

Abstract
An increasing number of studies have documented the central role of T cell costimulation in autoimmunity. Here we show that the autoimmune diabetes-prone nonobese diabetic (NOD) mouse strain, deficient in B7-2 costimulation, is protected from diabetes but develops a spontaneous autoimmune peripheral polyneuropathy. All the female and one third of the male mice exhibited limb paralysis with histologic and electrophysiologic evidence of severe demyelination in the peripheral nerves beginning at 20 wk of age. No central nervous system lesions were apparent. The peripheral nerve tissue was infiltrated with dendritic cells, CD4+, and CD8+ T cells. Finally, CD4+ T cells isolated from affected animals induced the disease in NOD.SCID mice. Thus, the B7-2–deficient NOD mouse constitutes the first model of a spontaneous autoimmune disease of the peripheral nervous system, which has many similarities to the human disease, chronic inflammatory demyelinating polyneuropathy (CIDP). This model demonstrates that NOD mice have “cryptic” autoimmune defects that can polarize toward the nervous tissue after the selective disruption of CD28/B7-2 costimulatory pathway.
Keywords: autoimmunity, peripheral neuropathy, B7-2 costimulation, animal model, NOD mice
Introduction
Engagement of the T cell receptor alone is insufficient to cause conventional activation of T cells and differentiation into effector cells. Rather, costimulatory signals are required generally to promote the expansion of antigen-reactive populations of T cells, migration of cells to sites of inflammation, and the production of soluble mediators of inflammation 1 2 3 4. One of the prominent costimulatory molecules, CD28, interacts with B7-1 (CD80) and B7-2 (CD86) on antigen-presenting cells 4. Drugs that block this critical costimulatory pathway have been shown to inhibit both the development and progression of many autoimmune diseases 5 6 7 8 9 10. Cytotoxic T lymphocyte antigen (CTLA)4-Ig, a soluble antagonist of the CD28/B7 pathway, completely inhibits the development of experimental autoimmune encephalitis and the onset of collagen-induced arthritis as well as other autoimmune diseases 8 9 10. B7-1 and B7-2 molecules have been shown to play distinct roles in several murine models of autoimmune diseases, including type I diabetes and experimental autoimmune encephalitis 6 7. We have previously reported that blockade of B7-1 in the spontaneous autoimmune NOD mouse strain induced diabetes with faster and higher incidence than untreated NOD animals 7. In contrast, in this study we show that blockade of B7-2 prevented diabetes; however, autoimmunity is redirected from the pancreas to the peripheral nerves, leading to the development of a CD4+ T cell–mediated spontaneous autoimmune peripheral neuropathy in these mice.
Materials and Methods
Mice.
B7-2 knockout (KO) mice 11 were backcrossed to NOD mice for nine generations and then intercrossed to generate KO mice. The genotypes were determined by PCR using B7-2–specific primers (sense: TAT-TTC-AAT-GGG-ACT-GCA-TAT-C, antisense: CGA-TCA-CTG-ACA-GTT-CTG-TTA). NOD mice were purchased from Taconic and then bred in the animal care facilities at The University of Chicago or UCSF. Experiments were complied with the standards set out in the guidelines of The University of Chicago and UCSF.
Histology and Immunohistochemistry.
Hematoxylin and eosin staining was performed on 5-μm sections of formalin-fixed tissues. Toluidine blue stained 1-μm sections were prepared from formalin fixed, glutaraldehyde postfixed nerve segments after plastic embedding. Immunohistochemistry was performed using the avidin-biotin method 12. Tissues were snap frozen in OCT and 5-μm cryostat sections were incubated with: rat mAbs to CD3 (L363–29B), CD4 (GK1.5), CD8 (3.155); hamster mAbs to dendritic cells (N418); or species- and isotype-specific nonreactive control mAbs overnight at 4°C. Room temperature incubation with biotinylated rabbit anti–rat or goat anti–hamster secondary Ab (Vector Laboratories) for 30 min was followed by a 1-h incubation with streptavidin-biotin complex (Vector Laboratories). Diaminobenzidine was used as the chromogen. Tease preparations of peripheral nerve were performed using published methods 13.
Fluorescence Immunohistochemistry.
Slides were stained using Tyramide Signal Amplification (TSA) Direct Kit (NEN Life Science Products) according to the manufacturer's instructions. Sections from each group were thawed, air-dried, fixed in acetone at room temperature, and rehydrated in 1× PBS. Nonspecific staining was blocked using anti-CD16/CD32, (Fcγ III/II receptor, 2.4G2; BD PharMingen), and an avidin/biotin blocking kit (Vector Laboratories) in addition to the blocking reagent provided by the TSA kit. Slides were stained with the following biotin-conjugated Abs: anti-CD4 (H129.19) and anti-CD80 (16–10A1) (BD PharMingen). Sections were counterstained with 4,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich) and then coverslipped with Vectashield mounting medium (Vector Laboratories). Slides were examined by epifluorescence using a chroma triple-band filter (Chroma Technology Corporation). Four serial sections from each sample per group were analyzed at 100× and 400×.
Electrophysiological Studies.
Studies were performed on sciatic nerves on mice anesthetized with Nembutal (80 mg/kg). Recording needle electrodes were placed subcutaneously in the footpad. The sciatic nerve was stimulated distally at the ankle and proximally at the sciatic notch by a 0.1-ms rectangular pulse using a pair of monopolar needle electrodes. Recordings were obtained on a TECA Neurostar electromyograph (TECA Corp.) with a filter setting of 2 Hz to 10 kHz. The distal or proximal latencies corresponding to the time laps between the distal or proximal stimulations and the beginning of the first compound muscle action potential were measured. Conduction velocities were calculated by dividing the distance between distal and proximal stimulation sites with the difference between the distal and proximal latencies. The peak-to-peak amplitude of compound muscle action potentials was measured. The comparison of this amplitude between normal and neuropathic mice was used to determine partial conduction block.
Adoptive Transfer.
CD4+ T cells (positive fraction) and CD4+ T cell–depleted cells (flow through) were separated on a VS+ column (Miltenyi Biotec) after staining with anti–CD4-biotin (GK1.5) and streptavidin microbeads (Miltenyi Biotec). Total cells or purified cells were stimulated with 1 μg/ml of anti-CD3 (145–2C11) and anti-CD28 (PV-1) for 48 h. IL-2 (20 U/ml) was added to the culture of the CD4+ T cell–depleted cells. 8–15 × 106 total cells and CD4+ T cell–depleted cells (<0.3% CD4+ T cells) and 4–6 × 106 CD4+ T cells (∼92% pure) were injected intravenously into 4–8-wk-old NOD.SCID mice.
Flow Cytometry Analysis.
Spleens and sciatic nerves were cut into small fragments and incubated in RPMI 1640 supplemented with 1.6 mg/ml collagenase (type IV; Sigma-Aldrich) and 200 μg/ml DNase I (Boehringer) at 37°C for 30 min. Cells were dissociated by repeated pipetting, reincubated at 37°C for 10 min, washed with PBS 3% serum, and resuspended in staining buffer (PBS, 3% serum, 0.02% azide). Cells were double stained with FITC-conjugated CD11b (M1/70) and PE-conjugated CD45 and biotin-conjugated CD11c (N418) mAbs followed by streptavidin-allophycocyanin staining and analyzed on a FACSCalibur™ (Becton Dickinson).
Results
This study was designed to explore the role of costimulation in the spontaneous autoimmune-prone NOD mouse strain, a prototypic murine model of type 1 Diabetes Mellitus 14. In these mice, insulitis first appears between 2–4 wk of age, and diabetes is detected beginning at 15 wk of age. Diabetes develops in >70% of female mice but occurs in only 30% of male mice. Elimination of B7-2 expression by breeding NOD mice onto the B7-2–deficient background 11 prevented the development of hyperglycemia in virtually 100% of both the female (Fig. 1 a) and male (data not shown) animals. Similar results were observed after anti–B7-2 Ab therapy (4; Fig. 1 c). However, beginning at 20 wk of age, the B7-2 KO NOD mice began to exhibit a symmetrical mild hind leg paralysis, slowly progressing to a generalized limb paralysis such that by 32 wk of age, 100% of female and 30% of the male B7-2 KO mice had succumbed to the disease (Fig. 1 b). The majority of affected animals exhibited full hind leg paralysis, moderate fore leg paralysis, and could only walk with difficulty (Fig. 1 b, inset). There was no evidence of central nervous system (CNS) disorders. The disease was never observed in B7-2 KO mice bred to the C57BL/6 or 129/Sv nonautoimmune mouse background (unpublished data). However, a similar paralytic disease was noted in one third of NOD mice treated with anti–B7-2 mAb but not in PBS-treated mice (Fig. 1 d) or NOD mice treated with isotypic control Abs (data not shown). These results support the important regulatory role of B7-2 in the development of this disease as two independent approaches, genetic modification of B7-2 in NOD mice and anti–B7-2 mAb treatment of wild-type (WT) NOD mice resulted in the same disease. Interestingly, the subset of NOD mice that became diabetic after the anti–B7-2 mAb therapy did not develop the peripheral neuropathy while none of the mice exhibiting the neuropathy became diabetic. The data suggest that elimination of the CD28/B7-2 interactions polarized the autoimmune disorder in the NOD mice from the previously described autoimmune diabetes to a spontaneous autoimmune peripheral polyneuropathy (SAPP).
Figure 1.

B7-2 blockade in NOD mice prevents diabetes but induces a peripheral neuropathy (a and b). B7-2–deficient mice (reference 14) were backcrossed to NOD mice for nine generations and then intercrossed to generate B7-2 WT (B7-2+/+) and B7-2 KO (B7-2−/−) NOD mice. B7-2+/+ (n = 10) and B7-2−/− (n = 10) females were checked weekly for blood glucose levels (a) and B7-2−/− females and males were checked weekly for clinical signs of neuropathy (hind leg weakness and inability to grasp wire bar lid of cages) (b). Inset: neuropathic mouse displaying deterioration of hind limb control and coordination. (c and d) WT NOD females were treated with 50 μg of anti–B7-2 blocking mAb (GL-1) every other day between 2 to 4 wk of age plus at weeks 5, 6, and 7 (n = 6) or with PBS (n = 5). Incidence of diabetes (c) and neuropathy (d) were evaluated at weekly intervals.
The neuropathic mice were examined histologically for evidence of an inflammatory infiltrate in the endocrine and nervous system tissues (Fig. 2). WT NOD mice exhibited significant cellular infiltrates and tissue destruction in the pancreas (Fig. 2 a) and salivary glands (data not shown) typically targeted in this autoimmune-prone strain of mice. In contrast, there was no infiltrate or pathology in the nervous system in these animals (Fig. 2c, Fig. e, Fig. g, and Fig. i). B7-2 KO animals affected with SAPP exhibited strongly reduced infiltrates in pancreatic islets (Fig. 2 b) and salivary glands (data not shown). It should be pointed out that peri-insulitis was present in some mice consistent with a retention of at least some autoreactive diabetic cells. However, insulitis in B7-2KO NOD mice appeared much later than in NOD mice and did not progress from peri-insulitis to severe insulitis. In sharp contrast, neuropathic B7-2KO NOD mice displayed severe inflammation of the peripheral nervous system. The dorsal and ventral spinal roots, the dorsal root ganglia, and peripheral nerves had mononuclear infiltrate (Fig. 2h and Fig. j, and data not shown). The skeletal muscles showed focal neurogenic atrophy in severely affected animals but no cellular infiltrates (data not shown). There were no readily detectable lesions in the brain or spinal cord (Fig. 2d and Fig. f). These results suggested that limb paralysis was a result of damage to peripheral nerves as opposed to a CNS or primary muscle disorder.
Figure 2.
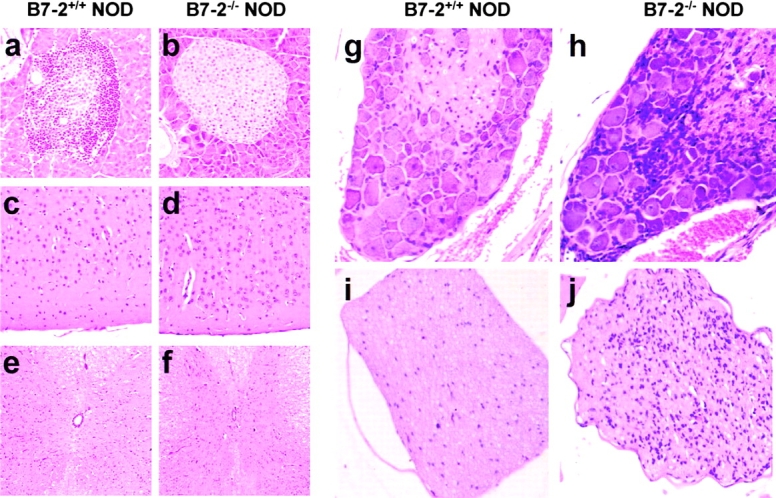
Inflammation in the peripheral nervous system in neuropathic B7-2−/− NOD mice. Hematoxylin and eosin staining on sections of pancreas (a and b), brain cortex (c and d), spinal cord (e and f), dorsal root ganglion (g and h), and sciatic nerve (i and j) in 25–30-wk-old B7-2−/− neuropathic females (b, d, f, h, and j) and age-matched B7-2+/+ control females (a, c, e, g, and i). (a–d and f–j) Original magnification: ×500. (e and f) Original magnification: ×250. B7-2+/+ mice, but not B7-2−/− mice, exhibit severe insulitis (a) whereas B7-2−/− mice, but not B7-2+/+ mice, exhibit inflammation in dorsal root ganglions (h) and nerves (j).
The nerve damage was confirmed by morphological analysis of mice with SAPP. As seen in Fig. 3, significant demyelination was detected in toluidine blue stained one micrometer cross sections from sciatic nerves (Fig. 3 a). Most significantly, on tease preparations, some fibers revealed an increase in the number of Nodes of Ranvier with irregular spacing and irregular thickness of myelin sheets, consistent with ongoing myelin repair by Schwann cells (Fig. 3 b). Electrophysiological studies performed on sciatic nerves revealed abnormalities indicative of a predominantly demyelinating neuropathy. There was prolongation of distal latencies, marked slowing of conduction velocity (17.9 ± 3.2 m/s in B7-2 KO NOD mice [n = 12] versus 50.5 ± 3.8 m/s in WT NOD mice [n = 8]; Student t test: P < 0.00001), and dispersion of compound muscle action potentials (Fig. 3 c). Partial conduction block was also observed in the more severe cases. Indeed, the peak-to-peak amplitudes of compound muscle action potentials were decreased suggesting some axonal damage secondary to the demyelination. This axonal damage was confirmed by a needle exam which revealed fibrillations and positive sharp waves in intrinsic foot muscles, but not in proximal muscles (data not shown).
Figure 3.
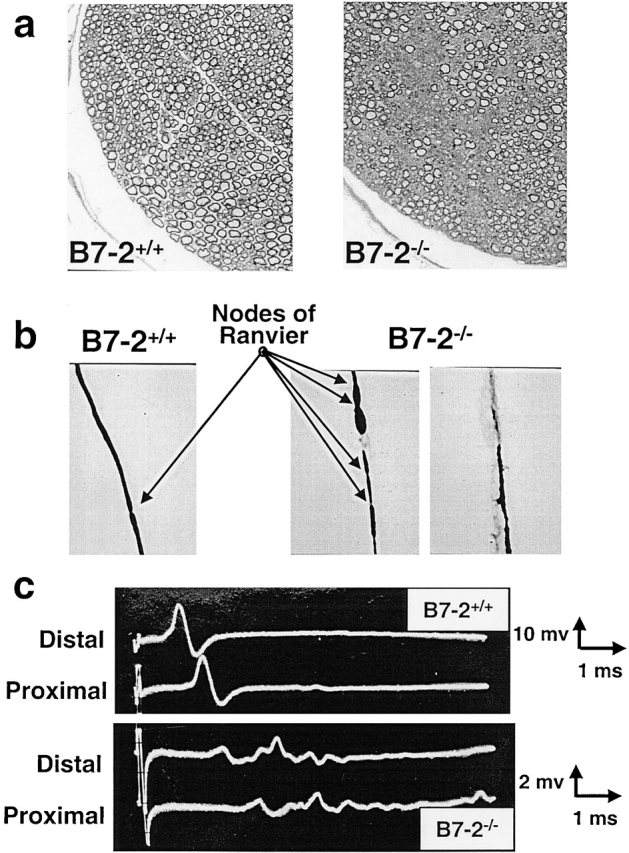
B7-2−/− neuropathic NOD mice have a demyelinating disease. (a) Toluidine blue staining on cross section of sciatic nerves showing destruction of myelin sheets in B7-2−/− neuropathic mice evidenced by lower sheet density (right panel). (b) Individual nervous fibers released by tease preparation of sciatic nerves showing clear evidence of demyelination in B7-2−/− mice (right panels). In addition some fibers have increased number of nodes of Ranvier (arrows) due to myelin sheet regeneration by newly dividing Schwann cells (middle panel). (c) Compound muscle action potentials were obtained upon distal (ankle) and proximal (thigh) stimulation of sciatic nerve. Examples of nerve conduction studies showing prolonged latencies, slowed conduction velocity, and dispersion of compound muscle action potentials in B7-2−/− neuropathic mice compared with controls (bottom panel).
Immunohistochemical studies of the dorsal root ganglia and peripheral nerves demonstrated an intense, predominantly mononuclear, cellular infiltrate composed of dendritic cells and scattered CD4+ T cells and CD8+ T cells (Fig. 4). The cellular infiltrate was detected throughout the sciatic and peripheral nerve tissue, extending to terminal nerve branches within skeletal muscle. The number of leukocytes was greatest in animals displaying the most severe disease. At no time were any immune cell infiltrates observed in nerve tissue derived from the CNS of B7-2 KO mice (data not shown). It remains uncertain whether this is due to a lack of target antigen in these CNS tissues or inaccessibility due to the intact blood–brain barrier in these animals. The histological results were consistent with an autoimmune etiology for the peripheral neuropathy; however, the results could not distinguish between a humoral versus cellular basis for the disease. Therefore, passive and adoptive transfer studies were performed. No NOD.SCID animals receiving sera from SAPP animals developed neuropathy within 20 wk after transfer (data not shown). Next, whole lymph node and spleen cells from neuropathic B7-2 KO NOD mice were cultured for 2 d in vitro with anti-CD3 and anti-CD28 mAbs and transferred into the NOD.SCID recipients. The transfer of 15 × 106 reactivated T cells from the SAPP mice induced a severe peripheral neuropathy (but not diabetes, data not shown) in 100% of NOD.SCID recipients within 7 wk of transfer (Fig. 5 a). By comparison, injection of reactivated T cells from age-matched nondiabetic WT NOD animals induced diabetes (data not shown) but not peripheral neuropathy (Fig. 5 a). These results suggest that SAPP is mediated by antigen-specific T cells. The individual role of CD4+ T cells versus CD8+ T cells in the pathogenesis of the disease was examined. The transfer of purified, reactivated, CD4+ T cells induced the disease in the NOD.SCID mice whereas CD4+ T-depleted cells did not (Fig. 5 b). In the mice transferred with purified CD4+ T cells, affected nerves revealed the presence of an abundant CD4+ T cell population but no CD8+ T cells (data not shown). Furthermore, SAPP was rapidly induced in NOD.SCID mice after transfer of CD8+ T cell–depleted preparations from affected mice (data not shown). These results highlight the necessary and sufficient role played by CD4+ T cells in the effector phase of this autoimmune disease; however, the results do not rule out that pathogenic CD8+ T cells or Abs may play a role in the disease or mediate it after longer periods of time.
Figure 4.

Nerves of B7-2−/− neuropathic NOD mice are highly infiltrated with CD4+, CD8+ T cells and dendritic cells. (Left panel) Frozen sections of sciatic nerve of a 30-wk-old B7-2−/− neuropathic female and an age match control mouse were immunostained for the presence of CD4+ and CD8+ T cells and dendritic cells (DC). No infiltrate was detected in B7-2+/+ mice (dark oblong structures are nuclei of Schwann cells) whereas B7-2−/− mice presented abundant infiltration (dark round structures are stained cells). (Right panel) Frozen sections of sciatic nerve of a 28-wk-old B7-2−/− neuropathic female and an age-matched B7-2−/− B6 male mouse were immunostained for the presence of CD4 (green, top panel) and B7-1 (green, bottom panel). No infiltrate or B7-1 staining was detected in a B7-2−/− B6 mouse whereas B7-2−/− NOD mice presented abundant infiltration and B7-1 expression.
Figure 5.

Essential role of CD4+ T cells in the neuropathy of B7-2−/− NOD mice. (a) Total spleen and lymph node cells from B7-2−/− neuropathic NOD mice or age match NOD controls were stimulated in vitro with anti-CD3 and anti-CD28 mAbs and injected into five NOD.SCID mice. Symptoms of neuropathy were then checked weekly. Inset: neuropathic NOD.SCID mouse displaying complete paralysis of hind limbs. (b) CD4+ T cells, CD4-depleted cells, and whole cells, purified from spleen and lymph nodes of B7-2−/− neuropathic NOD mice, were stimulated in vitro with anti-CD3 and anti-CD28 mAbs and injected into 14, 9, and 16 NOD.SCID mice, respectively. Data are from three independent experiments.
B7-1 has been shown to be a critical costimulatory molecule in experimental autoimmune encephalomyelitis, an autoimmune disease of the CNS 6 15. We thus analyzed levels of B7-1 expression in the sciatic nerves (and spleen) of B7-2 KO mice by flow cytometry (Fig. 6). B7-1 expression was easily detected in sciatic nerves of neuropathic B7-2 KO mice. This expression was strictly limited to infiltrating CD45-positive cells and could not be detected in the absence of a mononuclear infiltrate in 12–14-wk-old B7-2 KO or B7-2 WT mice (Fig. 6). Furthermore, expression of B7-1 in the nerves of neuropathic animals was high on CD11b-positive and CD11c-positive cells (Fig. 6, and data not shown), suggesting that antigen-presenting cells infiltrating the peripheral nerves expressed high levels of B7-1. B7-1 expression could be readily detected on some splenocytes of B7-2 KO animals, although this expression was lower than observed on the infiltrating CD11b-positive cells of sciatic nerves (data not shown). In contrast B7-1 was not detected in the spleen of WT NOD mice (data not shown). Finally, we compared CD4 and B7-1 staining by immunohistochemistry. As noted in Fig. 4 (right panel), incubation with inflamed sciatic nerve revealed B7-1 staining of the infiltrating cells; however, the staining pattern for B7-1 and CD4 does not appear to be overlapping. In fact, two-color analyses suggested a significant overlap of mononuclear infiltrate and B7-1 staining in this tissue (data not shown). Importantly, there was no B7-1 staining in the B7-2 KO B6 mouse consistent with the correlation of B7-1 expression and the disease. Finally, it should be emphasized that there was no infiltrate in the B7-2 KO mice on the B6 background consistent with the absence of any disease in these animals.
Figure 6.

Expression of B7-1 in B7-2KO NOD mice. Sciatic nerve tissues from 28-wk-old neuropathic B7-2 KO or 12–14-wk-old B7-2 KO and B7-2 WT females were digested with collagenase. Indicated cell subpopulations (CD45− cell population in B7-2 WT and young B7-2 KO mice) were then analyzed for B7-1 expression (shaded histograms) by flow cytometry. White histograms show the staining with the isotypic control Ab.
Discussion
Increasing evidence suggests that a variety of generalized mechanisms (i.e., decreased regulatory mechanisms and cytokine production, altered thymic selection, expression of specific MHC class II haplotypes, etc.) may play a critical role in the increased autoimmune disease incidence observed in certain genetic settings 14 16 17. Genes of susceptibility linked to multiple autoimmune diseases have been identified in both NOD mice and human 14 17 18 19 20. For instance, the DR3-DQB1*0201 haplotype in humans has been associated with multiple autoimmune diseases in humans including type 1 diabetes, Addison's disease, Graves' disease, and Hashimoto's thyroiditis 21. Moreover a single individual diabetic patient could present other autoimmune syndromes 17. Similarly, diabetic-prone NOD mice also exhibit immune reactions against multiple glands such as thyroid or salivary glands, T cell autoreactivity to CNS Ag and some features of nonorgan-specific autoimmune disease such as late-onset antinuclear Abs and hemolytic anemia 22 23 24. Thus, the development of SAPP in B7-2–deficient NOD mice, not observed in other mouse strains deficient in B7-2, could be related to the general susceptibility to autoimmunity of these mice. SAPP could share a subset of immunological and genetic attributes to those associated with the endocrine autoimmunity in this mouse strain.
Importantly, the regulatory role of B7-2 in protection from cryptic SAPP development was demonstrated in two independent models: in genetically modified B7-2 KO mice and in WT animals treated with blocking anti–B7-2 mAb. The lower incidence of neuropathy in the latter model is likely due to incomplete penetrance of the Ab into relevant tissues and the limited time of treatment. In addition, the induction of neuropathy after B7-2 disruption did not simply result from the prevention of diabetes. Indeed, more than 150 treatments preventing diabetes have been reported. None of them has been described to induce peripheral neuropathy.
Another significant finding in our study was the polarization of the autoimmune response in the B7-2 KO NOD mice, which develop SAPP but are free from the development of diabetes, pointing to the importance of immune dysregulation in the shift from one autoimmune disease to another. The lack of diabetes in these mice did not seem to be due to a lack of islet-reactive diabetogenic T cells. Indeed, in preliminary results we have observed that splenocytes from B7-2 KO mice could induce diabetes when transferred into NOD.SCID mice (data not shown). However, this required the depletion from the innoculum of the CD4+CD25+ T cells, which regulate autoimmune diabetes 25.
The development of SAPP in B7-2 KO NOD mice was unexpected. A recent study showed the presence of myelin-reactive T cells in the spleen of WT NOD mice 24. Such auto-reactive T cells, upon migration in peripheral nerves, could play a role in SAPP. In addition to T cells, peripheral nerves of neuropathic B7-2 KO mice were infiltrated with high number of myeloid cells identified by expression of CD11c and/or CD11b, most of them being myeloid type dendritic cells (data not shown). The high levels of B7-1 expression on these professional antigen-presenting cells likely amplified the activation of myelin-reactive T cells, leading to destruction of myelin sheets and development of SAPP. Whether B7-1 upregulation played a role in initiating and sustaining the destructive inflammatory infiltrate or conversely was a result of the local inflammatory milieu is currently under investigation. However, the striking overexpression of B7-1 in B7-2KO mice compared with NOD mice suggests that B7-1 played an active role in the development of SAPP in B7-2 KO mice. In this regard, it is interesting to note that B7-1 was selectively upregulated in nervous tissue of mice with experimental allergic encephalomyelitis 26. In this murine model, blockade of B7-1 using Fab fragments of anti–B7-1 mAbs reduced IFN-γ production and prevented disease whereas blockade with anti–B7-2 exacerbated disease 6 15. Interestingly, treatment with intact anti–B7-1 mAbs exacerbated the pathology for unknown reasons 27. Similarly, treatment of B7-2 KO NOD mice with this reagent resulted in a rapid and profound induction of diabetes and neuropathy (data not shown). Thus, future experiments will focus on the use of Fab fragments of anti–B7-1 mAbs to avoid potential complications of an “activating” form of the blocking reagent. Finally, the absence of B7-1 expression in the nerves before infiltration and the restriction of B7-1 expression to hematopoietic cells later in the course of the disease does not rule out a major role for resident nerve cells such as Schwann cells in the development of the inflammation in the nerves. However, it is unlikely that the nerve cells directly stimulate (costimulate) the immune response to initiate disease in these animals. Rather, the upregulation of B7-1 in the spleen of B7-2 KO NOD mice could increase the activation of myelin reactive T cells, identified in WT NOD mice 24, which then migrated into the peripheral nerves.
The shift from one autoimmune disease to another is reminiscent of multiple studies in mice and humans that showed concomitant disappearance of one autoimmune syndrome and worsening of a second. NOD mice treated with complete Freund's adjuvant are protected from diabetes but at the same time they can develop lupus 23. Multiple sclerosis patients treated with anti-CD52 Ab showed improvement of their neurologic disease but fully one third of them developed Graves' disease (unpublished data). These results suggest that autoimmune-prone individuals have immune dysfunctions that can manifest as distinct disease entities perhaps dependent upon the costimulatory milieu. For instance, differential expression and ligation of B7-2 and B7-1 at distinct anatomical sites may lead to different outcomes.
Importantly, SAPP has many similarities to human autoimmune demyelinating neuropathies such as chronic inflammatory demyelinating polyneuropathy (CIDP) and Guillain-Barré syndrome, although the slow progressive course in SAPP suggests that the immune mechanisms are more similar to those of CIDP 28. This new autoimmune model could help understand the pathophysiology of some autoimmune diseases of the nervous system such as CIDP. The essential role of CD4+ T cell in the effector phase of SAPP could indicate a critical role of these cells in CIDP.
SAPP adds to the very short list of models of spontaneous autoimmune disease mouse models including diabetes, lupus, rheumatoid arthritis, and colitis. In these models, self-tolerance breakdown probably results from chronic stimulation of autoreactive lymphocytes. The pathophysiology of these diseases could be fundamentally different to that observed in experimental autoimmune models in which self-tolerance breakdown is rapid and results from strong immunization with a specific autoantigen. In this regard, CD28 ligation blockade reduced severity or prevented various experimental autoimmune diseases whereas it exacerbated spontaneous autoimmune diabetes 8 9 10 25 29. Similarly, B7-2 ligation blockade promotes SAPP in NOD mice. Although these findings raise concerns about the use of costimulatory blockade in the patient setting, they provide a model for analyzing the genetic and immunological basis for autoimmunity that may lead to new insights into these complex disorders.
Acknowledgments
We thank Shihong Li, Julie Auger, Greg Szot, and Lori Favero for expert technical support.
This research was supported by a National Institutes of Health grant no. P01 DK49799. B. Salomon was supported by a Juvenile Diabetes Foundation fellowship no. 16245-01-01. H. Bour-Jordan was supported by a Juvenile Diabetes Foundation Fellowship no. 3-1999-132.
Footnotes
Abbreviations used in this paper: CIDP, chronic inflammatory demyelinating polyneuropathy; CNS, central nervous system; KO, knockout; SAPP, spontaneous autoimmune peripheral polyneuropathy; WT, wild-type.
B. Salomon's present address is Laboratoire de Biologie et Thérapeutique des Pathologies Immunitaires, Centre National de la Recherche Scientifique ESA 7087, CERVI, Hôpital de la Pitié-Salpêtrière, Paris 75013, France.
References
- Jenkins M.K., Schwartz R.H. Antigen presentation by chemically modified splenocytes induces antigen-specific T cell unresponsiveness in vitro and in vivo. J. Exp. Med. 1987;165:302–319. doi: 10.1084/jem.165.2.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linsley P.S., Brady W., Grosmaire L., Aruffo A., Damle N.K., Ledbetter J.A. Binding of the B cell activation antigen B7 to CD28 costimulates T cell proliferation and interleukin 2 mRNA accumulation. J. Exp. Med. 1991;173:721–730. doi: 10.1084/jem.173.3.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding F.A., McArthur J.G., Gross J.A., Raulet D.H., Allison J.P. CD28-mediated signalling co-stimulates murine T cells and prevents induction of anergy in T-cell clones. Nature. 1992;356:607–609. doi: 10.1038/356607a0. [DOI] [PubMed] [Google Scholar]
- Salomon B., Bluestone J.A. Complexities of CD28/B7CTLA-4 costimulatory pathways in autoimmunity and transplantation. Annu. Rev. Immunol. 2001;19:225–252. doi: 10.1146/annurev.immunol.19.1.225. [DOI] [PubMed] [Google Scholar]
- Finck B.K., Linsley P.S., Wofsy D. Treatment of murine lupus with CTLA4Ig. Science. 1994;265:1225–1227. doi: 10.1126/science.7520604. [DOI] [PubMed] [Google Scholar]
- Kuchroo V.K., Das M.P., Brown J.A., Ranger A.M., Zamvil S.S., Sobel R.A., Weiner H.L., Nabavi N., Glimcher L.H. B7-1 and B7-2 costimulatory molecules activate differentially the Th1/Th2 developmental pathwaysapplication to autoimmune disease therapy. Cell. 1995;80:707–718. doi: 10.1016/0092-8674(95)90349-6. [DOI] [PubMed] [Google Scholar]
- Lenschow D.J., Ho S.C., Sattar H., Rhee L., Gray G., Nabavi N., Herold K.C., Bluestone J.A. Differential effects of anti-B7-1 and anti-B7-2 monoclonal antibody treatment on the development of diabetes in the nonobese diabetic mouse. J. Exp. Med. 1995;181:1145–1155. doi: 10.1084/jem.181.3.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tada Y., Nagasawa K., Ho A., Morito F., Ushiyama O., Suzuki N., Ohta H., Mak T.W. CD28-deficient mice are highly resistant to collagen-induced arthritis. J. Immunol. 1999;162:203–208. [PubMed] [Google Scholar]
- Chang T.T., Jabs C., Sobel R.A., Kuchroo V.K., Sharpe A.H. Studies in B7-deficient mice reveal a critical role for B7 costimulation in both induction and effector phases of experimental autoimmune encephalomyelitis. J. Exp. Med. 1999;190:733–740. doi: 10.1084/jem.190.5.733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girvin A.M., Dal Canto M.C., Rhee L., Salomon B., Sharpe A., Bluestone J.A., Miller S.D. A critical role for B7/CD28 costimulation in experimental autoimmune encephalomyelitisa comparative study using costimulatory molecule- deficient mice and monoclonal antibody blockade. J. Immunol. 2000;164:136–143. doi: 10.4049/jimmunol.164.1.136. [DOI] [PubMed] [Google Scholar]
- Borriello F., Sethna M.P., Boyd S.D., Schweitzer A.N., Tivol E.A., Jacoby D., Strom T.B., Simpson E.M., Freeman G.J., Sharpe A.H. B7-1 and B7-2 have overlapping, critical roles in immunoglobulin class switching and germinal center formation. Immunity. 1997;6:303–313. doi: 10.1016/s1074-7613(00)80333-7. [DOI] [PubMed] [Google Scholar]
- Hsu S.M., Raine L., Fanger H. Use of avidin-biotin-peroxidase complex (ABC) in immunoperoxidase techniquesa comparison between ABC and unlabeled antibody (PAP) procedures. J. Histochem. Cytochem. 1981;29:577–580. doi: 10.1177/29.4.6166661. [DOI] [PubMed] [Google Scholar]
- Dyck P.J., Giannini C., Lais A. Peripheral Neuropathy. 3rd ed. W.B. Saunders, ; Philadelphia, PA: 1993. [Google Scholar]
- Todd J.A. From genome to aetiology in a multifactorial disease, type 1 diabetes. Bioessays. 1999;21:164–174. doi: 10.1002/(SICI)1521-1878(199902)21:2<164::AID-BIES10>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Miller S.D., Vanderlugt C.L., Lenschow D.J., Pope J.G., Karandikar N.J., Dal Canto M.C., Bluestone J.A. Blockade of CD28/B7-1 interaction prevents epitope spreading and clinical relapses of murine EAE. Immunity. 1995;3:739–745. doi: 10.1016/1074-7613(95)90063-2. [DOI] [PubMed] [Google Scholar]
- McDevitt H.O. The role of MHC class II molecules in susceptibility and resistance to autoimmunity. Curr. Opin. Immunol. 1998;10:677–681. doi: 10.1016/s0952-7915(98)80088-5. [DOI] [PubMed] [Google Scholar]
- Griffiths M.M., Encinas J.A., Remmers E.F., Kuchroo V.K., Wilder R.L. Mapping autoimmunity genes. Curr. Opin. Immunol. 1999;11:689–700. doi: 10.1016/s0952-7915(99)00038-2. [DOI] [PubMed] [Google Scholar]
- Becker K.G., Simon R.M., Bailey-Wilson J.E., Freidlin B., Biddison W.E., McFarland H.F., Trent J.M. Clustering of non-major histocompatibility complex susceptibility candidate loci in human autoimmune diseases. Proc. Natl. Acad. Sci. USA. 1998;95:9979–9984. doi: 10.1073/pnas.95.17.9979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker K.G. Comparative genetics of type 1 diabetes and autoimmune diseasecommon loci, common pathways? Diabetes. 1999;48:1353–1358. doi: 10.2337/diabetes.48.7.1353. [DOI] [PubMed] [Google Scholar]
- Encinas J.A., Wicker L.S., Peterson L.B., Mukasa A., Teuscher C., Sobel R., Weiner H.L., Seidman C.E., Seidman J.G., Kuchroo V.K. QTL influencing autoimmune diabetes and encephalomyelitis map to a 0.15-cM region containing Il2. Nat. Genet. 1999;21:158–160. doi: 10.1038/5941. [DOI] [PubMed] [Google Scholar]
- Huang W., Connor E., Rosa T.D., Muir A., Schatz D., Silverstein J., Crockett S., She J.X., Maclaren N.K. Although DR3-DQB1*0201 may be associated with multiple component diseases of the autoimmune polyglandular syndromes, the human leukocyte antigen DR4-DQB1*0302 haplotype is implicated only in beta-cell autoimmunity. J. Clin. Endocrinol. Metab. 1996;81:2559–2563. doi: 10.1210/jcem.81.7.8675578. [DOI] [PubMed] [Google Scholar]
- Krug J., Williams A.J., Beales P.E., Doniach I., Gale E.A., Pozzilli P. Parathyroiditis in the non-obese diabetic mouse—a new finding. J. Endocrinol. 1991;131:193–196. doi: 10.1677/joe.0.1310193. [DOI] [PubMed] [Google Scholar]
- Baxter A.G., Horsfall A.C., Healey D., Ozegbe P., Day S., Williams D.G., Cooke A. Mycobacteria precipitate an SLE-like syndrome in diabetes-prone NOD mice. Immunology. 1994;83:227–231. [PMC free article] [PubMed] [Google Scholar]
- Winer S., Astsaturov I., Cheung R.K., Gunaratnam L., Kubiak V., Cortez M.A., Moscarello M., O'Connor P.W., McKerlie C., Becker D.J., Dosch H.-Michael. Type I diabetes and multiple sclerosis patients target islet plus central nervous system autoantigens; nonimmunized nonobese diabetic mice can develop autoimmune encephalitis. J. Immunol. 2001;166:2831–2841. doi: 10.4049/jimmunol.166.4.2831. [DOI] [PubMed] [Google Scholar]
- Salomon B., Lenschow D.J., Rhee L., Ashourian N., Singh B., Sharpe A., Bluestone J.A. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 2000;12:431–440. doi: 10.1016/s1074-7613(00)80195-8. [DOI] [PubMed] [Google Scholar]
- Karandikar N.J., Vanderlugt C.L., Eagar T., Tan L., Bluestone J.A., Miller S.D. Tissue-specific up-regulation of B7-1 expression and function during the course of murine relapsing experimental autoimmune encephalomyelitis. J. Immunol. 1998;161:192–199. [PubMed] [Google Scholar]
- Vanderlugt C.L., Karandikar N.J., Lenschow D.J., Dal Canto M.C., Bluestone J.A., Miller S.D. Treatment with intact anti-B7-1 mAb during disease remission enhances epitope spreading and exacerbates relapses in R-EAE. J. Neuroimmunol. 1997;79:113–118. doi: 10.1016/s0165-5728(97)00108-2. [DOI] [PubMed] [Google Scholar]
- Hartung H.P., van der Meche F.G., Pollard J.D. Guillain-Barre syndrome, CIDP and other chronic immune-mediated neuropathies. Curr. Opin. Neurol. 1998;11:497–513. doi: 10.1097/00019052-199810000-00013. [DOI] [PubMed] [Google Scholar]
- Lenschow D.J., Herold K.C., Rhee L., Patel B., Koons A., Qin H.Y., Fuchs E., Singh B., Thompson C.B., Bluestone J.A. CD28/B7 regulation of Th1 and Th2 subsets in the development of autoimmune diabetes. Immunity. 1996;5:285–293. doi: 10.1016/s1074-7613(00)80323-4. [DOI] [PubMed] [Google Scholar]