

Abstract
A somatic mutation in the X-linked phosphatidylinositol glycan class A (PIGA) gene causes the loss of glycosyl phosphatidylinositol (GPI)-linked proteins on blood cells from patients with paroxysmal nocturnal hemoglobinuria. Because all blood cell lineages may be affected it is thought that the mutation occurs in a hematopoietic stem cell. In transgenic mice, germline transmission of an inactive Piga gene is embryonic lethal. To inactivate the murine Piga gene in early hematopoiesis we therefore chose conditional gene inactivation using the Cre/loxP system. We expressed Cre recombinase under the transcription regulatory sequences of the human c-fes gene. FES-Cre inactivated PIGA in hematopoietic cells of mice carrying a floxed Piga allele (LF mice). PIGA− cells were found in all hematopoietic lineages of definitive but not primitive hematopoiesis. Their proportions were low in newborn mice but subsequently increased continuously to produce for the first time mice that have almost exclusively PIGA− blood cells. The loss of GPI-linked proteins occurred mainly in c-kit+CD34+Lin− progenitor cells before the CFU-GEMM stage. Using bone marrow reconstitution experiments with purified PIGA− cells we demonstrate that LF mice have long-term bone marrow repopulating cells that lack GPI-linked proteins, indicating that recombination of the floxed Piga allele occurs in the hematopoietic stem cell.
Keywords: hematopoiesis, stem cell, c-fes, paroxysmal nocturnal hemoglobinuria, conditional gene inactivation
Introduction
Paroxysmal nocturnal hemoglobinuria (PNH) is believed to be a disease of the hematopoietic stem cell (HSC) 1 2. In patients with PNH, a somatic mutation occurs in the X-linked PIGA (phosphatidylinositol glycan class A) gene 3 4. PIGA encodes a protein subunit of the α-1-6-N-acetylglucosaminyltransferase, an enzyme essential in the biosynthesis of glycosyl phosphatidylinositol (GPI) molecules 5. Therefore, in patients with PNH, a proportion of blood cells lacks all surface proteins that use a GPI-anchor molecule to attach to the cell membrane (PIGA− cells) 6. All blood cell lineages may be affected, including erythrocytes, granulocytes, monocytes, platelets, and lymphocytes. Molecular analysis of PIGA gene mutations in the affected blood cells demonstrated that PIGA− cells are of clonal origin, indicating that the mutation occurs in a pluripotent hematopoietic progenitor cell, possibly the HSC 3 4.
Targeted mutagenesis of the murine Piga gene demonstrated that inactivation of PIGA in murine embryonic stem cells is lethal in very early embryonic development 7 8. To limit PIGA inactivation to hematopoietic cells we therefore employed conditional gene inactivation using the Cre/loxP system 9 10 11 and expressed the Cre recombinase under the DNA sequences necessary for the transcription of the human c-fes gene.
Human c-fes encodes a 92-kD nonreceptor protein-tyrosine kinase (FES) preferentially expressed in hematopoietic progenitor cells in the bone marrow and during myeloid differentiation 12. FES expression remains high during myelomonocytic differentiation 13, but decreases upon maturation along the erythroid lineage 14. C-fes messenger RNA has not been detected in lymphoid cells. High level FES expression is also documented in vascular endothelial cells 15. Inactivation of c-fes in mice resulted in functional abnormalities of myelomonocytic cells, and also suggested a role in B cell homeostasis 16 17. During embryonic development high levels of c-fes messenger RNA were found in cells enriched for murine liver stem cells 14 18.
A 13-kb EcoRI genomic DNA fragment encompassing the human c-fes gene 19 contains all regulatory DNA sequences required for a locus control region (LCR) with myelomonocyte-specific expression in transgenic animals 20 21. We used the c-fes LCR to express the Cre recombinase. Fes-cre transgenic mice were crossbred with mice carrying a floxed Piga allele. FES-Cre–mediated recombination of the floxed Piga allele caused a time-dependent increase in the proportion of PIGA− cells in all blood cell lineages and generated for the first time mice that have almost all blood cells deficient in GPI-linked proteins. Progenitor analysis and bone marrow transplantation experiments demonstrate that Piga inactivation in these mice occurs in HSCs of the bone marrow.
Materials and Methods
Mice.
The production of loxPiga (L) mice was described previously (see Fig. 1 A) 22. The fes-cre (F) mice (see Fig. 1 B) were generated by using the 13-kb EcoRI fragment encompassing human c-fes 19. An NdeI site in the noncoding region of exon 19 was first converted to a unique EcoRV site by digestion with NdeI, recessing the 3′ end with T4 DNA polymerase, and then annealing and ligating with the adapter linkers 5′-CCA GCT CAT AGA TAT C-3′ and 5′-TGT CAG CAT AGA TAT C-3′. The poliovirus type 2 internal ribosome entry site (IRES) between nucleotides 125 and 720 (Lansing strain; GenBank/EMBL/DDBJ accession no. M12197) was PCR amplified using primers containing SmaI (5′-CAC AAG CTT CCC GGG CAA GTT CAA TAG GAG-3′) and EcoRV sites (5′-AGA CCC GGG ATA TCT GGT AAT TCC AAT AGG TG-3′). The IRES-containing PCR fragment was digested with SmaI and EcoRV and cloned into the unique EcoRV site in the 3′ noncoding region of the c-fes gene within the pECE expression plasmid 23 to produce pEFOR-5. The CreN coding sequence from pTZ-CreN was excised as a HindIII to EcoI fragment, blunt ended with Klenow fragment, and cloned into the EcoRV site of pEFOR-5 to produce pEFOR-CreN. The fes-cre fragment was excised as an EcoRI fragment and injected into CBA/ca × C57Bl/6 F1 fertilized eggs (E. Lacy, Transgenic Core Facility, Memorial Sloan-Kettering Cancer Center, New York, NY). Founder mice were identified by Southern blot, and double transgenic mice (LF mice) by PCR analysis. Three F founder lines (nos. 16, 31, and 40) were evaluated. Tissue distribution of Piga gene inactivation was similar, but the kinetics were copy number dependent. Thus, for the following experiments, line 31 with the highest copy number (30 copies) was used. Initial experiments were performed in a mixed genetic background and later confirmed with mice in a C57Bl/6 n ≥ 10 background. F and L littermates were used as controls. All experiments involving animals were approved by the Animal Studies Committee of Washington University.
Figure 1.

Fes-cre and loxPiga mice. (A) Genomic structure of the wt Piga, loxPiga, and ΔPiga allele (loxPiga after recombination). Two loxP sites (triangles) and the coding region for lacZ were introduced into the Piga locus by homologous recombination in embryonic stem (ES) cells (reference 22). In the loxPiga configuration, PIGA function is not impaired (black boxes) and LacZ is not expressed (empty lacZ box). However, after Cre-mediated excision of the DNA sequence between the two loxP sites, Piga becomes inactive (empty Piga boxes) and lacZ falls under the endogenous Piga promoter and is expressed (black lacZ box). (B) Transgenic construct of fes-cre. Cre with a nuclear localization signal (CreN) was cloned into the 3′ untranslated region of human c-fes gene. The expression of fes-cre is maintained by an IRES, resulting in a bicistronic message under the control of the human c-fes LCR. E, EcoRI; RV, EcoRV.
DNA Amplification and Southern Blot Analysis.
Genotyping by PCR and Southern blot were performed as described 24. The intensity of the cre hybridization signal was used to identify probable fes-cre homozygous animals. For the isolation of tissue DNA animals were perfused with PBS.
Flow Cytometric Analysis.
Flow cytometric analysis was performed as described 8. Monoclonal antibodies used against GPI-linked proteins were CD24(M1/69), Gr-1(RB6-8C5), and CD48(HM48-1). Lineage specificity was analyzed by the use of antibodies against CD11b(M1/70), B220(RA3-6B2), TcR(H57-597), CD4(GK1.5), CD8(53-6.7), Ter119(TER-119), CD71(C2), c-kit(2B8), and CD34(RAM34). A mixture of PE-labeled antibodies against CD3(145-2C11), CD4, CD5(53-7.3), CD8, B220, CD11b, and Ter119 was used to exclude lineage-committed bone marrow cells. CD45.2(104) was used to determine the CD45 allotype. Reticulocyte counts and blood values were determined as described 22.
Analysis of β-Gal Expression.
Anesthetized mice were perfused with 2% paraformaldehyde, 0.2% glutaraldehyde in PBS. Small organ pieces were postfixed for 60 min and stained for lacZ expression as described by Sanes et al. 25. Incubation was for 24 h at 37°C.
Clonogenic Assays.
Fetal liver cells from embryonic day (E)12.5 or E18.5 embryos and adult bone marrow cells were plated in methylcellulose (MethoCult™ M3434; StemCell Technologies Inc.). Colonies were counted after 7–10 d. Visual inspection, light scatter characteristics, and the expression of CD71 and CD11b were used to characterize individual colonies.
Aerolysin Treatment.
Mouse femora were flushed with HBSS containing 2% isogenic mouse serum. 6 × 106 cells in a final volume of 500 μl 2% mouse serum-HBSS were incubated for 30 min at 37°C with no, 10 nM, or 50 nM aerolysin (Thomas Buckley, University of Victoria, Victoria, Canada). In preliminary in vitro experiments 10 and 50 mM concentrations demonstrated efficient lysis of PIGA+ but not PIGA− hematopoietic progenitor cells.
Bone Marrow Transplantation.
All mice were in C57Bl/6 background. We used the CD45.2/45.1 allotype system to distinguish donor and recipient blood cells. Recipient mice and bone marrow cells for radioprotection were CD45.1 (B6.SJL-Ptprca Pep3b/BoyJ mice; The Jackson Laboratory), and donor cells for aerolysin treatment were CD45.2. Recipient mice were irradiated with a single dose of 10 Gy the day before marrow injection. 106 aerolysin-treated or sham-treated cells (as counted before treatment) and 0.5 × 106 untreated radioprotection cells in a total volume of 300 μl HBSS were injected into lateral tail veins.
Results
Generation of fes-cre Mice.
To target Cre-mediated Piga gene inactivation to early hematopoietic cells we expressed the Cre recombinase under the LCR of the human c-fes gene (20; Fig. 1 B). In double transgenic mice, Cre-mediated recombination of the loxPiga allele deletes part of Piga exon 2, which abolishes the function of PIGA (22; Fig. 1 A). Piga maps to the X chromosome and is subject to X chromosome inactivation 24. Thus, in males the inactivation of the one Piga allele is sufficient to cause the loss of GPI-linked proteins on the cell surface. In females the inactivation of the Piga allele when it is on the active X-chromosome will lead to the loss of GPI-linked proteins on the cell surface (due to random X-inactivation this is expected to occur in about half of the cells). LF mice were born with the expected frequency and were indistinguishable from their littermates that only inherited one transgene. 16 LF mice (9 males and 7 females) were monitored over a time period of 12–16 mo.
LF Mice Have Increasing Numbers of Circulating PIGA− Blood Cells.
To monitor the proportion of PIGA− cells in peripheral blood, we analyzed the expression of GPI-linked proteins by flow cytometry. Analysis of red cells 1 to 3 d after birth showed a small but distinct proportion of PIGA− cells (average 1.4%, range 0.1–12.5%). Over the following months, the proportion of PIGA− red cells increased continuously in all LF animals and by the age of 16 mo the proportion of PIGA− red blood cells reached up to 94% (average 52%, 5–94%, n = 10; Fig. 2 A). A similar continuous increase of PIGA− cells was found in white blood cells. At the age of 1 mo, the proportion of PIGA− cells was 27% in granulocytes (11–49%), 14% in B cells (5–22%), and 20% in T cells (7–34%). At 16 mo the proportions of PIGA− cells increased to 69% (41–97%) in granulocytes, 56% (26–83%) in B cells, and 77% (64–87%) in T cells. No PIGA− blood cells were detected in control mice (≤1%, data not shown). In female mice the proportion of PIGA− blood cells was always lower than in male mice. This is consistent with random X chromosome inactivation in female somatic cells causing only half of the cells to express the recombined Piga gene (Fig. 2a and Fig. b). Fig. 2 B shows the relative level of PIGA− cells for all four blood cell lineages and the proportion of PIGA− reticulocytes measured at the age of 4 (45%, 34–57%), 8 (53%, 24–74%), and 16 (66%, 33–97%) mo. The proportion of PIGA− red blood cells was significantly lower than PIGA− reticulocytes in all animals analyzed, which is consistent with the shortened half-life of PIGA− red cells in circulation 22. We have previously shown that PIGA− red cells from mice mosaic for a nonfunctional Piga gene have an increased sensitivity to complement 22. Similarly, also PIGA− red cells from LF mice showed an increased lysis when exposed to complement (data not shown), suggesting that the decreased half-life of PIGA− red cells is due to the action of complement. Within the white blood cells the percentage of PIGA− cells was consistently lower in B cells than in any other cells.
Figure 2.
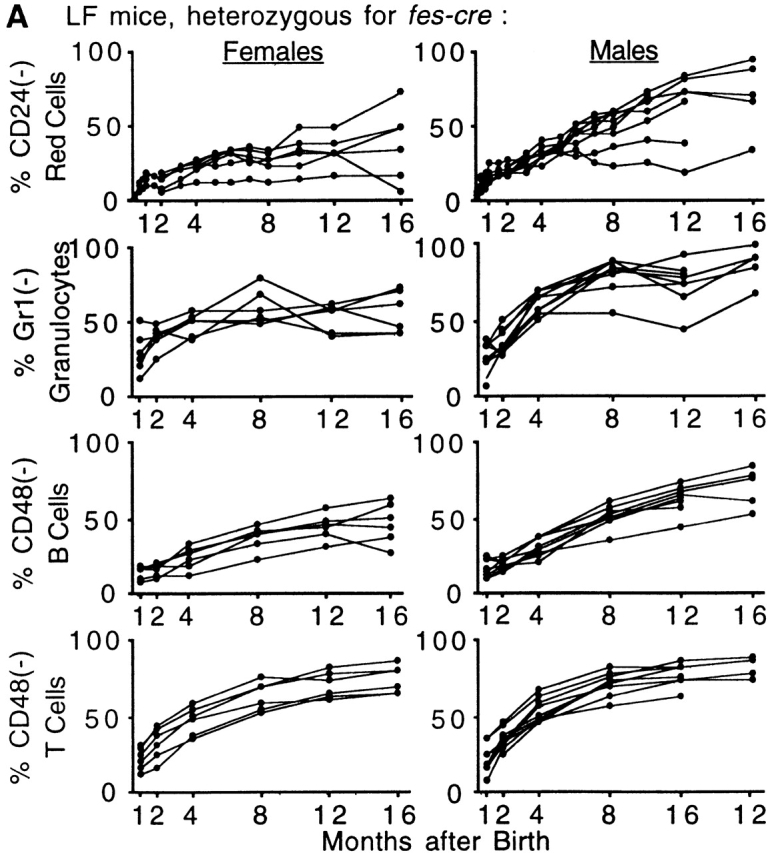



Proportions of PIGA− cells in peripheral blood from LF and LFF mice. (A) 16 mo follow-up of the proportion of PIGA− blood cells in LF mice (females n = 6, males n = 9). (B) Comparison of the relative number of PIGA− cells in different blood cell lineages in LF mice. Each data point represents the average value from six female and nine male mice. (C) Proportion of PIGA− blood cells in three male LFF mice. The gray-shaded area represents the proportions of PIGA− cells between the 10th and 90th percentile in male LF animals for comparison. (D) Flow cytometric analysis of peripheral blood cells from an 8-mo-old male LFF mouse with 100% PIGA− red cells, 99% PIGA− granulocytes, 93% PIGA− B cells, and 94% PIGA− T cells. Most of the few remaining PIGA+ B and T cells were CD44high and CD62Llow (data not shown), suggesting that they are mainly long lived memory cells. Monoclonal antibodies against CD24, GR-1, and CD48 were used as markers for GPI-linked proteins. Antibodies identifying the blood cell lineage were CD11b for granulocytes, B220 for B cells, and TcR for T cells. Red blood cells were identified by their light scatter characteristics. The top left quadrant shows the proportion of PIGA− cells for each blood cell lineage.
Fes-cre Homozygosity Accelerates Rate of Piga Recombination.
To test whether an increase in the fes-cre transgene copy number will accelerate the rate of Cre-mediated loxPiga recombination we crossbred female LF mice with male mice homozygous for fes-cre. In 12 mice positive for loxPiga and homozygous for fes-cre (LFF mice) the proportions of PIGA− red blood cells were higher and the increase faster than in LF mice. A similar rapid increase of PIGA− cells was also found within granulocytes, B cells, and T cells. These data indicate that the rate of loxPiga recombination in LFF mice is accelerated (Fig. 2 C). For the first time we were able to generate mice with almost 100% of PIGA− blood cells (Fig. 2 D).
FES-Cre–mediated Recombination of the loxPiga Allele in Fetal Hematopoiesis.
To test whether recombination of the loxPiga allele occurs also in primitive hematopoiesis, we collected genotyped fetuses from timed pregnancies. At day E12.5, 90% of circulating red blood cells are nucleated primitive red blood cells derived from yolk sac erythropoiesis. Analysis of GPI-linked proteins on primitive red cells did not reveal a significant difference between control, LF, and LFF fetuses, suggesting that FES-Cre does not mediate recombination of the loxPiga allele in hematopoietic progenitors of primitive red cells. However, in fetuses of E18.5 whose circulating red blood cells are derived from definitive hematopoiesis we found a distinct population of PIGA− red blood cells in 5 out of 16 LF and LFF fetuses (Fig. 3). Clonogenic progenitor assays similarly revealed no PIGA− colonies from fetal liver cells of 12 E12.5 LF fetuses. In contrast, cultures from fetal liver cells from all five E18.5 LF fetuses with PIGA− red cells grew PIGA− colonies with differentiation into the erythroid and myelomonocytic lineage (data not shown), indicating that progenitors with a recombined loxPiga allele reside in fetal livers of E18.5 but not in fetal livers of E12.5 embryos.
Figure 3.

PIGA− blood cells during embryogenesis. Percentage of PIGA− red blood cells in LF (•) and LFF (○) fetuses at embryonic day E12.5 and E18.5. Circulating red blood cells were analyzed for the expression of CD24. The horizontal lines represent the means.
Tissue Distribution of PIGA− Cells in LF Mice.
The proportion of PIGA− cells found in bone marrow, spleen, and lymph nodes from four LF mice are summarized in Fig. 4 A. Recombination of the loxPiga allele in nonhematopoietic tissues was determined by Southern blot analysis. Representative Southern blots of a 2- and 8-mo-old male LF mouse are shown in Fig. 4 B. The strongest hybridization signal specific for ΔPiga was detected in hematopoietic organs including bone marrow, spleen, thymus, and lymph nodes. Nonhematopoietic organs, including liver, lung, kidney, gut, brain, and cerebellum, showed no or only a faint hybridization signal for ΔPiga. A slightly stronger ΔPiga hybridization signal was detected in heart and muscle tissue and in older animals also in lung, kidney, and brain.
Figure 4.

Distribution of PIGA− cells in different organs. (A) Proportion of PIGA− red, myeloid, B, and T cells in peripheral blood, spleen, lymph nodes, and bone marrow in LF mice as determined by flow cytometry. The horizontal line indicates the mean of the proportion of PIGA− cells in four 6-mo-old male LF mice, each value is represented by an individual dot. RBC, red blood cells; Reti, reticulocytes; Sp, spleen; BM, bone marrow; PB, peripheral blood. (B) LoxPiga recombination in various organs. Southern blot analysis was used to determine degree of loxPiga recombination in different organs of male LF mice. The top panel represents a 2-mo-old and the bottom panel an 8-mo-old LF mouse. DNA was digested with PstI, and blots hybridized with a Sac-Bam Piga DNA probe. The 10.4-kb fragment corresponds to loxPiga and the 8.1-kb fragment to ΔPiga. Sp, spleen; Th, thymus; Ln, node; Bm, bone marrow; Li, liver; H, heart; Lu, lung; K, kidney; G, gut; M, muscle; T, testis; B, brain; C, cerebellum.
All mice were healthy until the age of about 12 mo. There were no obvious signs of anemia, hemoglobinuria, or thrombosis. In mice with a high proportion of PIGA− red cells the hemoglobin values, although still within normal limits, were lower then in control mice, whereas the reticulocyte counts were higher. The absolute neutrophil, B, and T cell counts did not differ between mice with a high proportion of PIGA− blood cells and normal mice at all times analyzed (see also Table ). At the age of 12 mo, male LF mice developed neuromuscular symptoms including weight loss, decreased overall activity, general muscular weakness, and a wide gait. Neuromuscular symptoms were observed in all male LF mice but not in female LF mice nor in control mice. Histological examination of brain and skeletal muscle from the affected male mice showed no obvious pathological changes. We cannot exclude selective neuronal loss or damage to a small subpopulation of central or peripheral neurons; however, no global abnormalities in nervous system structure were found to account for the observed symptoms. Neuromuscular symptoms also developed in LF mice transplanted with normal bone marrow cells, but did not occur in wild-type (wt) mice transplanted with PIGA− bone marrow cells. This suggests that neuromuscular symptoms were not due to the high number of PIGA− blood cells. To test for loxPiga recombination in individual neurons we assayed sections of the brain from a 12-mo-old LF male for β-galactosidase activity. Small groups of neuronal cells with blue staining nuclei were found particularly in the thalamus (data not shown), indicating that PIGA inactivation occurs in specialized neurons.
Table 1.
Blood Values in Control, LF, and LFF Mice at 8 Mo of Age
| Animal | Hemoglobin | Retis | Platelets | PMN | B cells | T cells |
|---|---|---|---|---|---|---|
| g/dl | % | 103/μl | 103/μl | 103/μl | 103/μl | |
| Controls (n = 11) | 14.6 ± 0.7 | 4.1 ± 0.7 | 1,244 ± 114 | 1.9 ± 1.0 | 3.2 ± 1.4 | 1.8 ± 0.5 |
| LF males (n = 11) | 13.5 ± 1.4 | 5.2 ± 1.7 | 1,520 ± 165 | 1.8 ± 0.8 | 4.0 ± 1.3 | 2.2 ± 0.8 |
| LFF males (n = 11) | 12.7 ± 1.2 | 6.2 ± 1.5 | 1,184 ± 561 | 1.2 ± 1.0 | 3.7 ± 2.5 | 2.2 ± 1.2 |
Some LF mice older than 12 mo developed hemangiomas and B cell lymphomas in spleen and lymph nodes. Identical tumors were also observed in F mice from two different founder lines, indicating that they were caused by the expression of FES-Cre.
Recombination of the loxPiga Allele in LF Mice Occurs in c-kit+CD34+Lin− Progenitor Cells before the GEMM Stage.
To identify the earliest cell stage of differentiation that lacks GPI-linked proteins due to an inactive Piga gene, we analyzed the c-kit+Lin− and CD34+Lin− bone marrow cells for the expression of GPI-linked proteins. In control mice, only 7% (2–18%, n = 4) of c-kit+Lin− cells and 10% (2–19%, n = 3) of CD34+Lin− cells were deficient in CD24. In contrast, in four LF mice the proportion of CD24-negative cells was 75% (71–79%) in c-kit+Lin− and 91% (83–96%) in CD34+Lin− bone marrow cells (Fig. 5). Interestingly, the proportion of c-kit+Lin− and CD34+Lin− progenitor cells in all four LF mice was larger than in control animals. This might be explained by an increased and left shifted erythropoiesis in LF mice due to the shortened half-life of PIGA− red cells 22. Experiments investigating hemolysis in LF mice are currently ongoing. To further determine the stage of hematopoietic differentiation in which FES-Cre–mediated recombination occurs, we next performed progenitor culture assays. Table summarizes the expression of GPI-linked proteins in 313 individual CFUs grown from bone marrow cells of seven LF animals. The majority of colonies consisted of either PIGA+ cells or PIGA− cells. Only 4 out of 75 CFU-GEMM colonies were mixed colonies with cells expressing and cells lacking GPI-linked proteins. These results suggest that recombination of the loxPiga allele occurs mostly before the CFU-GEMM stage. As for the few mixed colonies, we cannot determine conclusively whether loxPiga recombination occurred during the maturation of the individual colony, or if they are due to the accidental close seeding of two progenitor cells. The finding that in all four mixed CFU-GEMMs the proportion of PIGA− cells were the same in myeloid and erythroid cells supports the latter.
Figure 5.

Proportions of PIGA− bone marrow progenitor cells. Bone marrow mononuclear cells were stained with antibodies against c-kit or CD34 and an antibody mixture to exclude lineage-committed cells. CD24 was used as a marker for GPI-linked proteins. The c-kit+Lin− and CD34+Lin− progenitors from LF (top panel) and control (bottom panel) mice are displayed in the top left quadrant of the first and third dot blot, respectively. The second and fourth dot blot in each panel is gated for lineage negative cells (Lin−). PIGA− c-kit+Lin− progenitors (LF 79%, control 3%) are displayed in the top left quadrant of the second dot blot, PIGA− CD34+Lin− progenitors (LF 88%, control 2%) are displayed in the upper left quadrant of the forth dot blot.
Table 2.
PIGA Phenotype of CFUs in Clonogenic Assays
| Colonies | BFU-E | CFU-GM | CFU-G | CFU-GEMM | Total CFU |
|---|---|---|---|---|---|
| PIGA+ | 25 | 46 | 58 | 39 | 168 |
| PIGA− | 24 | 45 | 36 | 32 | 137 |
| Mixed | 1 | 1 | 2 | 4 | 8 |
| Total | 50 | 92 | 96 | 75 | 313 |
BFU-E, burst-forming unit erythroid; G, granulocytes; GEMM, granulocytes, erthyrocytes, megakaryocytes, monocytes.
PIGA− Cells in LF Mice Have Long-Term Repopulating Abilities.
We next tested whether LF mice have PIGA− hematopoietic progenitor cells that have long-term bone marrow repopulating potential. To enrich for PIGA− cells we incubated bone marrow cells with the toxin aerolysin that selectively lyses wt cells 26. First, we transplanted aerolysin-treated and sham-treated bone marrow cells from wt mice (Fig. 6 A). In these experiments, the treated cells were from a CD45.2+ mouse and were transplanted along with untreated cells from a CD45.1+ mouse to ensure survival. The ability of the treated cells to engraft is monitored by measuring the engrafted CD45.2 cells. 1 and 3 mo after transplantation only low numbers of donor-derived aerolysin-treated cells were detected in the peripheral blood of recipient mice (1 mo: average 7.2%, range 5.6–10.0%; 3 mo: average 4.2%, 3.4–5.5%, n = 4). In contrast, in two mice transplanted with sham-treated marrow cells, 62 and 69% of white blood cells were donor derived, which is consistent with the ratio of transplanted donor cells and cells injected for radioprotection. These findings demonstrate that aerolysin efficiently lyses HSCs from wt mice. Next, we transplanted aerolysin- and sham-treated bone marrow cells from a female LF mouse. 1 mo after transplantation, analysis of recipient mice that have received bone marrow cells treated with aerolysin revealed 45% donor chimerism (32–55%) with 93% (89–96%, n = 6) of PIGA− white blood cells (Fig. 6 B). 3 mo after transplantation high donor chimerism (72%, 65–83%) and the high proportion of PIGA− cells (97%, 95–97%) persisted. Mice receiving sham-treated LF bone marrow cells showed 59 and 80% of donor chimerism, with only 43 and 53% donor-derived PIGA− cells 1 and 3 mo after transplantation (Fig. 6 B). Finally, in secondary transplantation experiments, the donor chimerism was stable (average 90%, range 87–94%, n = 3), and 99.3, 99.4, and 99.6% of LF donor-derived cells were deficient in GPI-linked proteins 4 mo after transplantation.
Figure 6.
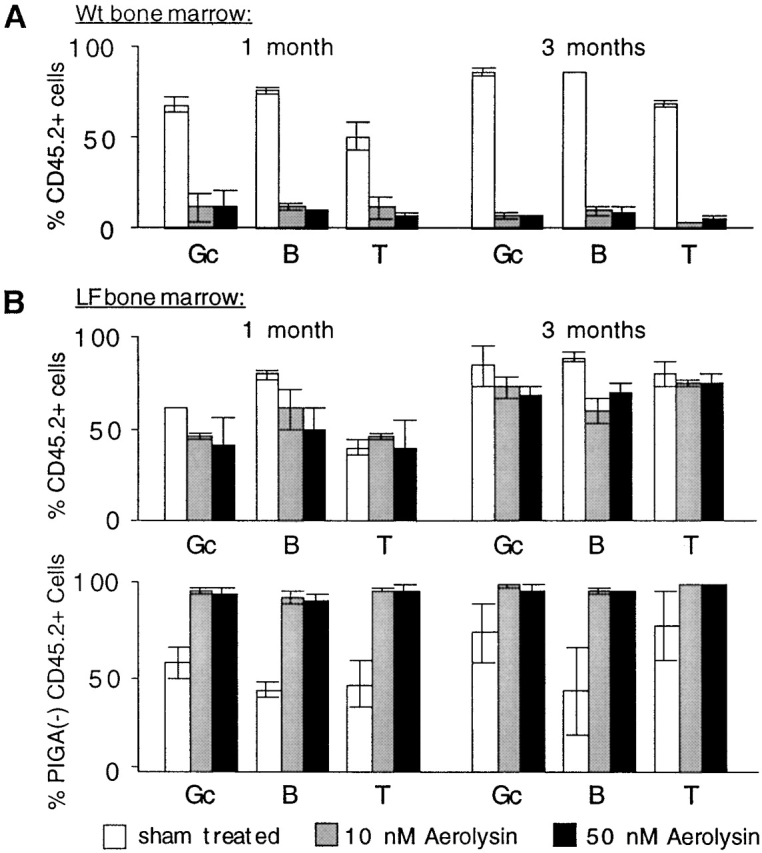
Bone marrow reconstitution using aerolysin-treated bone marrow cells. (A) Donor chimerism after aerolysin treatment of wt bone marrow cells. 106 bone marrow cells of a CD45.2+ wt mouse were incubated with aerolysin and injected into lethally irradiated recipient mice of the CD45.1 allotype. 10 and 50 nM concentrations of aerolysin were used. 0.5 × 106 nonirradiated, congenic CD45.1+ marrow cells were given simultaneously for radioprotection. Peripheral blood cells of recipients (n = 4) were analyzed by flow cytometry. Columns represent the proportion of donor-derived chimerism (percentage of CD45.2+ cells). (B) Bone marrow reconstitution with PIGA− cells. 106 marrow cells of a female CD45.2+ LF mouse (red blood cells 19%, granulocytes 37%, B cells 19%, and T cells 52% PIGA− cells) were incubated with aerolysin and transplanted into lethally irradiated CD45.1+ recipient mice (n = 6). 0.5 × 106 nonirradiated, CD45.1+ marrow cells were coinjected for radioprotection. The top panel represents the proportion of donor-derived, CD45.2+ cells. The bottom panel shows the proportion of donor-derived CD45.2+ cells that lack GPI-anchored surface molecules. Shown are the means with standard deviations. Gc, granulocytes; B, B cells; T, T cells.
Discussion
In patients with PNH, a somatic mutation of the PIGA gene occurs in a multipotent progenitor cell that probably is a HSC 3 4. To further study the pathogenesis and pathophysiology of this disorder and to develop a possible cure, a mouse model would be invaluable. Here we report on the generation of a mouse line in which a somatic mutation leads to PIGA− cells in all blood cell lineages. In contrast to our previously reported mice which were mosaic for cells with an inactive Piga gene and had only very low numbers of PIGA− cells in peripheral blood limiting the possibility of functional studies 22, we have now produced animals that have almost exclusively PIGA− blood cells. Analysis of hematopoietic progenitors demonstrates that in our LF mice inactivation of the Piga gene occurs in the HSC.
We chose to express the Cre recombinase under the c-fes LCR because of known high expression of FES in early myeloid progenitor cells 12 13 and the convenience of using an LCR for the expression of a transgene. As the Piga gene is on the X chromosome, only a single Cre-mediated recombination event is required to cause the loss of GPI-linked proteins on the cell surface. In the case of females, the loss only occurs when the recombined allele is on the active X chromosome. Guided by previous expression of human FES by the c-fes LCR in transgenic animals 20 21, we expected a predominantly myelo-monocyte–specific expression of the PIGA− phenotype in LF animals. Thus, the constant time-dependent increase of PIGA− cells in all blood cell lineages including the erythroid lineage (Fig. 2a and Fig. b) was a surprise. In LF mice the loss of GPI-linked proteins occurred for the majority of hematopoietic cells in c-kit+CD34+Lin− progenitor cells before the CFU-GEMM stage of differentiation. No additional Piga gene inactivation was found in vitro in clonogenic progenitors during myeloid differentiation (Table ). The increased proportion of PIGA− granulocytes in peripheral blood compared with the bone marrow (Fig. 4 A), however, might indicate low level Piga gene inactivation in vivo during myeloid differentiation. Alternative explanations would be early release of PIGA− granulocytes or a prolonged survival in circulation. Functional studies of PIGA− blood cells are ongoing.
Southern blot analysis suggests that FES-Cre–mediated Piga gene inactivation occurs predominantly, although not exclusively, in hematopoietic cells. Old LF mice have stronger ΔPiga specific hybridization signals in nonhematopoietic organs compared with young LF animals, suggesting the slow accumulation of a small proportion of PIGA− cells in various tissues. In some organs, such as the lung and gut, this might be due to increasing lymphocytic infiltrates in older animals. In other tissues, such as the heart, this is more likely due to loxPiga recombination in non-hematopoietic cells, for example the vascular endothelium. However, we did not detect β-galactosidase activity in endothelial nuclei (data not shown). In contrast, recombination of the loxPiga allele was found in neuronal cells, causing a neuromuscular deficit in aging male LF mice. We can not exclude that low level of loxPiga recombination occurs in additional cells that due to the loss of GPI-linked proteins die and thus are not accounted for. However, necropsy of 24 mice did not reveal any macroscopic or histological abnormalities.
Two characteristics define a HSC. First, they have the potential to differentiate into all blood cell lineages. Second, they can generate more stem cells, a process of self-renewal. To test whether PIGA− HSCs exist in LF mice, bone marrow reconstitution experiments were designed to allow purified donor-derived PIGA− hematopoiesis to be evaluated over the course of several months. In the presence of a radioprotective dose of adult bone marrow cells, aerolysin-treated marrow cells competed for hematopoiesis in irradiated recipient mice. Aerolysin is a pore-forming toxin from Aeromonas hydrophilia that needs to bind to GPI-anchored surface proteins of target cells to develop lytic activity 26. Wt aerolysin-treated cells were efficiently out-competed, indicating that aerolysin efficiently lyses wt bone marrow stem cells (Fig. 6 A). In contrast, blood cells derived from aerolysin treated cells of LF mice were found at levels up to 45% for more than 3 mo and were almost exclusively of the PIGA− phenotype (Fig. 6 B). The proportion of LF-derived cells corresponds to the proportion of radioprotective cells and aerolysin-resistant PIGA− hematopoietic cells transplanted. This demonstrates that LF mice have PIGA− bone marrow cells that efficiently compete for hematopoiesis in an irradiated recipient mouse. Long-term repopulation ability of PIGA− cells was demonstrated by secondary transplantation experiments leading to persistent PIGA− hematopoiesis in secondary recipient mice. These findings prove unambiguously that LF mice have PIGA− HSCs and that recombination of the loxPiga allele must occur in HSCs. Inactivation of PIGA in blood cells of LF mice is the first clear demonstration of transgene expression in HSCs. Interestingly, loxPiga recombination was not found in red blood cells derived from primitive hematopoiesis, suggesting that FES-Cre causes recombination of the loxPiga allele in HSCs from definitive but not from primitive hematopoiesis.
Our findings of FES-Cre–mediated loxPiga recombination differ significantly from the myelo-monocyte–specific transgene expression of the human c-fes LCR reported previously 20 21 27. We can think of several explanations. First, Cre-mediated recombination of the loxPiga allele is irreversible and thus is very sensitive in tracing Piga gene inactivation in a small population of progenitor cells that might be easily missed in the analysis of a temporarily expressed transgene. Second, a certain threshold level of Cre might be required for successful recombination of the loxPiga allele, which might only be reached in a small number of stem cells, but not in rapidly dividing myeloid progenitors. Third, the loss of Piga gene function in end-differentiated, short lived, nondividing cells might not become apparent due to the long half-life of many GPI-linked proteins 28 29. Although transgene expression in mice via a human LCR not necessarily mirrors human or murine gene expression in vivo, our findings suggest that FES in addition to its previously described expression pattern might also be expressed in HSCs of definitive hematopoiesis. The isolation of murine c-fes messenger RNA in cells enriched for murine liver stem cells 18 and the defect in bone marrow HSCs identified in PU.1-deficient mice 30, PU.1 being the main transcription factor that regulates c-fes gene expression 31, support this hypothesis.
LF mice with almost 100% of PIGA− blood cells have lower hemoglobin levels and higher reticulocyte counts compared with normal mice. However, in contrast to patients with PNH, LF mice do not develop obvious anemia, hemoglobinuria, or thrombosis. Investigations are currently ongoing to test whether the comparatively modest decrease in hemoglobin levels is due to the low hemolytic activity of complement in C57Bl/6 mice or rather caused by the expression of an additional complement regulatory protein in mice, Crry, which is a transmembrane protein and thus is not affected by the lack of GPI-anchor molecules. White blood cell values and platelet counts in LF mice were normal (Table ), indicating that PIGA− HSCs are able to maintain normal blood cell values. Thus, our LF mice demonstrate that pancytopenia, in particular thrombocytopenia, which are often found in patients with PNH, are not due to the lack of GPI-linked proteins, but rather caused by the underlying bone marrow failure that accompanies or precedes PNH. In contrast to our previous mouse model for PNH 22, FES-Cre–mediated recombination of the loxPiga allele provides us with mice that due to a somatic mutation in HSCs in the bone marrow have high percentages of PIGA− blood cells in all blood cell lineages. In this respect, LF mice mimic accurately the situation of PIGA− cells in patients with PNH. Mice with almost exclusively PIGA− blood cells promise to be a powerful new tool to investigate the functional consequences caused by the loss of GPI-linked proteins in hematopoiesis, immunosurveillance, and leukemogenesis. In addition, the possibility to easily and reproducibly target gene expression to HSCs using the human c-fes LCR promises new and exciting possibilities in stem cell biology.
Acknowledgments
We thank Kevin A. Roth for his expertise in neuropathological studies and Joshua R. Sanes for advice in the β-gal staining. Aerolysin was a generous gift of T. Buckley, University of Victoria, Victoria, Canada. We are grateful to Daniel C. Link and Timothy Graubert for discussions, and Philip Mason for reading the manuscript.
This work was supported by grant RO1-HL-56678 from the National Institutes of Health, the Mallinckrodt Foundation, Howard Hughes Medical Institute, American Cancer Society IRG I#N-36-39, and the McDonnell Foundation. M. Bessler is awarded the Junior Faculty Award of the American Society of Hematology. P. Keller is supported by the Novartis Foundation and the EMDO Stiftung, Switzerland.
Footnotes
Abbreviations used in this paper: GPI, glycosyl phosphatidylinositol; HSC, hematopoietic stem cell; IRES, internal ribosome entry site; LCR, locus control region; PIGA, phosphatidylinositol glycan class A; PNH, paroxysmal nocturnal hemoglobinuria; wt, wild-type.
References
- Rosse W.F. Paroxysmal nocturnal hemoglobinuria as a molecular disease. Medicine. 1997;76:63–93. doi: 10.1097/00005792-199703000-00001. [DOI] [PubMed] [Google Scholar]
- Dacie J.V., Luzzatto L. Paroxysmal nocturnal haemoglobinuria. In: Wetherall D.J., Warrell A., Ledigham L., editors. Oxford Textbook of Medicine. 3rd edition, Vol. 3. Oxford University Press; Oxford, UK: 1996. pp. 3449–3452. [Google Scholar]
- Takeda J., Miyata T., Kawagoe K., Iida Y., Endo Y., Fujita T., Takahashi M., Kitani T., Kinoshita T. Deficiency of the GPI anchor caused by a somatic mutation of the PIG-A gene in paroxysmal nocturnal hemoglobinuria. Cell. 1993;73:703–711. doi: 10.1016/0092-8674(93)90250-t. [DOI] [PubMed] [Google Scholar]
- Bessler M., Mason P.J., Hillmen P., Miyata T., Yamada N., Takeda J., Luzzatto L., Kinoshita T. Paroxysmal nocturnal haemoglobinuria (PNH) is caused by somatic mutations in the PIG-A gene. EMBO J. 1994;13:110–117. doi: 10.1002/j.1460-2075.1994.tb06240.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe R., Inoue N., Westfall B., Taron C.H., Orlean P., Takeda J., Kinoshita T. The first step of glycosylphosphatidylinositol biosynthesis is mediated by a complex of PIG-A, PIG-H, PIG-C and GPI1. EMBO J. 1998;17:877–885. doi: 10.1093/emboj/17.4.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davitz M.A., Low M.G., Nussenzweig V. Release of decay-accelerating factor (DAF) from the cell membrane by phosphatidylinositol-specific phospholipase C (PIPLC). Selective modification of a complement regulatory protein. J. Exp. Med. 1986;163:1150–1161. doi: 10.1084/jem.163.5.1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawagoe K., Kitamura D., Okabe M., Taniuchi I., Ikawa M., Watanabe T., Kinoshita T., Takeda J. Glycosylphosphatidylinositol-anchor-deficient miceimplications for clonal dominance of mutant cells in paroxysmal nocturnal hemoglobinuria. Blood. 1996;87:3600–3606. [PubMed] [Google Scholar]
- Rosti V., Tremml G., Soares V., Pandolfi P.P., Luzzatto L., Bessler M. Murine embryonic stem cells without pig-a gene activity are competent for hematopoiesis with the PNH phenotype but not for clonal expansion. J. Clin. Invest. 1997;100:1028–1036. doi: 10.1172/JCI119613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer B., Henderson N. Site-specific DNA recombination in mammalian cells by the Cre recombinase of bacteriophage P1. Proc. Natl. Acad. Sci. USA. 1988;85:5166–5170. doi: 10.1073/pnas.85.14.5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobe C.G., Nagy A. Conditional genome alteration in mice. Bioessays. 1998;20:200–208. doi: 10.1002/(SICI)1521-1878(199803)20:3<200::AID-BIES3>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Rajewsky K., Gu H., Kuhn R., Betz U.A., Muller W., Roes J., Schwenk F. Conditional gene targeting. J. Clin. Invest. 1996;98:600–603. doi: 10.1172/JCI118828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman R.A., Gabrilove J.L., Tam J.P., Moore M.A., Hanafusa H. Specific expression of the human cellular fps/fes-encoded protein NCP92 in normal and leukemic myeloid cells. Proc. Natl. Acad. Sci. USA. 1985;82:2379–2383. doi: 10.1073/pnas.82.8.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smithgall T.E., Yu G., Glazer R.I. Identification of the differentiation-associated p93 tyrosine protein kinase of HL-60 leukemia cells as the product of the human c-fes locus and its expression in myelomonocytic cells. J. Biol. Chem. 1988;263:15050–15055. [PubMed] [Google Scholar]
- Care A., Mattia G., Montesoro E., Parolini I., Russo G., Colombo M.P., Peschle C. c-fes expression in ontogenetic development and hematopoietic differentiation. Oncogene. 1994;9:739–947. [PubMed] [Google Scholar]
- Greer P., Haigh J., Mbamalu G., Khoo W., Bernstein A., Pawson T. The Fps/Fes protein-tyrosine kinase promotes angiogenesis in transgenic mice. Mol. Cell. Biol. 1994;14:6755–6763. doi: 10.1128/mcb.14.10.6755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senis Y., Zirngibl R., McVeigh J., Haman A., Hoang T., Greer P.A. Targeted disruption of the murine fps/fes proto-oncogene reveals that Fps/Fes kinase activity is dispensable for hematopoiesis. Mol. Cell. Biol. 1999;19:7436–7446. doi: 10.1128/mcb.19.11.7436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackenmiller R., Kim J., Feldman R.A., Simon M.C. Abnormal Stat activation, hematopoietic homeostasis, and innate immunity in c-fes-/- mice. Immunity. 2000;13:397–407. doi: 10.1016/s1074-7613(00)00039-x. [DOI] [PubMed] [Google Scholar]
- Phillips R.L., Ernst R.E., Brunk B., Ivanova N., Mahan M.A., Deanehan J.K., Moore K.A., Overton G.C., Lemischka I.R. The genetic program of hematopoietic stem cells. Science. 2000;288:1635–1640. doi: 10.1126/science.288.5471.1635. [DOI] [PubMed] [Google Scholar]
- Roebroek A.J., Schalken J.A., Verbeek J.S., Van den Ouweland A.M., Onnekink C., Bloemers H.P., Van de Ven W.J. The structure of the human c-fes/fps proto-oncogene. EMBO J. 1985;4:2897–2903. doi: 10.1002/j.1460-2075.1985.tb04020.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer P., Maltby V., Rossant J., Bernstein A., Pawson T. Myeloid expression of the human c-fps/fes proto-oncogene in transgenic mice. Mol. Cell. Biol. 1990;10:2521–2527. doi: 10.1128/mcb.10.6.2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heydemann A., Warming S., Clendenin C., Sigrist K., Hjorth J.P., Simon M.C. A minimal c-fes cassette directs myeloid-specific expression in transgenic mice. Blood. 2000;96:3040–3048. [PubMed] [Google Scholar]
- Tremml G., Dominguez C., Rosti V., Zhang Z., Pandolfi P.P., Keller P., Bessler M. Increased sensitivity to complement and a decreased red blood cell life span in mice mosaic for a nonfunctional Piga gene. Blood. 1999;94:2945–2954. [PubMed] [Google Scholar]
- Ellis L., Clauser E., Morgan D.O., Edery M., Roth R.A., Rutter W.J. Replacement of insulin receptor tyrosine residues 1162 and 1163 compromises insulin-stimulated kinase activity and uptake of 2- deoxyglucose. Cell. 1986;45:721–732. doi: 10.1016/0092-8674(86)90786-5. [DOI] [PubMed] [Google Scholar]
- Keller P., Tremml G., Rosti V., Bessler M. X inactivation and somatic cell selection rescue female mice carrying a Piga-null mutation. Proc. Natl. Acad. Sci. USA. 1999;96:7479–7483. doi: 10.1073/pnas.96.13.7479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes J.R., Johnson Y.R., Kotzbauer P.T., Mudd J., Hanley T., Martinou J.C., Merlie J.P. Selective expression of an acetylcholine receptor-lacZ transgene in synaptic nuclei of adult muscle fibers. Development. 1991;113:1181–1191. doi: 10.1242/dev.113.4.1181. [DOI] [PubMed] [Google Scholar]
- Diep D.B., Nelson K.L., Raja S.M., Pleshak E.N., Buckley J.T. Glycosylphosphatidylinositol anchors of membrane glycoproteins are binding determinants for the channel-forming toxin aerolysin. J. Biol. Chem. 1998;273:2355–2360. doi: 10.1074/jbc.273.4.2355. [DOI] [PubMed] [Google Scholar]
- Olson M.C., Scott E.W., Hack A.A., Su G.H., Tenen D.G., Singh H., Simon M.C. PU. 1 is not essential for early myeloid gene expression but is required for terminal myeloid differentiation. Immunity. 1995;3:703–714. doi: 10.1016/1074-7613(95)90060-8. [DOI] [PubMed] [Google Scholar]
- Moss D.J., White C.A. Solubility and posttranslational regulation of GP130/F11 - a neuronal GPI-linked cell adhesion molecule enriched in the neuronal membrane skeleton. Eur. J. Cell. Biol. 1992;57:59–65. [PubMed] [Google Scholar]
- Hollander N. Membrane dynamics of the phosphatidylinositol-anchored form and the transmembrane form of the cell adhesion protein LFA-3. J. Biol. Chem. 1992;267:5663–5667. [PubMed] [Google Scholar]
- Fisher R.C., Lovelock J.D., Scott E.W. A critical role for PU.1 in homing and long-term engraftment by hematopoietic stem cells in the bone marrow. Blood. 1999;94:1283–1290. [PubMed] [Google Scholar]
- Heydemann A., Juang G., Hennessy K., Parmacek M.S., Simon M.C. The myeloid-cell-specific c-fes promoter is regulated by Sp1, PU.1, and a novel transcription factor. Mol. Cell. Biol. 1996;16:1676–1686. doi: 10.1128/mcb.16.4.1676. [DOI] [PMC free article] [PubMed] [Google Scholar]