

Abstract
Dendritic cells (DCs) play a central role in the immune system as they drive activation of T lymphocytes by cognate interactions. However, as DCs express high levels of major histocompatibility complex class I, this intimate contact may also result in elimination of DCs by activated cytotoxic T lymphocytes (CTLs) and thereby limit induction of immunity. We show here that immature DCs are indeed susceptible to CTL-induced killing, but become resistant upon maturation with anti-CD40 or lipopolysaccharide. Protection is achieved by expression of serine protease inhibitor (SPI)-6, a member of the serpin family that specifically inactivates granzyme B and thereby blocks CTL-induced apoptosis. Anti-CD40 and LPS-induced SPI-6 expression is sustained for long periods of time, suggesting a role for SPI-6 in the longevity of DCs. Importantly, T helper 1 cells, which mature DCs and boost CTL immunity, induce SPI-6 expression and subsequent DC resistance. In contrast, T helper 2 cells neither induce SPI-6 nor convey protection, despite the fact that they trigger DC maturation with comparable efficiency. Our data identify SPI-6 as a novel marker for DC function, which protects DCs against CTL-induced apoptosis.
Keywords: granzyme, CTL, survival, CD40, LPS
Introduction
Dendritic cells (DCs) are highly specialized APCs that are pivotal in the induction of immunity 1. In their immature state, DCs can efficiently capture and process antigen, but only through maturation these cells acquire the capacity to prime naive T cells 1. DC maturation can be triggered by a number of natural stimuli, including infectious agents, inflammatory cytokines, and ligation of the CD40 receptor. This latter signal is delivered to the DCs through cognate interaction with CD40L-expressing CD4+ Th cells, and can be mimicked by agonistic anti-CD40 Ab 2 3 4. Maturation converts DCs from antigen-scavengers into cells that present a high density of peptide–MHC complexes and costimulatory molecules 1. In addition, the maturation process coincides with migration towards the lymphoid organs, an event that involves expression of the chemokine receptor CCR7 5. When fully matured, DCs will stimulate naive CD8+ CTLs and provide these cells with their “license to kill” 2 3 4 6. This endows the CTLs with the capacity to induce apoptosis in their target cells, which occurs via two distinct mechanisms. The first cytotoxic mechanism involves exocytosis of cytotoxic granules. Apoptosis via this pathway is due to the combined actions of the pore-forming molecule perforin and the cytotoxic protease granzyme (Gr)B 7 8. After secretion by the CTLs, this Gr binds to the mannose-6-phosphate receptor and enters the target cell via receptor-mediated endocytosis 9 10. Perforin then serves to release GrB from the endosomes into the cytoplasm of the target cell 10 11. Once delivered into the cytosol, GrB initiates the apoptotic signaling cascade through cleavage of several substrates, including Bid, inhibitor of caspase-activated DNase, caspase-3, -7, and -8 12 13 14 15. Although CTLs release other Grs, in particular GrA, GrB is the key factor in the rapid induction of DNA fragmentation and the absence of GrB greatly hampers the capacity of a CTL to induce apoptosis via the perforin pathway 8 9. The second pathway used by a CTL to kill its target cell is cross-linking of death receptors on the target cell. This mainly involves CD95L (APO-1L, FasL; references 16 17 18), which has been shown to induce a caspase cascade that will lead to the destruction of the target cell 19.
It is important to note that both these cytotoxic mechanisms are induced rapidly upon activation of naive CTLs 20 21. Therefore, DCs that present the relevant MHC–peptide complexes qualify as potential targets and are at risk of being eliminated by the CTLs they have activated. Such an event would seriously limit the capacity of DCs to prime CTL immunity. Recently, it was proposed that DCs are protected from premature CTL-mediated elimination due to downregulation of cell surface expression of the CCR7 homing receptor on activated CTLs 22 23. This would limit lymphoid localization of active CTLs and thereby prevent a fatal interaction with DCs. However, recent data suggest that this mechanism does not provide sufficient protection, as injected populations of APCs were cleared from the lymph nodes through a CTL-dependent mechanism (B. Ludewig, University of Zurich, personal communication; references 24 and 25). Furthermore, virus-induced acquired immune suppression in mice infected with lymphocytic choriomeningitis virus was shown to involve CTL-dependent elimination of APCs in peripheral lymphoid organs 26. This indicates that DCs do become exposed to the cytolytic machinery of activated CTLs in vivo. Therefore, we performed a detailed analysis of the consequences of interaction between DCs and effector CTLs, using several defined DC cultures of murine and human origin. We find that DCs effectively protect themselves against the actions of CTLs by expression of the GrB-inhibitor serine protease inhibitor (SPI)-6/PI-9, provided that they receive the appropriate maturation signals.
Materials and Methods
Mice, Cell Lines, and Reagents.
Female C57BL/6Kh (B6, H-2b) and DO11.10 TCR transgenic mice (provided by Dr. M. Wauben, Utrecht University, Utrecht, Netherlands) were kept at the LUMC animal facility and used at 6–10 wk of age in accordance with national legislation and under supervision of the animal experimental committee of the University of Leiden. OVA-specific Th1 and Th2 cells were obtained from DO11.10 TCR transgenic mice. Th1 cells were generated as described previously 27 28. Th2 cells were generated from D011.10 mice in a similar fashion as Th1 cells except that the medium was supplemented with 20 ng/ml IL-4 (Peprotech), 10 μg/ml anti–IL-12 (C17.8, a gift from Dr. A. van Halteren, LUMC), and 10 μg/ml anti–IFN-γ (XMG1.2) instead of recombinant IL-12 and anti-IL4.D1, a long-term growth factor–dependent murine DC cell line was a gift from Dr. P. Ricciardi-Castagnoli and cultured as described previously 29. Primary bone marrow DCs were generated by culturing bone marrow isolates from C57BL/6Kh mice 30 in D1 culture medium 29. These bone marrow DC cultures are standardly >80% CD11c+ of which <10% is mature as measured by B7-2 expression. Spleen cell DCs were isolated directly from homogenized spleen (C57BL6/Kh) by cell sorting using anti-CD11c/FITC and anti-B7-2/PE to determine the maturation status. Human monocyte–derived DCs were generated using a standard protocol with IL-4 and GM-CSF as described previously 31. The E1B-specific CTL clone 100B6 was cultured routinely as described previously 32. Retroviral transduction of D1 cells was performed as described with an LZRS-based vector encoding either enhanced green fluorescent protein (EGFP) or vsv-tagged SPI-6 plus EGFP 33. Tranduced cells were sorted twice on the basis of EGFP expression. The anti-CD40 (FGK45) antibody was a gift from Dr. A. Rolink (Basel Institute for Immunology, Basel, Switzerland) and used at 50 μg/ml. LPS of Escherichia coli was obtained from Difco Laboratories and used at 10 μg/ml for murine DCs and at 1 μg/ml for human DCs. Murine and human leucine zipper CD40L was a gift of Dr. M. Kubin (Immunex) and used at 1 μg/ml. IL-10 was added to the D1 cultures at 50 ng/ml while IFN-γ was used at 10 IU/ml. CD40L blocking experiments were performed with 50 μg/ml moAb MR-1, IL-10 was blocked using 5 μg/ml moAb JES5-2A5 (BD PharMingen), and IL-4 was blocked using 10 μg/ml of the moAb 11B11 (a gift from Dr. A van Halteren).
Cell Surface Staining, Western Blot Analysis, and ELISA.
FACS® analysis for B7-2 was performed with GL-1 (BD PharMingen). Western blot analysis was performed with polyclonal rabbit anti–mitogen-activated protein kinase (MAPK) as a control antibody 34 or with MoAb17 generated against PI-9, which is specific for PI-9 (human) 35 and efficiently crossreacts with (murine) SPI-6 (Fig. 2 b). Western blot analysis were developed using HRP-coupled secondary antibodies and ECL. The murine IL-12(p40) and human IL-12(p70) ELISA were performed as described previously 27 31. The murine IL-10 ELISA was performed using the ELISA kit from BD PharMingen.
Figure 2.

SPI-6 is expressed in D1 cells. (a) D1 cells were treated with Lz-CD40L, FGK45, or LPS as indicated and SPI-6 expression was determined with semiquantitative rtPCR or Northern blot analysis (two transcripts). Top panels represent SPI-6, while bottom panels represent a control rtPCR/Northern for GAPDH. (b) Cells were treated as under (a) and tested for the protein expression of SPI-6 on Western blot (top). Lysates from equal numbers of cells were applied on SDS-PAGE (0.5 × 106 cells) and equal protein loading was further checked by ponceau S staining. In addition, an antibody against MAPK was used as a control (bottom). A murine tumor cell line transduced with a retrovirus encoding SPI-6 and the non-transduced parental cell line (MBL2FasFLIP; MFF) were used as controls to ensure that the anti-PI-9 mAb specifically cross-reacted with SPI-6.
PCR and Northern Blot Analysis.
PCR of a 741-bp fragment of SPI-6 (from 737–1,477) was performed using GCTCCCAGATGAGGGTGT as forward and TCTCTCTAGCTCCATTAT as reverse primer. Resulting PCR product was sequenced and compared with the published sequence 36 to certify the identity of the product. A GAPDH control PCR (fragment length 232 bp, 384–615) was performed with GAGCCAACGGGTCATCATCT and GAGGGGCCATCCACAGTCTT as primers. Northern blot analysis was performed using 10 μg per lane of total RNA from either immature or FGK45-matured D1 cells. A [32P]-labeled probe was generated with random priming using the complete SPI-6 coding region as a template.
Just Another Method.
The just another medthod (JAM) assay was performed essentially as described previously 32 37 and detects DNA fragmentation, a hallmark in the induction of apoptosis 38. In short, D1 cells were labeled with 1 μCi of [3H]thymidine for 3 d during which they were treated with LPS for 24 h or FGK45 for 48 h, which both result in full maturation. Assays were performed with 1,000 target cells (1,000–2,000 cpm) per well and in the presence of cold thymidine (1.5 μM) for 6 h. Targets were loaded with peptide at 1 μg/ml just before the assay. In most experiments the E1B-specific CTL clone was used as effector population at the indicated E:T ratios. In a separate set of experiments we used in vivo–activated CTLs, which were obtained essentially as described previously 39. In short, splenocytes isolated from mice that were immunized 10 d earlier with a temperature-sensitive mutant of Adenovirus type 5 (Ad5ts125) were restimulated with 10% irradiated tumor cells (XhoC3) that express the Adenovirus E1A and E1B proteins. Restimulation was performed for 6 d after which the cultures were applied to a Ficoll gradient to isolate living cells. In this fashion, bulk CTL cultures were obtained that are specific for the E1B epitope and these were used at the indicated E:T ratios. Assays were left for 6 h at 37°C and all assays were performed in six-plo to minimize the variation. Where indicated the E1B-specific CTL clone was preincubated with concanamycin A (CMA; Sigma-Aldrich) 40 for 2 h at 30 nM. CMA was present throughout the assay.
CaspaTag Cytotoxicity Assay.
105 immature or FGK-matured (48 h) D1 cells where incubated for 3.5 h with the E1B-specific CTL clone at an E:T ratio of 3 in the absence or presence of E1B-peptide (1 μg/ml). After this initial incubation, CTLs were stained for 10 min at 37°C with anti-CD8α-APC (Ly-2; BD PharMingen). A further incubation at 37°C for 1 h was performed in the presence of CaspaTag (FAM-VAD-fmk; Intergen) according to the manufacturer's indications. CaspaTag irreversibly binds to active caspases and was used to detect the caspase activity in the target cell on a FACScan™ (Becton Dickinson) by gating out the CD8α1 CTLs and measuring the fluorensence intensity of the CaspaTag (fl-1) in the target cells.
Results
Mature DCs Resist CTL-induced Apoptosis.
The murine D1 cell line displays a stable phenotype typical for immature DCs unless it receives an activation signal through agonistic anti-CD40 Ab, LPS, or Th cells 27 29. This trigger results in fully matured D1 cells that are highly capable of CTL priming 27. To directly analyze the sensitivity of DCs for CTL-mediated killing, we measured the efficacy of a CTL clone to induce antigen-dependent apoptosis in both immature and mature D1 cells. Immature D1 cells, exogenously loaded with the relevant antigen, were highly susceptible to CTL-induced DNA fragmentation as measured in a JAM assay 37 38 (Fig. 1 a). This cytotoxicity was largely perforin-dependent as it was blocked by CMA (Fig. 1 b), a substance that specifically prevents perforin-dependent cytotoxicity 40. Interestingly, maturation of D1 with anti-CD40 Ab or LPS prevented CTL-induced apoptosis (Fig. 1 a). Notably, even though anti-CD40 or LPS treatment resulted in maturation of a large fraction of the D1 cells (70–80%), a small population remained immature. This most likely explains the residual killing that is detected in our cytotoxicity assay when using anti-CD40 or LPS-matured D1 cells as targets (Fig. 1 a). Insensitivity to CTL-mediated killing was not due to the lack of recognition by the CTLs, as both anti-CD40 and LPS-matured D1 cells express very high levels of MHC class I 27 29 and efficiently induce CTL activation as measured by the secretion of IFN-γ (data not shown). The observed difference was also not restricted to this particular CTL clone, as it was detected with a separate clone that contains a distinct peptide specificity (data not shown). Moreover, polyclonal CTL cultures, obtained by restimulation of splenocytes from Adenovirus-infected C57BL/6 mice, efficiently induced DNA fragmentation of D1 cells, but failed to kill anti-CD40–matured D1 cells (Fig. 1 c). This indicates that the lack of CTL-induced DNA fragmentation in anti-CD40–matured D1 cells is a general feature. Although DNA fragmentation is a very reliable marker of apoptosis, it is a rather late event that can be prevented through intervention with the apoptosis signaling cascade at several different points. Therefore, we analyzed one of the most proximal events in perforin-dependent apoptosis, which is the activation of caspases. Upon incubation with the E1B-specific CTL clone, we observed that a proportion of the immature D1 cells displayed caspase activity (Fig. 1 d). This caspase activity was dependent on the presence of the E1B peptide and was thus induced by the CTLs in an epitope-specific manner (Fig. 1 d). More importantly, CTL-induced caspase activation was not observed in D1 cells that had been matured with anti-CD40 (Fig. 1 d). This indicates that inhibition of CTL-induced apoptosis occurs already at a proximal point in the cascade of events that lead to target cell apoptosis and implies that a powerful mechanism must be in place to protect mature DCs against elimination by activated CTLs.
Figure 1.


Sensitivity of D1 cells for CTL-induced apoptosis. (a) D1 cells were labeled with [3H]thymidine and exogenously loaded with Adenovirus E1B peptide. Labeled cells were either left untreated (immature, circles), treated with LPS for 24 h (triangles), or anti-CD40 for 48 h (FGK45, squares) and subsequently incubated at different E:T ratios with E1B-specific CTL clone for 6 h. (b) D1 cells were treated as indicated under a, and incubated at different E:T ratios with E1B-specific CTL clone for 6 h in the absence (white) or presence (black) of 30 nM CMA. (c) Splenocytes from C57BL/6 mice immunized with Adenovirus type 5 were once restimulated in vitro and subsequently used as effectors at the indicated E:T ratios. D1 cells (circles) or anti-CD40-matured D1 cells (squares) were labeled with [3H]thymidine and exogenously loaded with the E1B peptide. The specific amount of thymidine released in a, b, and c, which is a measure for CTL-induced apoptosis, is then calculated and given as a percentage of the maximal releasable amount. (d). Immature or anti-CD40–matured D1 cells were incubated with the E1B-specific CTL clone in the absence (light gray) or presence (dark gray) of the E1B peptide and analyzed for their caspase activity with the use of CaspaTag. The percentage of cells that stained positive for caspase activity is given.
The Antiapoptotic Protein SPI-6 Is Induced during DC Maturation.
Perforin-dependent target cell apoptosis is mainly due to the activity of the serine protease GrB 7 8. Therefore, we investigated whether DCs resistance involves blockade of this protease. In this respect, it is interesting to note that CTLs were previously reported to express a GrB inhibitor, which was suggested to provide CTLs with a protective mechanism against their own lytic machinery 36 41. This inhibitor, which is called PI-9 in human and SPI-6 in mouse, belongs to the SPI (serpin) family and specifically binds and inactivates GrB in vitro 36 41 42. To directly determine whether this GrB-inhibitory serpin is expressed in DCs and is related to their resistance from CTL-mediated apoptosis, we analyzed the expression levels of SPI-6 in D1 cells. Both semiquantitative PCR and Northern blot analysis revealed very low expression of SPI-6 mRNA in immature D1 cells (Fig. 2 a). In accordance, only low levels of SPI-6 protein could be detected (Fig. 2 b). Importantly, strong induction of SPI-6 mRNA was observed mainly during CD40-induced maturation (Fig. 2 a), in line with a rapid and strong increase of SPI-6 at the protein level (Fig. 2 b). Similarly, LPS-induced maturation resulted in increased levels of SPI-6 protein (Fig. 2 b).
As the D1 cells represent a long-term cell line, we verified our observations using primary DCs. Immature CD11c+ DCs, generated from murine bone marrow, were found to express little SPI-6 (Fig. 3 b). Treatment with CD40L resulted in maturation of these DCs (Fig. 3 a) and, concomitantly, in a marked increase in SPI-6 expression (Fig. 3 b). As was found for D1 cells, bone marrow DCs were highly sensitive to CTL-induced apoptosis in their immature state, but insensitive after maturation (Fig. 3 c).
Figure 3.
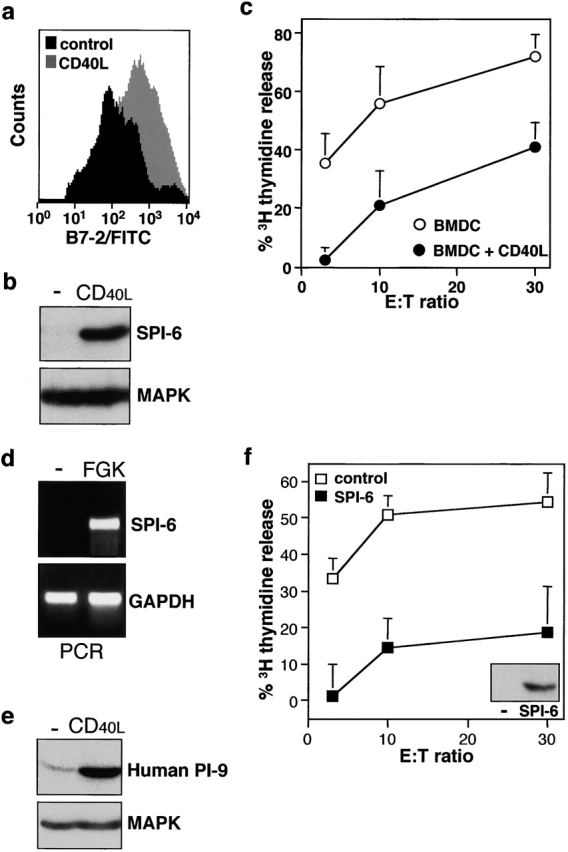
SPI-6 is expressed in primary DCs and prevents cytotoxicity. Primary bone marrow DCs were either left untreated or treated for 48 h with Lz-CD40L and tested for (a) B7-2 expression by FACS® analysis, (b) SPI-6 expression using Western blot analysis, and (c) CTL sensitivity as described in Fig. 1 a. (d) Spleen cell DCs were isolated by FACS® sorting using CD11c and B7-2 expression as markers. Cells were isolated either from untreated mice (sorted for immature DC, B7-low) or from FGK45-injected mice (sorted for mature DCs, B7-high) and SPI-6 as well as GAPDH expression was determined directly by rtPCR. (e) Primary human monocyte–derived DCs were treated with Lz-CD40L or LPS for 24 h and tested for PI-9 expression (i.e., the human homologue of SPI-6) by Western blot analysis. In all cases lysates from equal numbers of cells were applied on SDS-PAGE (0.5 × 106 cells) and equal protein loading was further checked by ponceau S staining and control Western for MAPK (bottom). (f) D1 cells transduced with control retrovirus (white squares) or SPI-6-encoding retrovirus (black squares) were tested for SPI-6 expression by Western blot (inset) and CTL sensitivity by DNA fragmentation.
SPI-6 induction is not confined to DC maturation in vitro, as two serial injections of anti-CD40 into mice resulted in the in vivo induction of SPI-6 mRNA in CD11c+ spleen cell DCs (Fig. 3 d). Furthermore, regulation of serpin expression is conserved between mouse and man. Immature human monocyte–derived DCs were found to express low basal levels of the human homologue of SPI-6, PI-9, whereas CD40-dependent maturation of these DCs resulted in greatly increased PI-9 expression (Fig. 3 e). Taken together our observations demonstrate the generality of this feature for both murine and human DCs and point at a role for SPI-6/PI-9 in the protection of mature DCs against elimination by CTLs.
Overexpression of SPI-6 Conveys Resistance to CTL-induced Apoptosis, but Is not a Consequence of DC Maturation persé.
To address the question whether there is indeed a causal relation between SPI-6/PI-9 expression in mature DCs and their insensitivity to CTL-induced apoptosis, we transduced the D1 cells with a retrovirus encoding SPI-6. In agreement with previous observations by Gasperi et al. 43, we found that retroviral gene-transduction does not change the immature state of D1 cells (data not shown). As a result, we obtained immature D1 cells overexpressing SPI-6 at levels comparable to that observed in anti-CD40–matured D1 cells (Fig. 3 f). These immature SPI-6 gene-transduced D1 cells were highly resistant to CTL-induced apoptosis (Fig. 3 f). This indicates that SPI-6 expression as observed in matured DC suffices to protect these cells from CTL attack and, therefore, can fully account for this resistant phenotype.
Maturation of DCs during the priming of T cell immune responses in vivo can occur via cognate interaction with Th cells 2 3 4. Classically, these Th cells can be divided into subsets on the basis of their cytokine profile 44. Th1 cells are IFN-γ–secreting cells that foster development of CTLs, while Th2 cells secrete IL-4 and IL-10 and skew the immune system towards humoral responses 44. This functional difference between Th subsets prompted us to analyze their effects on SPI-6 expression in DCs. We made use of TCR transgenic CD4+ T cells derived from the DO10.11 mouse strain, which are directed against the I-Ab–restricted OVA epitope 323–339 (ISQAVHAAHAEINEAGR). These CD4+ T cells can develop into Th1 or Th2 type cells by in vitro culturing in the presence of antigen in combination with respectively IL-12 and anti–IL-4 or IL-4, anti–IL-12, and anti–IFN-γ 29. Incubation of Th1 or Th2 cells with D1 cells (at a ratio of 1:20) resulted in maturation in an antigen-dependent fashion (Fig. 4 a). As was reported before 29, only Th1 cells had the capacity to induce IL-12 secretion by DCs, a cytokine that feeds back into Th1 development and reportedly facilitates CTL formation 45. Importantly, also the expression of SPI-6 was only induced by Th1 cells, while Th2 cells remained without effect (Fig. 4 b). Controls reveal that the SPI-6 expression detected in this experiment was derived from the D1 cells and not the Th1 cells (Fig. 4 b).
Figure 4.


Th1 induces, while Th2 and IL-10 block SPI-6 induction in D1. (a) 5 × 104 OVA-specific Th1 and Th2 were incubated with 106 D1 cells for 48 h. Maturation of OVA peptide–loaded D1 cells was measured by B7-2 expression. (b–d) 106 D1 cells were incubated with 5 × 104 Th1 or Th2 cells, peptide, anti-CD40L, and anti–IL-10 as indicated and tested for IL-12p40 secretion by ELISA and SPI-6 expression by Western blotting after 24 h. Lysates from equivalent numbers of cells were applied on SDS-PAGE (0.5 × 106 cells). Equal protein loading was confirmed by ponceau S staining as well as by a control Western for MAPK (bottom). Please note that D1 cells were incubated with Th cells at a ratio of 20:1, resulting in a 20-fold lower protein content in the samples that contained Th cells only (right lanes). (e) Anti-CD40 (FGK) or Th1-induced SPI-6 expression in D1 cells was determined after 48 h in the absence or presence of IL-10 (left). The right panel represents SPI-6 expression in D1 cells after incubation with Th2 cells in the presence or absence of IFN-γ, anti–IL-10, and anti–IL-4 as indicated. As a control the Th1-induced SPI-6 expression is shown on the far right. (f) DNA fragmentation of Th1- (circles) or Th2 (triangles)-matured D1 cells (E1B-peptide loaded) was tested with the E1B-specific CTL clone for 6 h as described in the legend to Fig. 1 a.
Interestingly, the immune-regulatory cytokine IL-10 has previously been reported to reduce IL-12 secretion by DCs 29 and is mainly secreted by Th2 cells. Also our Th2 cells produce large quantities of this cytokine (244.2 ng/ml per million cells), while our Th1 cells produce relatively little IL-10 when activated by DCs (4.6 ng/ml per million cells). Therefore, we analyzed whether IL-10 secreted by the Th2 cell is responsible for the block in SPI-6 induction. Sequestering IL-10 with a blocking antibody almost completely restored the capacity of Th1 cells to induce SPI-6 expression in the presence of Th2 cells (Fig. 4 d). Moreover, exogenously added recombinant IL-10 partially prevented anti-CD40 as well as Th1-induced SPI-6 expression (Fig. 4 e). Our data indicate that IL-10 is critically involved in the repression of SPI-6 induction by Th2 cells, but also suggest that IL-10 is not the sole factor responsible for this repression. In agreement with this notion, sequestering IL-10 was not sufficient to allow significant SPI-6 induction in the D1 cells upon incubation with Th2 cells (Fig. 4 e). Since Th2 cells are also known to produce IL-4, whereas Th1 cells secrete IFN-γ 29, we tested the involvement of these additional cytokines in Th-mediated regulation of SPI-6 expression. Our data reveal that only the combination of blocking antibodies to IL-10 and IL-4 together with exogenously added IFN-γ could enable induction of SPI-6 by Th2 cells (Fig. 4 e). Taken together, this suggests that the differential induction of SPI-6 by Th1 and Th2 cells is due to the overall cytokine profile of these T cell subsets in combination with the expression of CD40L (Fig. 4 c).
Importantly, Th2-matured D1 were rapidly killed upon incubation with CTLs (Fig. 4 f). This indicates that maturation without SPI-6 induction results in a sensitive phenotype. In contrast, Th1-induced maturation did result in SPI-6 expression as well as in resistance to CTL-induced apoptosis (Fig. 4 f). Therefore, our data reveal a second prominent difference, in addition to IL-12 secretion, in the way Th1 and Th2 cells modulate DC function. This coordinate regulation of SPI-6 and IL-12 expression is in agreement with the notion that both play a role in Th1/CTL-dependent immunity. Moreover, the differential effect of Th1 and Th2 on SPI-6 expression indicates that CTL resistance is not necessarily linked to DC maturation, which was also apparent from the fact that immature SPI-6 gene-transduced D1 cells are highly resistant to CTL killing (Fig. 3 f).
Exhausted DCs Maintain High Levels of SPI-6/PI-9.
SPI-6–mediated resistance of mature DCs to CTL-induced killing is expected to positively affect the life span of DCs, whereas the induction of T cell memory was reported to require persistence of these APCs 46 47. Newly matured DCs were shown to efficiently drive activation of polarized effector T cells, which involves secretion of IL-12. At later time points, the same DCs become “exhausted,” lose production of IL-12 and preferentially prime nonpolarized memory-type T cells 47. In view of these findings one would predict that efficient induction of T cell memory requires survival of exhausted DCs. Since we have observed that DCs protection is mediated by high expression of SPI-6/PI-9, we investigated the expression levels of this inhibitor of DC death in newly matured versus exhausted DCs. In accordance with the results reported by Langekamp et al. 47, we found that human monocyte–derived DCs treated with LPS for 6 h, but not those treated for 24 or 48 h, secrete IL-12 upon a secondary stimulation with CD40L (Fig. 5 a). This confirms the exhausted nature of DCs treated for longer periods of time with LPS. However, treatment of these exhausted DCs with CD40L did result in increased maturation as determined by B7-2 expression (Fig. 5 b), indicating that these DCs are responsive to a secondary stimulus, but are exhausted in terms of IL-12 secretion. Importantly, when analyzing the expression of PI-9 we found that it was rapidly induced and remained high for at least 48 h upon CD40L or LPS treatment (Fig. 5 c). This indicates that PI-9 is expressed in both newly matured and exhausted human DCs and implies that mature DCs are protected from CTL-induced apoptosis even when in an exhausted state.
Figure 5.
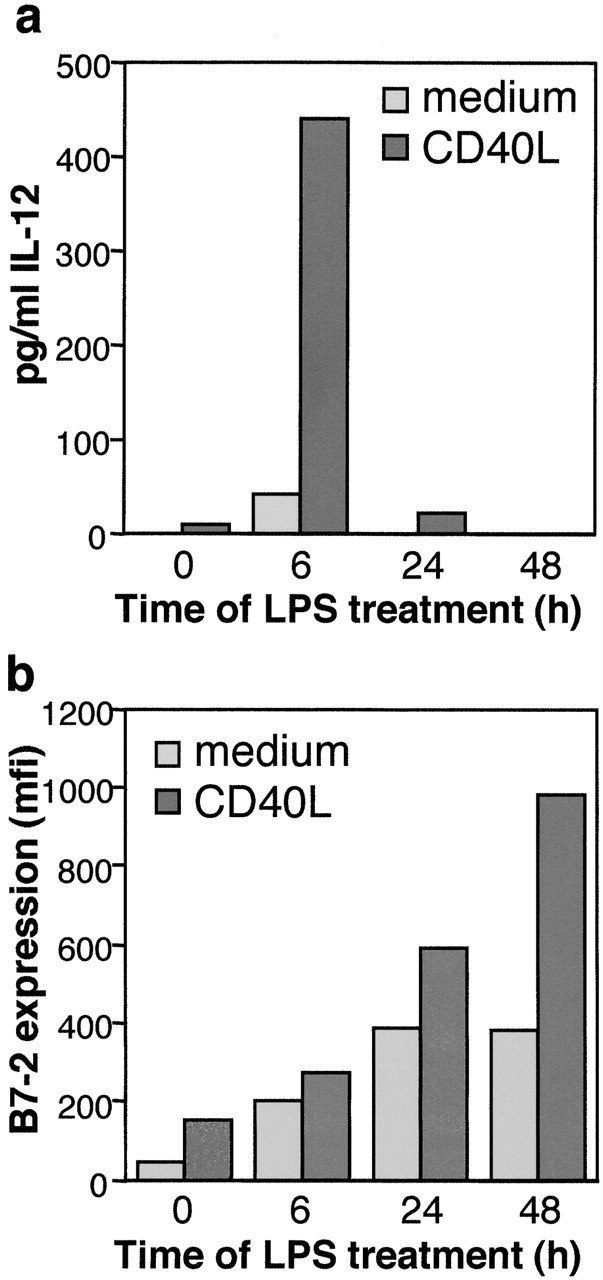

SPI-6/PI-9 is highly expressed in exhausted DCs. Primary human monocyte–derived DCs were treated for the indicated times with 100 ng/ml LPS after which the medium was refreshed. After a secondary incubation for 24 h in the absence (light gray) or presence (dark gray) of Lz-CD40L at 1 μg/ml, (a) IL12p70 secretion was analyzed by ELISA and (b) B7-2 expression was analyzed by FACS®. (c) In addition, PI-9 expression was determined at the indicated times after either LPS or Lz-CD40L treatment of human DCs. D1 cells were treated for different periods of time with LPS or FGK45 as indicated. (d) IL-12p40 secretion by the D1 cells was determined by ELISA. To analyze the IL-12 production in the final 24 h of incubation, the culture medium was refreshed for the last 24 h. (e) SPI-6 expression was determined in parallel by Western blot analysis. Lysates from equal numbers of cells were applied on SDS-PAGE (0.5 × 106 cells) and equal protein loading was checked by ponceau S staining and control Western blot analysis for MAPK (bottom).
We found that this feature is conserved between mouse and man as LPS treatment of D1 cells strongly, but transiently induced the secretion of IL-12p40. Secretion was mainly observed during the first 24 h and decreased significantly afterwards (Fig. 5 d), whereas maturation as measured by B7-2 expression increased in time (Fig. 2 b). Anti-CD40–induced IL-12p40 secretion by D1 was less pronounced, but also transient (Fig. 5 d). Similar results were obtained using primary bone marrow DCs (data not shown). This indicates that also murine DCs reach a state of exhaustion when considering IL-12 secretion. Nevertheless, SPI-6 expression steadily increased up to 72 h after the addition of LPS or anti-CD40 and remained high for at least another 24 h (Fig. 5 e). Taken together, our data argue for a role for SPI-6/PI-9 in the protection of DCs against CTL-induced apoptosis and suggest that SPI-6/PI-9–mediated DC survival may be of particular importance for the induction of proper T cell memory.
Discussion
We have shown that expression of the serpin SPI-6/PI-9 in DCs is upregulated upon DC activation by LPS treatment, CD40 triggering, or Th1 cells. Expression of SPI-6/PI-9 is tightly linked to resistance of DCs to CTL-induced apoptosis. The causal connection between SPI-6 overexpression and resistance to CTL-mediated apoptosis is illustrated particularly well by the fact that retroviral transduction of DCs with the SPI-6 gene, although not affecting the immature state of the cells, does elicit resistance to killing. In a parallel study involving immunohistochemical staining of human lymphoid organs, we found expression of PI-9 in DCs 35. Our present work reveals that expression of SPI-6/PI-9 is dependent on the maturation status and shows that it is of functional significance to DC survival.
The need for DCs to protect themselves from CTL-mediated eradication can be deduced from three studies that have demonstrated CTL-dependent clearance of injected DCs in mice (B. Ludewig, University of Zurich, personal communication; references 24 and 25). Monitoring of primary murine DC populations after reinjection into syngeneic mice revealed that at least a fraction of these DCs migrated to the lymph nodes, but also that these DCs were subsequently cleared from the lymph nodes in a CTL-dependent fashion. In one of these studies a role for perforin in DC clearance was postulated 24, whereas in a second study this process was shown to be independent of perforin (B. Ludewig, University of Zurich, personal communication). Importantly, in neither of these studies the maturation status of the DCs was determined. Therefore, we interpret these studies as a demonstration that DCs can indeed serve as targets for CTLs in vivo, without pinpointing the CTL effector mechanisms involved in relation to the DC maturation status.
The mechanism by which we find DCs to protect themselves from CTL-mediated apoptosis is also exploited for immune escape by certain viruses. Orthopoxviruses, in particular cowpox, rabbitpox, and vaccinia virus, use serpins to protect themselves from the host's immune response 48. One of these poxvirus serpins, SPI-2 (CrmA), was shown to target GrB as well as caspases 49. In agreement with our observations that SPI-6 can prevent cytotoxicity, SPI-2 in combination with SPI-1, can protect cells from alloreactive CTL-induced killing via both perforin and death receptor-dependent pathways in vitro 48. More importantly, rabbit poxvirus lacking either or both of the serpins are much less virulent as compared with the normal virus 48. This indicates that expression of GrB-inhibitory serpins can prevent CTL-induced killing not only in vitro but also in vivo.
Of significance with respect to the in vivo relevance of our observations is also the fact that we have used DNA fragmentation, a measure for apoptosis, as a read-out for cytotoxicity, rather than the chromium release assay. Recent experiments have revealed that this so called JAM assay more closely reflects the in vivo situation 9. In this particular study it was reported that cells lacking the mannose-6-phosphate receptor, which binds GrB and is essential for entry of GrB into the target cell, resist CTL-induced killing in vitro as measured by DNA fragmentation, while their receptor-transfected counterparts were fully CTL sensitive. However, both lines were equally sensitive to the actions of the CTLs in vitro when measured with a classical chromium-release assay. Grafting of these cells into mice revealed that the GrB receptor–deficient cells survive in vivo, despite massive infiltration of CTLs, which do eradicate the receptor-positive line 9. This work convincingly demonstrates that the in vitro chromium release overestimates the capacity of CTLs to lyse in vivo. In line with these observations, we have previously shown that the DNA fragmentation assay provides a better insight into the in vivo CTL-dependent cytotoxicity towards tumor cells than the classical chromium release assay 32. The effect of SPI-6 described here is most prominent when using apoptosis as a read-out, although it is detectable when CTL-induced chromium release is tested. However, this release is only marginally reduced upon maturation of D1 cells (data not shown). Nevertheless, others have shown that PI-9 (the human SPI-6) is capable of preventing chromium release to a reasonable extent when LAK cells are used as effector population 50. Apparently, SPI-6 can prevent DC lysis in vitro as well when tested under limiting conditions. However, the effect is clearly more prominent when apoptosis is taken as a readout, which as discussed more closely correlates to the in vivo sensitivity of the target cells.
It is important to note that, in addition to SPI-6/PI-9 overexpression, several other antiapoptotic mechanisms were found to increase the life span of DCs. For instance, both immature and LPS- or CD40-matured DCs are resistant to death receptor-induced death, which is achieved by the expression of the antiapoptotic protein cellular Fas-associated death domain–like IL-1β–converting enzyme (FLICE) inhibitory protein (c-FLIP; references 51 and 52). We have confirmed these observations for the D1 cell line (data not shown). As c-FLIP cannot prevent perforin-dependent killing 32 53, it cannot be responsible for the observed inhibition of cytotoxicity in mature DCs as reported here (Fig. 1). Instead, c-FLIP appears to serve as a more general survival mechanism that not only protects DCs from death receptor-dependent apoptosis by CTLs, but also by CD95L-positive Th cells. In agreement, recent evidence indicates that CD95 cross-linking on DCs during cognate interaction with Th cells triggers DC maturation rather than death 54.
The predominant Th-derived maturation signal, delivered to the CD40 receptor on DCs, was also found to promote DC survival. This involves upregulation of Bcl-2 55, a general regulator of apoptosis that prevents mitochondrial-dependent death induced by a variety of cellular stress signals 56. A third Th cell–derived signal for DCs involves TNF-related activation-induced cytokine expression (TRANCE), a member of the TNF superfamily that also promotes survival. It results in induction of the Bcl-2 homologue Bcl–XL 57, which protects DCs in a similar fashion as Bcl-2. Thus, DCs receive a mixture of signals that protect them against apoptosis. Importantly, unlike the antiapoptotic pathways represented by c-FLIP and Bcl-2/Bcl-XL, overexpression of SPI-6/PI-9 is not a general survival mechanism. Rather, it selectively offers protection of mature DCs against the actions of GrB and thereby allows DCs to survive in the hostile vicinity of activated CTLs. As such, it may be of particular relevance in the prevention of a premature decline in CTL immunity.
A prominent aspect of our findings is the differential regulation of SPI-6 expression in DCs by Th1 versus Th2 cells. The failure of Th2 cells to upregulate SPI-6, as well as their dominant inhibitory effect on Th1-induced SPI-6 expression, depends on the secretion of the immunoregulatory cytokine IL-10 (Fig. 4). It is tempting to speculate that this Th2/IL-10–mediated effect may result in a more rapid turnover of mature DCs in the T cell rich areas of the peripheral lymphoid organs and, thereby, favor skewing towards humoral responses. Importantly, the SPI-6/PI-9–dependent protection induced by Th1 cells (CD40) or LPS persists for a long period of time and recent evidence suggests that prolonged DC survival is required for the induction of T cell memory. At later timepoints after the induction of maturation DCs become exhausted and induce CCR7+ nonpolarized T cells. These T cells act as “central memory” cells 23, i.e., T cells without apparent effector function but with the capacity to home to the lymphoid organs. Our data strongly argue that SPI-6/PI-9 is involved in extending the life span of a DCs in the face of a CTL response. Therefore, SPI-6/PI-9 constitutes a novel functional marker for DCs, the expression of which coincides with LPS, anti-CD40, and Th1-induced DC maturation.
Acknowledgments
The authors would like to thank Drs. A. Rolink, P. Ricciardi-Castagnoli, M. Kubin, M. Wauben, and A. van Halteren for supplying cells, reagents, and mice. We are also grateful for the cytox help of Jurjen Velthuis. Furthermore, we would like to acknowledge the animal caretakers for their expert assistance.
This study was in part funded by a grant of the Dutch Cancer Society.
Footnotes
Abbreviations used in this paper: c-FLIP, cellular Fas-associated death domain–like IL-1β–converting enzyme-inhibitory protein; CMA, concanamycin A; DC, dendritic cell; EGFP, enhanced green fluorescent protein; Gr, granzyme; JAM, just another method; MAPK, mitogen-activated protein kinase; PI, protease inhibitor; SPI, serine PI.
References
- Banchereau J., Steinman R.M. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- Ridge J.P., Di Rosa F., Matzinger P. A conditional dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature. 1998;393:474–478. doi: 10.1038/30989. [DOI] [PubMed] [Google Scholar]
- Bennett S.R.M., Carbone F.R., Karamalis F., Flavell R.A., Miller J.F., Heath W.R. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature. 1998;393:478–480. doi: 10.1038/30996. [DOI] [PubMed] [Google Scholar]
- Schoenberger S.P., Toes R.E.M., van der Voort E.I.H., Offringa R., Melief C.J.M. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature. 1998;393:480–483. doi: 10.1038/31002. [DOI] [PubMed] [Google Scholar]
- Cyster J.G. Leukocyte migrationscent of the T zone. Curr. Biol. 2000;10:R30–R33. doi: 10.1016/s0960-9822(99)00253-5. [DOI] [PubMed] [Google Scholar]
- Lanzavecchia A. Licence to kill. Nature. 1998;393:413–414. doi: 10.1038/30845. [DOI] [PubMed] [Google Scholar]
- Heusel J.W., Wesselschmidt R.L., Shresta S., Russell J.H., Ley T.J. Cytotoxic lymphocytes require granzyme B for the rapid induction of DNA fragmentation and apoptosis in allogeneic target cells. Cell. 1994;76:977–987. doi: 10.1016/0092-8674(94)90376-x. [DOI] [PubMed] [Google Scholar]
- Shresta S., MacIvor D.M., Heusel J.W., Russell J.H., Ley T.J. Natural killer and lymphokine-activated killer cells require granzyme B for the rapid induction of apoptosis in susceptible target cells. Proc. Natl. Acad. Sci. USA. 1995;92:5679–5683. doi: 10.1073/pnas.92.12.5679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motyka B., Korbutt G., Pinkoski M.J., Heibein J.A., Caputo A., Hobman M., Barry M., Shostak I., Sawchuk T., Holmes C.F. Mannose 6-phosphate/insulin-like growth factor II receptor is a death receptor for granzyme B during cytotoxic T cell-induced apoptosis. Cell. 2000;103:491–500. doi: 10.1016/s0092-8674(00)00140-9. [DOI] [PubMed] [Google Scholar]
- Froelich J.C., Orth K., Turbov J., Gottleib R., Babior B., Shah G.M., Bleackley R.C., Dixit V.M., Hanna W. New paradigm for lymphocyte granule-mediated cytotoxicity. Target cells bind and internalize granzyme B, but an endosomolytic agent is necessary for cytosolic delivery and subsequent apoptosis. J. Biol. Chem. 1996;271:29073–29079. doi: 10.1074/jbc.271.46.29073. [DOI] [PubMed] [Google Scholar]
- Shi L., Mai S., Israels S., Browne K., Trapani J.A., Greenberg A.H. Granzyme B (GraB) autonomously crosses the cell membrane and perforin initiates apoptosis and GraB nuclear localization. J. Exp. Med. 1997;185:855–866. doi: 10.1084/jem.185.5.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapani J.A., Davis J., Sutton V.R., Smyth M.J. Proapoptotic functions of cytotoxic lymphocyte granule constituents in vitro and in vivo. Curr. Opin. Immunol. 2000;12:323–329. doi: 10.1016/s0952-7915(00)00094-7. [DOI] [PubMed] [Google Scholar]
- Thomas D.A., Du C., Xu M., Wang X., Ley T.J. DFF45/ICAD can be directly processed by granzyme B during the induction of apoptosis. Immunity. 2000;12:621–632. doi: 10.1016/s1074-7613(00)80213-7. [DOI] [PubMed] [Google Scholar]
- Sutton V.R., Davis J.E., Cancilla M., Johnstone R.W., Ruefli A.A., Sedelies K., Browne K.A., Trapani J.A. Initiation of apoptosis by granzyme B requires direct cleavage of bid, but not direct granzyme B-mediated caspase activation. J. Exp. Med. 2000;192:1403–1414. doi: 10.1084/jem.192.10.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heibein J.A., Goping I.S., Barry M., Pinkoski M.J., Shore G.C., Green D.R., Bleackley R.C. Granzyme B-mediated cytochrome c release is regulated by the bcl-2 family members bid and Bax. J. Exp. Med. 2000;192:1391–1402. doi: 10.1084/jem.192.10.1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagi D., Vignaux F., Ledermann B., Bürki K., Depreatere V., Nagata S., Hengartner H., Golstein P. Fas and perforin pathways as major mechanisms of T cell-mediated cytotoxicity. Science. 1994;265:528–530. doi: 10.1126/science.7518614. [DOI] [PubMed] [Google Scholar]
- Lowin B., Hahne M., Mattmann C., Tschopp J. Cytolytic T-cell cytotoxicity is mediated through perforin and Fas lytic pathways. Nature. 1994;370:650–652. doi: 10.1038/370650a0. [DOI] [PubMed] [Google Scholar]
- Berke G. The CTL's kiss of death. Cell. 1995;81:9–12. doi: 10.1016/0092-8674(95)90365-8. [DOI] [PubMed] [Google Scholar]
- Peter M.E., Scaffidi C., Medema J.P., Kischkel F., Krammer P.H. The death receptors. Results Probl. Cell. Differ. 1999;23:25–63. doi: 10.1007/978-3-540-69184-6_3. [DOI] [PubMed] [Google Scholar]
- Liu C.-C., Rafii S., Granelli-Piperno A., Trapani J.A., Ding-E Young J. Perforin and serine esterase expression in stimulated human T cells. J. Exp. Med. 1989;170:2105–2118. doi: 10.1084/jem.170.6.2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oehen S., Brduscha-Riem K. Differentiation of naive CTL to effector and memory CTLcorrelation of effector function with phenotype and cell division. J. Immunol. 1998;161:5338–5346. [PubMed] [Google Scholar]
- Potsch C., Vohringer D., Pircher H. Distinct migration patterns of naive and effector CD8 T cells in the spleencorrelation with CCR7 receptor expression and chemokine reactivity. Eur. J. Immunol. 1999;29:3562–3570. doi: 10.1002/(SICI)1521-4141(199911)29:11<3562::AID-IMMU3562>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Sallusto F., Lenig D., Förster R., Lipp M., Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–712. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- Loyer V., Fontaine P., Pion S., Hetu F., Roy D.C., Perreault C. The in vivo fate of APCs displaying minor H antigen and/or MHC differences is regulated by CTLs specific for immunodominant class I-associated epitopes. J. Immunol. 1999;163:6462–6467. [PubMed] [Google Scholar]
- Hermans I.F., Ritchie D.S., Yang J., Roberts J.M., Ronchese F. CD8+ T cell-dependent elimination of dendritic cells in vivo limits the induction of antitumor immunity. J. Immunol. 2000;164:3095–3101. doi: 10.4049/jimmunol.164.6.3095. [DOI] [PubMed] [Google Scholar]
- Odermatt B., Eppler M., Leist T.P., Hengartner H., Zinkernagel R.M. Virus-triggered acquired immunodeficiency by cytotoxic T-cell-dependent destruction of antigen-presenting cells and lymph follicle structure. Proc. Natl. Acad. Sci. USA. 1991;88:8252–8256. doi: 10.1073/pnas.88.18.8252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuurhuis D.H., Laban S., Toes R.E.M., Ricciardi-Castagnoli P., Kleijmeer M.J., van der Voort E.I., Rea D., Offringa R., Geuze H.J., Melief C.J.M., Ossendorp F. Immature dendritic cells acquire CD8+ cytotoxic T lymphocyte priming capacity upon activation by T helper cell-independent and -dependent stimuli. J. Exp. Med. 2000;192:145–150. doi: 10.1084/jem.192.1.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ria F., Penna G., Adorini L. Th1 cells induce and Th2 inhibit antigen-dependent IL-12 secretion by dendritic cells. Eur. J. Immunol. 1998;28:2003–2016. doi: 10.1002/(SICI)1521-4141(199806)28:06<2003::AID-IMMU2003>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Winzler C., Rovere P., Rescigno M., Granucci F., Penna G., Adorini L., Zimmermann V.S., Davoust J., Ricciardi-Castagnoli P. Maturation stages of mouse dendritic cells in growth factor-dependent long-term cultures. J. Exp. Med. 1997;185:317–328. doi: 10.1084/jem.185.2.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inaba K., Inaba M., Romani N., Aya H., Deguchi M., Ikehara S., Muramatsu S., Steinman R.M. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J. Exp. Med. 1992;176:1693–1702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea D., Schagen F.H., Hoeben R.C., Mehtali M., Havenga M.J., Toes R.E.M., Melief C.J.M., Offringa R. Adenoviruses activate human dendritic cells without polarization toward a T-helper type 1-inducing subset. J. Virol. 1999;7:10245–10253. doi: 10.1128/jvi.73.12.10245-10253.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema J.P., de Jong J., van Hall T., Melief C.J.M., Offringa R. Immune escape of tumors in vivo by expression of cellular FLICE inhibitory protein. J. Exp. Med. 1999;190:1033–1038. doi: 10.1084/jem.190.7.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heemskerk M.H., Hooijberg E., Ruizendaal J.J., van der Weide M.M., Kueter E., Bakker A.Q., Schumacher T.N., Spits H. Enrichment of an antigen-specific T cell response by retroviral transduced human dendritic cells. Cell. Immunol. 1999;195:10–17. doi: 10.1006/cimm.1999.1520. [DOI] [PubMed] [Google Scholar]
- Medema J.P., Sark M.W., Backendorf C., Bos J.L. Calcium inhibits epidermal growth factor-induced activation of p21ras in human primary keratinocytes. Mol. Cell. Biol. 1994;14:7078–7085. doi: 10.1128/mcb.14.11.7078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bladergroen B.A., Strik M.C.M., Bovenschen N., van Berkum O., Scheffer G.L., Meijer C.J.L.M., Hack C.E., Kummer J.A. The granzyme B inhibitor, protease inhibitor 9, is mainly expressed by dendritic cells and at immune-privileged sites. J. Immunol. 2001;166:3218–3225. doi: 10.4049/jimmunol.166.5.3218. [DOI] [PubMed] [Google Scholar]
- Sun J., Ooms L., Bird C.H., Sutton V.R., Trapani J.A., Bird P.I. A new family of 10 murine ovalbumin serpins includes two homologs of proteinase inhibitor 8 and two homologs of the granzyme B inhibitor (proteinase inhibitor 9) J. Biol. Chem. 1997;272:15434–15441. doi: 10.1074/jbc.272.24.15434. [DOI] [PubMed] [Google Scholar]
- Matzinger P. The JAM test. A simple assay for DNA fragmentation and cell death. J. Immunol. Methods. 1991;14:185–192. doi: 10.1016/0022-1759(91)90325-a. [DOI] [PubMed] [Google Scholar]
- Nagata S. Apoptotic DNA fragmentation. Exp. Cell Res. 2000;256:12–18. doi: 10.1006/excr.2000.4834. [DOI] [PubMed] [Google Scholar]
- Toes R.E., Offringa R., Blom R.J., Melief C.J.M., Kast W.M. Peptide vaccination can lead to enhanced tumor growth through specific T-cell tolerance induction. Proc. Natl. Acad. Sci. USA. 1996;93:7855–7860. doi: 10.1073/pnas.93.15.7855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataoka T., Shinohara N., Takayama H., Takaku K., Kondo S., Yonehara S., Nagai K. Concanamycin A, a powerful tool for characterization and estimation of contribution of perforin- and Fas-based lytic pathways in cell-mediated cytotoxicity. J. Immunol. 1996;156:3678–3686. [PubMed] [Google Scholar]
- Sun J., Bird C.H., Sutton V., McDonald L., Coughlin P.B., De Jong T.A., Trapani J.A., Bird P.I. A cytosolic granzyme B inhibitor related to the viral apoptotic regulator cytokine response modifier A is present in cytotoxic lymphocytes. J. Biol. Chem. 1996;271:27802–27809. doi: 10.1074/jbc.271.44.27802. [DOI] [PubMed] [Google Scholar]
- Bird P.I. Serpins and regulation of cell death. Results Probl. Cell Differ. 1998;24:63–89. doi: 10.1007/978-3-540-69185-3_4. [DOI] [PubMed] [Google Scholar]
- Gasperi C., Rescigno M., Granucci F., Citterio S., Matyszak M.K., Sciurpi M.T., Lanfrancone L., Ricciardi-Gastagnoli P. Retroviral gene transfer, rapid selection, and maintenance of the immature phenotype in mouse dendritic cells. J. Leukoc. Biol. 1999;66:263–267. doi: 10.1002/jlb.66.2.263. [DOI] [PubMed] [Google Scholar]
- Mosmann T.R., Coffman R.L. TH1 and TH2 cellsdifferent patterns of lymphokine secretion lead to different functional properties. Annu. Rev. Immunol. 1989;7:145–173. doi: 10.1146/annurev.iy.07.040189.001045. [DOI] [PubMed] [Google Scholar]
- Trinchieri G. Interleukin 12a proinflammatory cytokine with immunoregulatory functions that bridge innate resistance and antigen-specific adaptive immunity. Annu. Rev. Immunol. 1995;13:251–276. doi: 10.1146/annurev.iy.13.040195.001343. [DOI] [PubMed] [Google Scholar]
- Kundig T.M., Bachmann M.F., Oehen S., Hoffmann U.W., Simard J.J., Kalberer C.P., Pircher H., Ohashi P.S., Hengartner H., Zinkernagel R.M. On T cell memoryarguments for antigen dependence. Immunol. Rev. 1996;150:63–90. doi: 10.1111/j.1600-065x.1996.tb00696.x. [DOI] [PubMed] [Google Scholar]
- Langekamp A., Messi M., Lanzavecchia A., Sallusto F. Kinetics of dendritic cell activationimpact on priming of TH1, TH2 and nonpolarized T cells. Nat. Immunol. 2000;1:311–316. doi: 10.1038/79758. [DOI] [PubMed] [Google Scholar]
- Macen J.L., Garner R.S., Musy P.Y., Brooks M.A., Turner P.C., Moyer R.W., McFadden G., Bleackley R.C. Differential inhibition of the Fas- and granule-mediated cytolysis pathways by the orthopoxvirus cytokine response modifier A/SPI-2 and SPI-1 protein. Proc. Natl. Acad. Sci. USA. 1996;93:9108–9113. doi: 10.1073/pnas.93.17.9108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komiyama T., Quan L.T., Salvesen G.S. Inhibition of cysteine and serine proteinases by the cowpox virus serpin CRMA. Adv. Exp. Med. Biol. 1996;389:173–176. doi: 10.1007/978-1-4613-0335-0_21. [DOI] [PubMed] [Google Scholar]
- Bird C.H., Sutton V.R., Sun J., Hirst C.E., Novak A., Kumar S., Trapani J.A., Bird P.I. Selective regulation of apoptosisthe cytotoxic lymphocyte serpin proteinase inhibitor 9 protects against granzyme B-mediated apoptosis without perturbing the Fas cell death pathway. Mol. Cell. Biol. 1998;18:6387–6398. doi: 10.1128/mcb.18.11.6387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashany D., Savir A., Bhardwaj N., Elkon K.B. Dendritic cells are resistant to apoptosis through the Fas (CD95/APO-1) pathway. J. Immunol. 1999;163:5303–5311. [PubMed] [Google Scholar]
- Willems F., Amraoui Z., Vanderheyde N., Verhasselt V., Aksoy E., Scaffidi C., Peter M.E., Krammer P.H., Goldman M. Expression of c-FLIP(L) and resistance to CD95-mediated apoptosis of monocyte-derived dendritic cellsinhibition by bisindolylmaleimide. Blood. 2000;95:3478–3482. [PubMed] [Google Scholar]
- Kataoka T., Schroter M., Hahne M., Schneider P., Irmler M., Thome M., Froelich C.J., Tschopp J. FLIP prevents apoptosis induced by death receptors but not by perforin/granzyme B, chemotherapeutic drugs, and γ irradiation. J. Immunol. 1998;161:3936–3942. [PubMed] [Google Scholar]
- Rescigno M., Piguet V., Valzasina B., Lens S., Zubler R., French L., Kindler V., Tschopp J., Ricciardi-Castagnoli P. Fas engagement induces the maturation of dendritic cells, the release of interleukin (IL)-1β, and the production of interferon γ in the absence of IL-12 during dendritic cell-T cell cognate interactiona new role for Fas ligand in inflammatory responses. J. Exp. Med. 2000;192:11–19. doi: 10.1084/jem.192.11.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorck P., Banchereau J., Flores-Romo L. CD40 ligation counteracts Fas-induced apoptosis of human dendritic cells. Int. Immunol. 1997;9:365–372. doi: 10.1093/intimm/9.3.365. [DOI] [PubMed] [Google Scholar]
- Chao D.T., Korsmeyer S.J. BCL-2 familyregulators of cell death. Annu. Rev. Immunol. 1998;16:395–419. doi: 10.1146/annurev.immunol.16.1.395. [DOI] [PubMed] [Google Scholar]
- Wong B.R., Josien R., Lee S.Y., Sauter B., Li H.L., Steinman R.M., Choi Y. TRANCE (tumor necrosis factor [TNF]-related activation-induced cytokine), a new TNF family member predominantly expressed in T cells, is a dendritic cell-specific survival factor. J. Exp. Med. 1997;186:2075–2080. doi: 10.1084/jem.186.12.2075. [DOI] [PMC free article] [PubMed] [Google Scholar]