., Dickens M., Davis R.J. Nuclear accumulation of NFAT4 opposed by the JNK signal transduction pathway. Science. 1997;278:1638–1641. doi: 10.1126/science.278.5343.1638. [DOI] [PubMed] [Google Scholar]
- Mohit A.A., Martin J.H., Miller C.A. p49I3F12 kinase, a novel MAP kinase expressed in a subset of neurons in the human nervous system. Neuron. 1995;14:67–78. doi: 10.1016/0896-6273(95)90241-4. [DOI] [PubMed] [Google Scholar]
- Kallunki T., Su B., Tsigelny I., Sluss H.K., Dérijard B., Moore G., Davis R.J., Karin M. JNK2 contains a specificity-determining region responsible for efficient c-Jun binding and phosphorylation. Genes Dev. 1994;8:2996–3007. doi: 10.1101/gad.8.24.2996. [DOI] [PubMed] [Google Scholar]
- Gupta S., Barrett T., Whitmarsh A.J., Cavanagh J., Sluss H.K., Dérijard B., Davis R.J. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO (Eur. Mol. Biol. Organ.) J. 1996;15:2760–2770. [PMC free article] [PubMed] [Google Scholar]
- Sabapathy K., Hu Y., Kallunki T., Schreiber M., David J.P., Jochum W., Wagner E.F., Karin M. JNK2 is required for efficient T-cell activation and apoptosis but not for normal lymphocyte development. Curr. Biol. 1999;9:116–125. doi: 10.1016/s0960-9822(99)80065-7. [DOI] [PubMed] [Google Scholar]
- Yang D.D., Conze D., Whitmarsh A.J., Barrett T., Davis R.J., Rincon M., Flavell R.A. Differentiation of CD4+ T cells to Th1 cells requires MAP kinase JNK2. Immunity. 1998;9:575–585. doi: 10.1016/s1074-7613(00)80640-8. [DOI] [PubMed] [Google Scholar]
- Dong C., Yang D.D., Wysk M., Whitmarsh A.J., Davis R.J., Flavell R.A. Defective T cell differentiation in the absence of JNK1. Science. 1998;282:2092–2095. doi: 10.1126/science.282.5396.2092. [DOI] [PubMed] [Google Scholar]
- Rincon M., Whitmarsh A., Yang D.D., Weiss L., Derijard B., Jayaraj P., Davis R.J., Flavell R.A. The JNK pathway regulates the in vivo deletion of immature CD4+CD8+ thymocytes. J. Exp. Med. 1998;188:1817–1830. doi: 10.1084/jem.188.10.1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishina H., Fischer K.D., Radvanyi L., Shahinian A., Hakem R., Rubie E.A., Bernstein A., Mak T.W., Woodgett J.R., Penninger J.M. Stress-signaling kinase Sek1 protects thymocytes from apoptosis mediated by CD95 and CD3. Nature. 1997;385:350–353. doi: 10.1038/385350a0. [DOI] [PubMed] [Google Scholar]
- Nishina H., Bachmann M., Oliveira-dos-Santos A.J., Kozieradzki I., Klaus D., Fischer K.D., Odermatt B., Wakeham A., Shahinian A., Takimoto H. Impaired CD28-mediated interleukin 2 production and proliferation in stress kinase SAPK/ERK1 kinase (SEK1)/mitogen-activated protein kinase kinase 4 (MKK4)-deficient T lymphocytes. J. Exp. Med. 1997;186:941–953. doi: 10.1084/jem.186.6.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin S., Bevan M.J. Antigen-specific and nonspecific deletion of cortical thymocytes caused by antigen injection. Eur. J. Immunol. 1997;27:2726–2736. doi: 10.1002/eji.1830271037. [DOI] [PubMed] [Google Scholar]
- Berberich I., Shu G., Siebelt F., Woodgett J.R., Kyriakis J.M., Clark E.A. Cross-linking CD40 on B cells preferentially induces stress-activated protein kinases rather than mitogen-activated protein kinases. EMBO (Eur. Mol. Biol. Organ.) J. 1996;15:92–101. [PMC free article] [PubMed] [Google Scholar]
- Sabapathy K., Jochum W., Hochedlinger K., Chang L., Karin M., Wagner E.F. Defective neural tube morphogenesis and altered apoptosis in the absence of both JNK1 and JNK2. Mech. Dev. 1999;89:115–124. doi: 10.1016/s0925-4773(99)00213-0. [DOI] [PubMed] [Google Scholar]
- Kuan C.Y., Yang D.D., Roy D.S., Davis R.J., Rakic P., Flavell R. The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain development. Neuron. 1999;22:667–676. doi: 10.1016/s0896-6273(00)80727-8. [DOI] [PubMed] [Google Scholar]
- Dong C., Yang D.D., Tournier C., Whitmarsh A.J., Xu J., Davis R.J., Flavell R. JNK is required for effector T cell function but not for T-cell activation. Nature. 2000;405:91–94. doi: 10.1038/35011091. [DOI] [PubMed] [Google Scholar]
- Karin M., Liu Z.G., Zandi E. AP-1 function and regulation. Curr. Opin. Cell Biol. 1997;9:240–246. doi: 10.1016/s0955-0674(97)80068-3. [DOI] [PubMed] [Google Scholar]
- Angel P., Hattori K., Smeal T., Karin M. The jun proto-oncogene is positively autoregulated by its product, Jun/AP-1. Cell. 1988;55:875–885. doi: 10.1016/0092-8674(88)90143-2. [DOI] [PubMed] [Google Scholar]
- Schorle H., Holtschke T., Hunig T., Schimpl A., Horak I. Development and function of T cells in mice rendered interleukin-2 deficient by gene targeting. Nature. 1991;352:621–624. doi: 10.1038/352621a0. [DOI] [PubMed] [Google Scholar]
- Suzuki H., Kundig T.M., Furlonger C., Wakeham A., Timms E., Matsuyama T., Schmits R., Simard J.J., Ohashi P.S., Griesser H. Deregulated T cell activation and autoimmunity in mice lacking interleukin-2 receptor β. Science. 1995;268:1472–1476. doi: 10.1126/science.7770771. [DOI] [PubMed] [Google Scholar]
- Willerford D.M., Chen J., Ferry J.A., Davidson L., Ma A., Alt F.W. Interleukin-2 receptor α chain regulates the size and content of the peripheral lymphoid compartment. Immunity. 1995;3:521–530. doi: 10.1016/1074-7613(95)90180-9. [DOI] [PubMed] [Google Scholar]
- Chow C.W., Rincon M., Cavanagh J., Dickens M., Davis R.J. Nuclear accumulation of NFAT4 opposed by the JNK signal transduction pathway. Science. 1997;278:1638–1641. doi: 10.1126/science.278.5343.1638. [DOI] [PubMed] [Google Scholar]
- Swat W., Fujikawa K., Ganiatis S., Yang D., Xavier R.J., Harris N.L., Davidson L., Ferrini R., Davis R.J., Labow M.A. SEK1/MKK4 is required for maintenance of a normal peripheral lymphoid compartment but not for lymphocyte development. Immunity. 1998;8:625–634. doi: 10.1016/s1074-7613(00)80567-1. [DOI] [PubMed] [Google Scholar]