

Abstract
Interleukin (IL)-13 is a key mediator of tissue fibrosis caused by T helper cell type 2 inflammation. We hypothesized that the fibrogenic effects of IL-13 are mediated by transforming growth factor (TGF)-β. To test this hypothesis we compared the regulation of TGF-β in lungs from wild-type mice and CC10-IL-13 mice in which IL-13 overexpression causes pulmonary fibrosis. IL-13 selectively stimulated TGF-β1 production in transgenic animals and macrophages were the major site of TGF-β1 production and deposition in these tissues. IL-13 also activated TGF-β1 in vivo. This activation was associated with decreased levels of mRNA encoding latent TGF-β–binding protein-1 and increased mRNA encoding urinary plasminogen activator, matrix metalloproteinase (MMP)-9, and CD44. TGF-β1 activation was abrogated by the plasmin/serine protease antagonist aprotinin. It was also decreased in progeny of crosses of CC10-IL-13 mice and MMP-9 null mice but was not altered in crosses with CD44 null animals. IL-13–induced fibrosis was also significantly ameliorated by treatment with the TGF-β antagonist soluble TGFβR-Fc (sTGFβR-Fc). These studies demonstrate that IL-13 is a potent stimulator and activator of TGF-β1 in vivo. They also demonstrate that this activation is mediated by a plasmin/serine protease- and MMP-9–dependent and CD44-independent mechanism(s) and that the fibrogenic effects of IL-13 are mediated, in great extent, by this TGF-β pathway.
Keywords: lung, plasmin, matrix metalloproteinase-9, CD44, asthma
Introduction
Tissue fibrosis is a well-documented consequence of Th2 cytokine-dominated inflammatory responses. This is nicely illustrated in asthma where Th2-dominated inflammation is the cornerstone of the disorder 1 2 and airway remodeling with subepithelial fibrosis are believed to be adverse consequences of this inflammatory response (for a review, see reference 3). Th2 inflammation also plays a central role in the pathogenesis of a variety of other fibrotic disorders including progressive systemic sclerosis 4 5 6, idiopathic pulmonary fibrosis (IPF; references 5 6 7 8), radiation-induced pulmonary fibrosis 9, chronic lung allograft rejection 10, bleomycin lung 11, and hepatic fibrosis 12 13 14 15 . In vitro polarized Th2 cells also induce tissue fibrosis when passively transferred and activated in vivo 16. In contrast, Th1 cytokine-dominated responses eventuate in tissue fibrosis less frequently and IFN-γ, the prototypic Th1 cytokine, has a variety of antifibrotic effector properties and has been shown to be a useful antifibrotic agent in murine and human pulmonary and renal fibrotic disorders 8 17 18 19. As a result of these findings, the “type 2 cytokine hypothesis of fibrosis” has been formulated which suggests that fibrosis occurs when cytokine balance shifts in a Th2 (type 2) direction 19. Surprisingly, the mechanism(s) by which Th2 inflammation generates tissue fibrosis is poorly understood. In addition, although it is assumed in this hypothesis that Th2 cytokines induce fibrosis via pathways that are independent of known mediators of fibrosis such as TGF-β, the validity of this assumption has not been adequately assessed.
IL-13 is a pleiotropic cytokine produced by a gene on chromosome 5 at q31 that is elaborated in significant quantities by appropriately stimulated Th2 cells 20 21. A variety of lines of evidence have demonstrated that IL-13 is an important mediator in the pathogenesis of asthma 22 23 24 25. IL-13 has also been implicated in the pathogenesis of hepatic fibrosis, progressive systemic sclerosis, pulmonary fibrosis, and nodular sclerosing Hodgkin's disease 4 12 13 26 27 28. In these diseases IL-13 is felt to contribute to the pathogenesis of tissue fibrosis. This is based on studies from our laboratory that demonstrated that the transgenic overexpression of IL-13 in the murine airway causes subepithelial airway fibrosis that is similar, in many ways, to the fibrotic response seen in remodeled human asthmatic airways 3 25. It is also based on studies that demonstrate that IL-13 is the primary cytokine responsible for hepatic fibrosis in murine schistosomiasis and that IL-13 is a potent stimulator of fibroblast proliferation and collagen production in vitro 12 13 29 30. Surprisingly, the mechanism of IL-13–induced fibrosis has not been further defined. In addition, the relationship between IL-13 and TGF-β cytokines, which are also dysregulated in many of these disorders, have not been elucidated.
We hypothesized that IL-13 mediates its fibrogenic effects in the lung and other organs by altering TGF-β cytokine homeostasis. To test this hypothesis we characterized the regulation and role of TGF-β moieties in lungs from wild-type (WT) mice and transgenic CC10-IL-13 mice in which the overexpression of IL-13 causes airway and parenchymal fibrosis 25. These studies demonstrate that IL-13 selectively stimulates and activates TGF-β1. They also define the mechanism of this TGF-β1 activation and demonstrate that the fibrotic effects of IL-13 are mediated, in great extent, by this TGF-β1 induction and activation pathway.
Materials and Methods
Genetically Manipulated Mice.
CC10-IL-13 mice, mice with homozygous null mutations of matrix metalloproteinase (MMP)-9−/−, and mice with homozygous null mutations of CD44 (CD44−/−) were used in these studies. In the CC10-IL-13 mice, the Clara cell 10-kDa (CC10) promoter was used to constitutively overexpress IL-13 in a lung-specific fashion. The methods that were used to generate these mice in our laboratory and the organ specificity of transgene expression, levels of bronchoalveolar lavage (BAL) IL-13, and the phenotype of these animals have been described previously 25. The MMP-9−/− mice were also generated in our laboratories 31. MMP-9 is inactivated in these mice as a result of the insertion of NEO genes in exon-2 of each MMP-9 allele. CD44−/− mice were obtained from T. Mak (Amgen Institute, Toronto, Canada; reference 32). CC10-IL-13 mice with homozygous null mutations of MMP-9 (CC10-IL-13/MMP-9−/−) or CD44 (CC10-IL-13/CD44−/−) were generated by breeding CC10-IL-13 transgene (+) mice with MMP-9−/− or CD44−/− animals. Unless otherwise indicated, WT littermates (+/+) were used as negative controls.
BAL.
After anesthesia, a median sternotomy was performed and the trachea was exposed by blunt dissection. A 22-gauge catheter was inserted into the trachea, and BAL was performed by instilling 1 ml of PBS into the trachea and gently aspirating back. This was repeated twice. The BAL samples from each animal were pooled and centrifuged. Differential cell counts were performed on the pellet and the supernatant was stored at −20°C until used.
Histologic Evaluation.
Histologic evaluation was performed as described previously 25 33 34. In brief, animals were killed, a median sternotomy was performed, and right heart perfusion was accomplished with calcium- and magnesium-free PBS to clear the pulmonary intravascular space. The heart and lung were then removed en bloc, fixed to pressure (25 cm) with neutral buffered formalin, incubated overnight in formalin, embedded in paraffin, sectioned, and stained. Hematoxylin and eosin, Mallory's Trichrome, and Sirius red staining were performed in the Research Pathology Laboratory at Yale University, New Haven, CT.
Treatment with Soluble TGF-β Type II Receptor-Fc and Aprotinin.
The role of TGF-β was assessed using soluble TGFβR-Fc (sTGFβR-Fc). This TGF-β antagonist contains a soluble TGF-β type II receptor attached to the Fc fragment of human IgG. The generation protocols and in vitro and in vivo efficacy of this construct have been described previously 35 36. It was administered to IL-13 transgene (+) and littermate control mice in PBS (2 mg/kg) by intraperitoneal injection every other day for 8 wk, starting when the mice were 4 wk old. An equivalent dose of pooled human IgG was used as a control.
Aprotinin from bovine lung (6618 KIU/mg) was purchased from ICN Inc. It was administered to IL-13 transgene (+) mice and littermate controls (500 μg/mouse) by intraperitoneal injection every day for 4 wk starting when the mice were 4 wk of age. An equivalent dose of PBS was administered according to the same schedule as a vehicle control.
mRNA Analysis.
mRNA levels were assessed using reverse transcription (RT)-PCR and ribonuclease protection assays as described previously by our laboratories 33 37. In these experiments, total lung RNA was obtained using TRIzol reagent (Life Technologies) as per the manufacturer's instructions followed by treatment with DNase I (Ambion, Inc.). In the RT-PCR assays, 0.5 μg of RNA was reverse transcribed to cDNA and gene-specific primers (Table ) were used to amplify selected regions of each target moiety. The primers and optimum conditions used in the reactions were designed according to published sequences. The whole reaction was performed in a 25 μl volume using an Access RT-PCR kit purchased from Promega according to the manufacturer's instructions. All primers were synthesized at the Yale Oligonucleotide Synthesis Laboratory. To verify that equal amounts of undegraded RNA were added in each RT-PCR reaction, β-actin was used as an internal standard. Amplified PCR products were detected using 1.2% agarose ethidium bromide gel electrophoresis, quantitated electronically, and confirmed by nucleotide sequencing. Ribonuclease protection assays were performed using the RiboQuant In Vitro Transcription kit (BD PharMingen) according to manufacturer's instructions.
Table 1.
Primer Sequences and RT-PCR Conditions
| Gene | s/as | Primer Sequences (5′ to 3′) | Anneal. Tm. | Cycle | Product Size (bp) | GenBank Accession Number |
|---|---|---|---|---|---|---|
| LTBP-1 | s | GGTGTGTGGATGTGAACGAG | 60°C | 30 | 447 | AF022889 |
| as | CTCCAAACAGCAAGCATTCA | |||||
| CD36 | s | GCAAAGAAGGAAAGCCTGTG | 60°C | 30 | 337 | L23108 |
| as | TCTACCATGCCAAGGAGCTT | |||||
| TSP-1 | s | GACGTCGATGAGTGCAAAGA | 60°C | 27 | 532 | M87276 |
| as | AGTCATACTGGGCTGGGTTG | |||||
| INT-β6 | s | AGGGGTGACTGCTATTGTGG | 60°C | 30 | 423 | AF115376 |
| as | GGCACCAATGGCTTTACACT | |||||
| uPA | s | CCTACAATGCCCACAGACCT | 60°C | 25 | 471 | NM-008873 |
| as | GCTGCTCCACCTCAAACTTC | |||||
| PAI-1 | s | TCATCAATGACTGGGTGGAA | 60°C | 30 | 539 | M33960 |
| as | CTGCTCTTGGTCGGAAAGAC | |||||
| MMP-9 | s | CCATGAGTCCCTGGCAG | 58°C | 30 | 505 | X72795 |
| as | ATGACAATGTCCGCTTCG | |||||
| CD-44 | s | GAGGATTCATCCCAACGCTA | 60°C | 25 | 499 | M27129 |
| as | GGTCACTCCACTGTCCTGGT | |||||
| β-actin | s | GTGGGCCGCTCTAGGCACCA | 65°C | 20 | 241 | X03672 |
| as | TGGCCTTAGGGTTCAGGGGG |
Anneal. Tm., annealing temperature; as, antisense; INT, integrin; s, sense; TSP, thrombospondin.
Quantification of TGF-β1.
The levels of immunoreactive TGF-β1 and TGF-β2 were determined with cytokine-specific commercial ELISA kits as per the manufacturer's instructions (R&D Systems).
Immunohistochemistry.
Immunostains for TGF-β1 was performed on paraffin-embedded tissues that had been fixed in either Streck's tissue fixative or neutral buffered 4% formalin. After sectioning the tissues were rehydrated, endogenous peroxidase was blocked by quenching with H2O2 and methanol and the tissues were treated in a microwave oven in the presence of citrate buffer (10 mM, pH 6.0) for 9 min (3 cycles of 3 min each). After preblocking with Dako blocking reagent (Dako) for 15 min, polyclonal rabbit anti–TGF-β1 antibody (Santa Cruz Biotechnology, Inc.) was applied at a 1:100 dilution. In experiments designed to see if an appropriate peptide competed for antibody binding, the anti–TGF-β1 antiserum was preincubated with murine TGF-β1 (R&D Systems) at a 1:1 molar ratio for 2 h before it was applied to the tissues. After overnight incubation at 4°C, the tissues were rinsed with PBS/0.1% Tween 20 and Envision (Dako) peroxidase coupled anti–rabbit secondary reagent was applied for 30 min. The slides were then washed, 3,3′diaminobenzidine was applied for 5 min, and the tissues were counterstained with hematoxylin.
In Situ Hybridization.
Lung tissue was fixed in formaldehyde and processed into paraffin. 5 μ sections were cut, deparaffinized, treated with proteinase K (20 μg/ml) for 20 min at 37°C followed by 0.1 M triethylnolamine/0.25% acetic anhydride, pH 8.0, for 10 min at room temperature, and then rinsed in PBS. The mouse TGF-β1 probe (containing the segment from the mouse TGF-β1 sequence between base pairs 1027–1465; GenBank Accession, NM-011577) was placed in a pBluescript II KS phagemid (Stratagene) between T7 and T3 promoter sites. Sense and antisense RNA probes were generated, labeled with a digoxigenin RNA labeling kit (Roche), denatured at 65°C, added to commercially available hybridization buffer (Ambion) at 6 ng/μl, and the hybridization mixture was incubated with tissue overnight at 52°C. The tissues were then washed twice with 4×SSC for 5 min at room temperature, twice with 2×SSC for 10 min at 37°C, and incubated with RNase A (10 μg/ml) for 45 min at 37°C. This was followed by 2 10-min washes in 2×SSC at room temperature and 3 20-min washes in 0.2×SSC at 50°C. Probe was detected by overnight incubation with sheep antibodies to digoxigenin-labeled with alkaline phosphatase (Roche) followed by 4-nitroblue tetrazolium chloride/5-bromo-4-chloro-3-indoyl-phosphate (BCIP/NBT) as described by the manufacturer.
TGF-β Bioassay.
To measure the bioactivity of TGF-β in BAL fluids, we used mink lung epithelial cells permanently transfected with a construct containing the TGF-β responsive human plasminogen activator inhibitor (PAI)-1 promoter fused to a luciferase reporter gene (TMLC, a gift from John Munger, NYU Medical Center, New York, NY). These cells were seeded into wells of 12-well tissue culture plates (105 cells per well per milliliter) in DMEM supplemented with 10% FCS and allowed to attach for 5 h. They were then washed and incubated in triplicate in mixtures containing 200 μl of BAL fluid and 800 μl of assay medium (DMEM with 2.5% FCS). These incubations were performed in the presence and absence of saturating quantities of neutralizing antibodies specific for TGF-β1, TGF-β2, or TGF-β3 (R&D Systems). The luciferase activities in these cells were measured 16 h later using the Luciferase Assay System (Promega) according to the manufacturer's instructions. The bioactivity attributed to TGF-β1 (TGF-β1 bioactivity) was defined as the difference in the luciferase activities of identical cells incubated in the absence and presence of anti–TGF-β1.
Western Blot Analysis.
BAL fluids were concentrated fivefold using Centricon 10 concentrators (Amicon). Equal volumes of test and control samples were fractionated on 15% SDS-PAGE gels under reducing conditions and transferred to PVDF membranes (Millipore). The membranes were then blocked with 5% nonfat dry milk, washed three times with PBS/0.1% Tween 20, and incubated for 1 h at room temperature in a 1:200 dilution of rabbit polyclonal anti–TGF-β1, anti–TGF-β2, or anti–TGF-β3 antibodies (Santa Cruz Biotechnology, Inc.). After washing with PBS/ 0.1% Tween 20, they were incubated for 1 h with a horseradish peroxidase-conjugated donkey anti–rabbit antibody (Amersham Pharmacia Biotech), rewashed in PBS/0.1% Tween 20, and protein bands were detected using the ECL Western blot detection system (Amersham Pharmacia Biotech).
Collagen Assay.
Total lung collagen content was determined by quantifying total soluble collagen using the Sircol Collagen Assay kit (Biocolor) according to the manufacturer's instructions. Briefly, lungs were homogenized in 5 ml of 0.5 M acetic acid containing 1 mg pepsin (Sigma-Aldrich)/10 mg tissue residue. Each sample was incubated for 24 h at 4°C with stirring. After centrifugation, 100 μl of each supernatant was assayed. 1 ml of Sircol dye reagent that specifically binds to collagen was then added to each sample and mixed for 30 min. After centrifugation the pellet was suspended in 1 ml of alkali reagent (0.5 M NaOH) included in the kit and optical density evaluated at 540 nm with a spectrophotometer. The values in the test samples were compared to the values obtained with collagen standard solutions provided by the manufacturer that were used to construct a standard curve.
Picosirius Red Staining and Evaluation.
Collagen content was assessed using Picosirius red staining. This approach was chosen because it accurately reflects organ collagen content assessed with hydroxyproline assays 38 39 and allows areas of localized collagen accumulation to be specifically evaluated. In these assays, sections of lung are stained with Sirius red as described previously 40. For examination of lung parenchyma, the tissue was examined under polarized light with an Olympus BH-2 microscope with an original magnification: 2× objective. This magnification allows virtually the entire lung section to be acquired for analysis using 4–6 fields. Image acquisition was performed with a DXC-970MD Sony video camera attached to a iXTV video capture card (IXMICRO) in a G3 Macintosh (Apple Computer) using software included with the card. Images are captured as grey scale TIFF encoded video clips. These are opened in NIH Image and averaged to reduce noise. Images are then subject to 2-D rolling ball background subtraction to make the background uniform. Areas of parenchyma are manually outlined to exclude airways and large vessels, then thresholded. The number of white versus total pixels are then recorded. The total number of white pixels versus total pixels examined is a reflection of collagen density.
For examination of airway fibrosis, images were acquired with an original objective: 4× objective. Only airways that were oval or circular and are outside the central bronchovascular sheath were analyzed. Each field was acquired as a regular color image as well as a polarized image. Images were opened within NIH Image. The airway was outlined using the color image to determine area and minor axis. The same airway was outlined in the polarized image and subject to thresholding to determine the number of white pixels. Data was expressed as area of collagen versus total airway area.
Statistics.
Normally distributed data are expressed as means ± SEM and assessed for significance by Student's t test or ANOVA as appropriate. Data that were not normally distributed were assessed for significance using the Wilcoxon rank sum test.
Results
Effect of IL-13 on TGF-β1 Production.
Pulmonary fibrosis was a prominent finding in CC10-IL-13 mice. As previously reported, subepithelial and adventitial airway fibrosis were readily appreciated in 1-mo-old animals 25. This response continued to progress over the life of the mouse. By the time the mice were 2–3 mo of age, parenchymal fibrosis was also appreciated (Fig. 1). To gain insight into the mechanisms of this fibrotic response, studies were undertaken to determine if IL-13 stimulated the production of TGF-β moieties in the murine lung. Our initial studies used ribonuclease protection assays to compare the levels of TGF-β1, TGF-β2, and TGF-β3 mRNA in lungs from transgene (−) and transgene (+) animals. As can be seen in Fig. 2, IL-13 caused an impressive increase in the levels of mRNA encoding TGF-β1. Comparable increases in the levels of mRNA encoding TGF-β2 were not appreciated. In addition, the levels of TGF-β3 in lungs from transgene (+) mice were lower than the levels in their transgene (−) littermate controls (Fig. 1).
Figure 1.
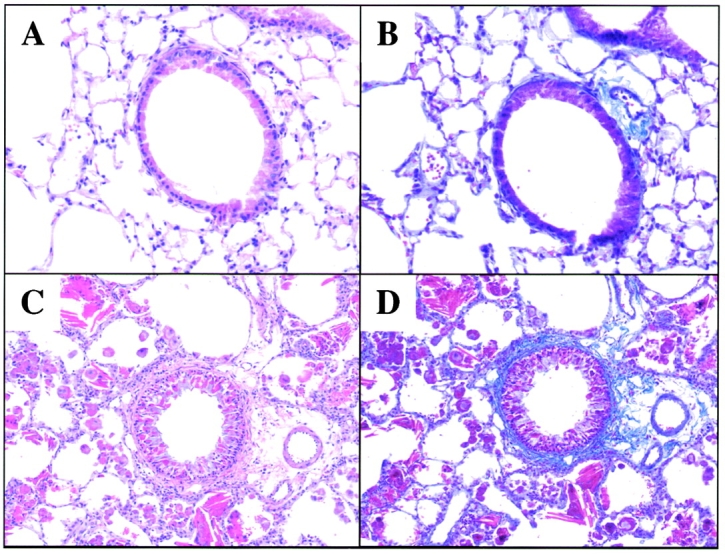
Histologic responses in CC10-IL-13 mice. H&E stains are used in A and C to compare the histologic features of WT littermate control mice and 3-mo-old CC10-IL-13 transgene (+) mice respectively. Trichrome stains are used in B and D to compare the collagen in WT littermate control mice and 3-mo-old transgene (+) animals respectively. All figures are at original magnification: 20×. Collagen stains blue in these panels.
Figure 2.

Effect of IL-13 on TGF-β cytokine mRNA. Whole lung RNA from IL-13 transgene (+) mice and transgene (−) littermate controls were obtained. The levels of mRNA encoding TGF-β1, TGF-β2, TGF-β3, and L32, the housekeeping control gene, were assessed using ribonuclease protection assays. Each lane represents an individual animal.
Studies were next undertaken to determine if the increased levels of TGF-β1 mRNA in lungs from transgene (+) mice were associated with increased levels of BAL fluid TGF-β1 protein. Using an ELISA assay that detects bioactive cytokine, TGF-β1 was not detected in significant quantities in acid-treated BAL fluids from transgene (−) animals (Fig. 3 A). Significantly increased levels of TGF-β1 were noted in acid-treated BAL from 1-mo-old transgene (+) animals and the levels of BAL TGF-β1 continued to increase as the animals aged (Fig. 3 A). In contrast, TGF-β2 was not detected by ELISA in BAL fluids from transgene (−) or (+) animals (data not shown). In accord with these findings, TGF-β1 monomers were readily detected in immunoblots performed with acid-treated BAL fluids from transgene (+) mice but were not readily apparent in BAL fluids from transgene (−) littermate controls (see Fig. 3 B). Comparable alterations in the levels of TGF-β2 and TGF-β3 proteins were not noted in comparisons of immunoblots from acid-activated BAL fluids from transgene (+) and (−) mice (data not shown). When viewed in combination, these findings demonstrate that IL-13 selectively stimulates TGF-β1 mRNA accumulation and protein production in the murine lung.
Figure 3.


Effect of IL-13 on TGF-β1 protein. In A, BAL fluids were obtained from 1–3-mo-old transgene (−) (black bars) and transgene (+) (hatched bars) mice and acid treated. The levels of total TGF-β1 in these fluids were characterized by ELISA. The noted values represent the mean ± SEM of a minimum of four animals. *P < 0.01, **P < 0.001 versus WT mice. In B, BAL fluids were obtained from 2-mo-old transgene (−) and (+) mice and acid activated. BAL TGF-β1 was then assessed by immunoblot analysis. Each lane represents an individual animal.
Localization of TGF-β 1 in IL-13 Transgenic Mice.
To gain insight into the mechanism(s) of TGF-β1 induction, immunohistochemistry and in situ hybridization (ISH) were used to localize and define the sites of TGF-β1 production in lungs from transgene (−) and transgene (+) animals. TGF-β1 protein and mRNA were readily apparent in airway epithelial cells in lungs from transgene (−) animals. Low levels of TGF-β, mRNA, and protein were also noted in some alveolar macrophages (Fig. 4 and Fig. 5 and data not shown). In lungs from IL-13 transgene (+) mice impressive increases in macrophage TGF-β1 mRNA and protein were readily apparent. Lower levels of TGF-β1 mRNA and protein could also be appreciated in type II alveolar epithelial cells, airway epithelial cells, and occasionally in eosinophils from these transgene (+) animals (Fig. 4 and Fig. 5 and data not shown). In these immunohistochemical evaluations, preincubation of our primary antibody with TGF-β1 peptide effectively abrogated the detection of TGF-β1 in these tissues (Fig. 4). In the ISHs, significant staining with the sense probe was not noted (Fig. 5). This demonstrates the specificity of these approaches. Thus, lung macrophages are the major sites of production and storage, and airway and alveolar epithelial cells and eosinophils are lesser sites of production and storage of TGF-β1 in lungs from IL-13 transgene (+) animals.
Figure 4.

Immunohistochemical localization of TGF-β1. Immunohistochemistry was used to localize the sites of TGF-β1 in WT transgene (−) mice and CC10-IL-13 transgene (+) animals (2–3 mo of age). Epithelial TGF-β1 can be appreciated in the transgene (−) animals (A, original magnification: 20×). Macrophage TGF-β1 is increased (C, original magnification: 20× and E, original magnification: 40×, black arrows), and alveolar type II epithelial cell (F, original magnification: 40×, white arrows) and eosinophil (F, 40×, black arrow) TGF-β1 can be appreciated in IL-13 transgene (+) animals. In all cases, preincubation with TGF-β peptide abrogated TGF-β1 staining (B and D, original magnification: 20×).
Figure 5.
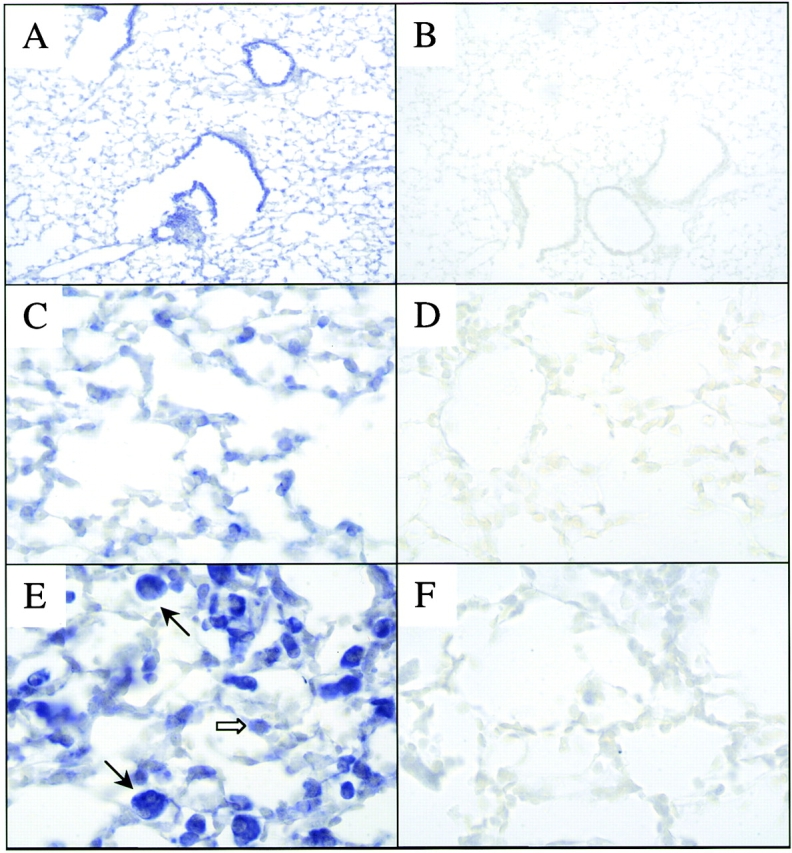
ISHs of TGF-β1. ISH was used to define the sites of TGF-β1 mRNA expression in transgene (−) and IL-13 transgene (+) mice (2–3 mo old). Epithelial TGF-β1 mRNA can be appreciated in the transgene (−) animals (A, original magnification: 10×). Enhanced macrophage (black arrows) and type II alveolar epithelial cell (white arrow) TGF-β1 mRNA can be seen in comparisons of parenchymal areas from IL-13 transgene (−) mice (C, original magnification: 40×) and transgene (+) mice (E, original magnification: 40×). The lack of staining with the sense probe is seen in B (original magnification: 10×, transgene [−]), D (original magnification: 40×, transgene [−]), and E (original magnification: 40×, transgene [+]).
Effect of IL-13 on TGF-β 1 Activation.
TGF-β cytokines are produced as biologically inactive moieties with the TGF-β1 dimer bound to its propeptide, latency-associated peptide (LAP). They exert their tissue effects after activation, a complex process that likely involves proteolysis in vivo and occurs in biologic fluids in vitro after local acidification 41 42 43 44 45. Studies were thus undertaken to determine if the TGF-β1 in BAL fluids from IL-13 transgene (+) mice was produced in a biologically inactive or spontaneously activated form. This was done by comparing the TGF-β1 bioactivities in BAL fluids from transgene (−) and transgene (+) mice using the TMLC/PAI-1 promoter-based luciferase bioassay that only responds to activated TGF-β moieties. As can be seen in Fig. 6, significant TGF-β1 bioactivity was not present in the untreated or acid-treated BAL fluids from transgene (−) mice. In contrast, impressive levels of spontaneous TGF-β1 bioactivity were present in untreated BAL fluids from IL-13 transgene (+) animals (Fig. 6 A). Latent TGF-β1 was also present since acidification revealed an impressive additional increase in the levels of TGF-β1 bioactivity in BAL fluids from transgene (+) animals (Fig. 6 B). When viewed in combination, these studies demonstrate that IL-13 increases the production and activates TGF-β1 in the murine lung.
Figure 6.
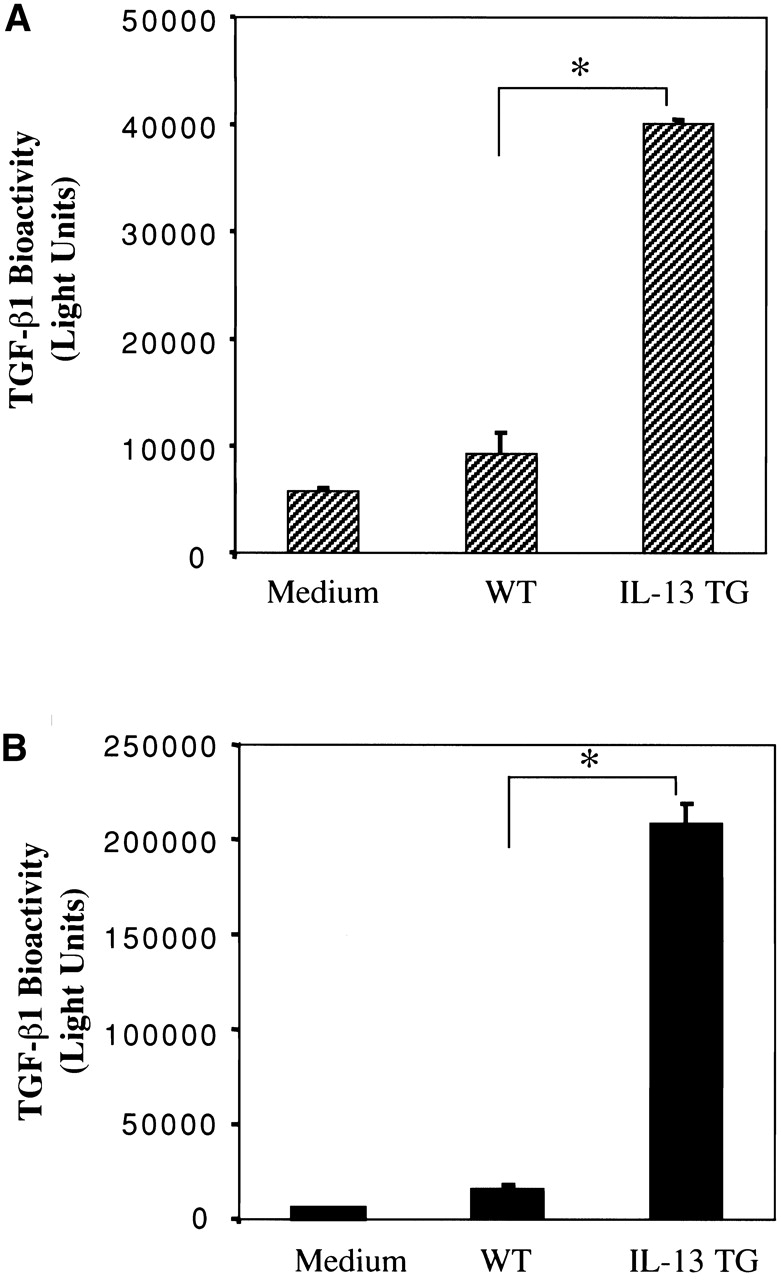
Spontaneous and total TGF-β1 bioactivity in BAL fluids from transgene (−) and transgene (+) mice. BAL fluids were obtained from WT transgene (−) animals and IL-13 transgene (+) (IL-13 TG) mice. The levels of TGF-β1 bioactivity in these fluids were assessed before (A, spontaneous activity) and after acid activation (B, total activity) using mink lung cells permanently transfected with constructs containing the PAI-1 promoter driving a luciferase reporter gene as described in Materials and Methods. The noted values represent the mean ± SEM of a minimum of four animals in each category. *P < 0.001 versus WT.
IL-13 Regulation of Processes Involved in TGF-β1 Activation.
Studies were next undertaken to elucidate the mechanism(s) by which IL-13 activated the TGF-β1 in lungs from transgene (+) mice. A variety of complex mechanisms of TGF-β1 activation have been described previously. They include pathways involving CD36 and thrombospondin-1 46 47, urokinase type plasminogen activator (uPA, and its inhibitor PAI-1) 41 43 46 48 49, the αVβ6 integrin 44, latent TGF-β–binding protein (LTBP)-1 50, and CD44 and MMP-9 51. To determine if these pathways were activated in IL-13 transgenic mice, we quantitated the levels of mRNA encoding key elements of these pathways in lungs from transgene (−) and transgene (+) animals. Similar levels of mRNA encoding CD36, thombospondin-1, the β6 integrin and PAI-1 were seen in whole lung RNA from transgene (−) and transgene (+) animals (Fig. 7). In contrast, an impressive decrease in the levels of mRNA encoding LTBP-1 and significant increases in the levels of mRNA encoding uPA, MMP-9, and CD44 were noted (Fig. 7).
Figure 7.

IL-13 regulation of processes involved in TGF-β1 activation. Whole lung mRNA was obtained from 2–3-mo-old transgene (−) and transgene (+) mice. The levels of mRNA encoding the noted moieties were evaluated via RT-PCR. Each lane represents an individual animal. The genotype of each animal is noted on the top of the figure.
Role of uPA/Serine Proteinases in TGF-β1 Activation.
Because the levels of mRNA encoding uPA were increased in IL-13 transgene (+) animals, studies were undertaken to determine if uPA (or related serine proteinases) played important roles in the activation of TGF-β1 in lungs from IL-13 transgene (+) mice. In these experiments we compared the levels of spontaneously bioactive and total TGF-β1 in BAL fluids from lungs from transgene (+) mice that had been treated with the plasmin/serine proteinase antagonist aprotinin or an appropriate vehicle control. Significant levels of spontaneously active or acid activatable TGF-β1 were not noted in BAL fluids from transgene (−) mice that received aprotinin or vehicle control (Fig. 8). Aprotinin did, however, totally abrogate the spontaneous activation of TGF-β1 in BAL fluids from IL-13 transgene (+) mice (Fig. 8). This decrease was out of proportion to the modest decrease in the levels of total TGF-β1 in the same BAL fluids (Fig. 8). These studies demonstrate that uPA-like serine proteinases, play a critical role in IL-13–induced TGF-β1 activation in this in vivo system.
Figure 8.
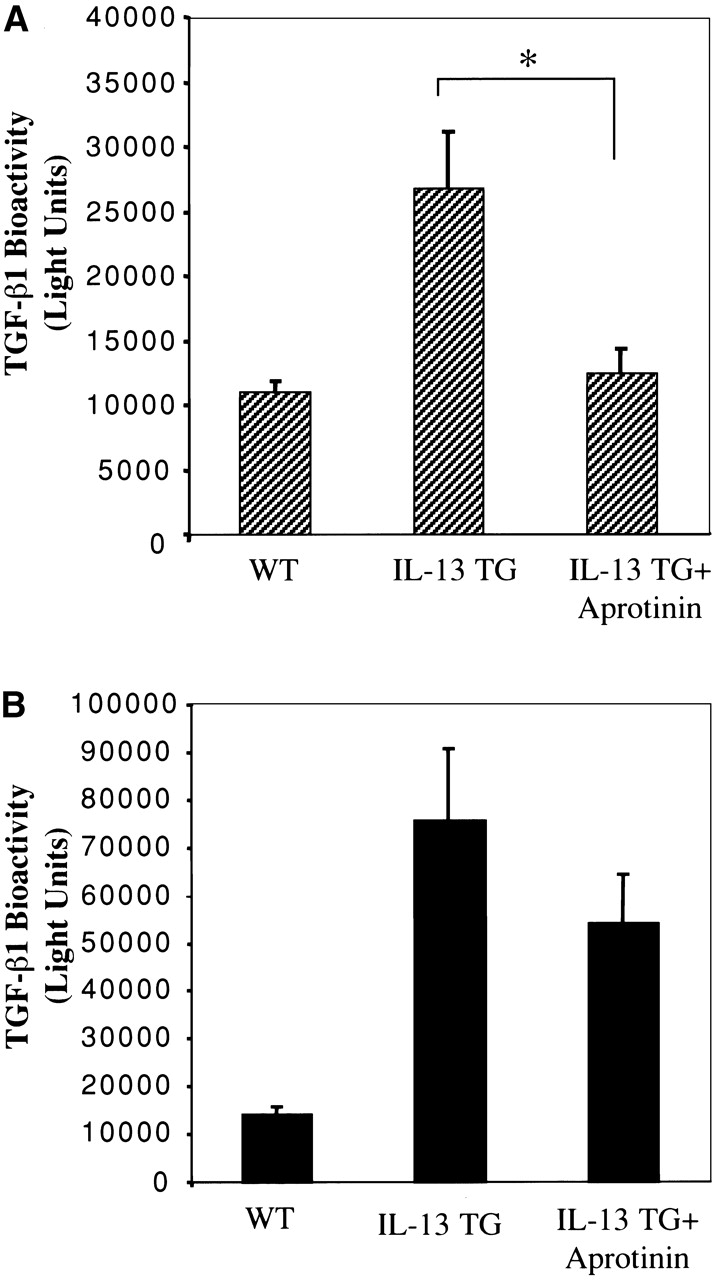
Effect of aprotinin on IL-13-induced TGF-β1 bioactivity. BAL fluids were obtained from transgene (+) mice treated from 1 mo of age to 2 mo of age with aprotinin (+ aprotinin) or vehicle control. The spontaneous (A) and acid-activatable (B) TGF-β1 bioactivity in these fluids are compared to that in BAL fluids from WT transgene (−) littermate control animals. The noted values represent the mean ± SEM of a minimum of four mice in each group. P < 0.01.
Role of MMP-9 and CD44 in IL-13-induced TGF-β1 Activation.
MMP-9 can activate TGF-β cytokines when presented on cell surface CD44 molecules 51. Thus, to define the role of MMP-9 in TGF-β1 activation, CC10-IL-13 transgene (+) mice were bred with mice with null mutations of MMP-9 or CD44. The spontaneous and total TGF-β1 bioactivity in BAL fluids from CC10-IL-13 mice with WT and null MMP-9 or CD44 loci were then compared. Spontaneously bioactive TGF-β1 was not noted in BAL fluids from WT transgene (−) littermate control animals or animals with null mutations of MMP-9 or CD44 (Fig. 9 and data not shown). As noted above, spontaneously bioactive TGF-β1 was noted in untreated BAL fluids from CC10-IL-13 transgene (+) mice with WT MMP-9 loci (Fig. 9). Interestingly, null mutations of MMP-9 caused a significant decrease in the levels of spontaneously active TGF-β1 in BAL fluids from CC10-IL-13 transgene (+) mice (Fig. 9 A). Overall, BAL fluids from CC10-IL-13/MMP-9−/− mice contained 57 ± 5% as much TGF-β1 bioactivity as BAL fluids from CC10-IL-13/MMP-9+/+ animals (P < 0.01). This decrease in TGF-β1 bioactivity was not due to a decrease in IL-13 elaboration or total TGF-β1 production since similar levels of BAL IL-13 and acid-activatable total TGF-β1were noted in transgene (+) mice with WT and null MMP-9 loci (Fig. 9 B and data not shown). CD44 also did not play an essential role in this MMP-9 effect since similar levels of spontaneously active TGF-β1 were noted in IL-13 transgenic (+) mice with WT and null CD44 loci (data not shown). These studies demonstrate that MMP-9 plays a critical role in IL-13–induced TGF-β1 activation, but not in IL-13–induced stimulation of TGF-β1 production in our transgenic system. They also demonstrate that these TGF-β1–activating effects of MMP-9 can be seen in the absence of CD44.
Figure 9.
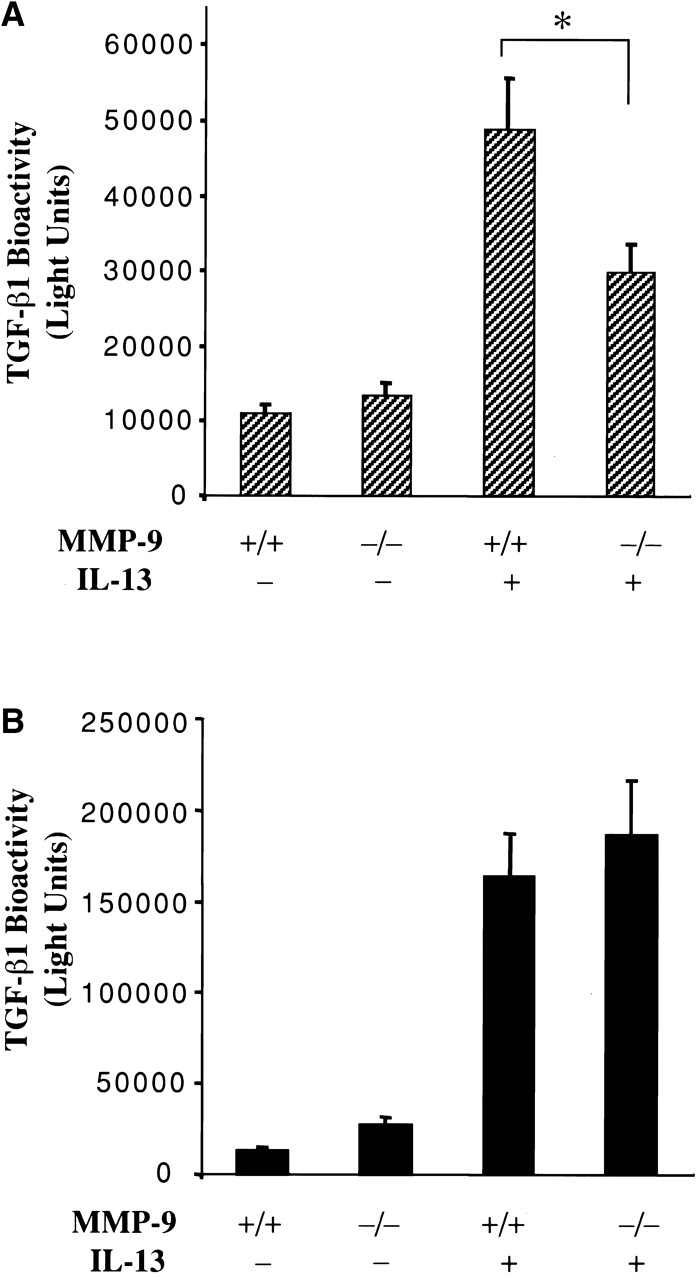
Role of MMP-9 in IL-13 activation and induction of TGF-β1. BAL fluids were obtained from transgene (−) and transgene (+) CC10-IL-13 mice with WT (+/+) and null (−/−) MMP-9 loci. The levels of spontaneous (A) and total (B) TGF-β1 bioactivity in these BAL fluids were assessed before and after acid activation respectively as described in Materials and Methods. The noted values represent the mean ± SEM of a minimum of four mice in each group. *P < 0.01.
Role of TGF-β1 and TGF-β1 Activation in IL-13–induced Fibrosis.
Qualitative histologic techniques (trichrome stains), quantitative biochemical approaches, and morphometric assessments of localized collagen deposition were then used to determine if TGF-β1 played a significant role in IL-13–induced airway and parenchymal fibrosis. In these studies we compared these collagen parameters in IL-13 transgene (+) mice that were treated with a TGF-β antagonist (sTGFβR-Fc), aprotinin (which blocks IL-13–induced TGF-β1 activation), or their appropriate controls. Similar amounts of collagen were noted in lungs from WT littermate control mice that received sTGFβR-Fc, aprotinin, or their respective controls (data not shown). As noted above, IL-13 caused an impressive increase in lung collagen content that could be easily appreciated with all three measurement techniques (Fig. 10 and data not shown). Histologic and biochemical assessments demonstrated that this increase in lung collagen was reduced almost to background levels in CC10-IL-13 mice that received sTGFβR-Fc (Fig. 10). In addition, the Sirius red morphometric evaluations demonstrated that this antifibrotic effect was readily apparent in both the airway and parenchymal tissue compartments where collagen was decreased 85.5 ± 9 and 81.3 ± 11% respectively after sTGFβR-Fc administration (P < 0.001 for both comparisons). Importantly, a significant decrease in collagen content was also seen after aprotinin administration (Fig. 10). Overall, these studies demonstrate, by multiple methods, that TGF-β1 is the major mediator of IL-13–induced airway and parenchymal fibrosis in our in vivo system. They also demonstrate that this fibrotic response requires TGF-β1 activation which is mediated, at least in part, by a uPA/serine protease-dependent mechanism.
Figure 10.
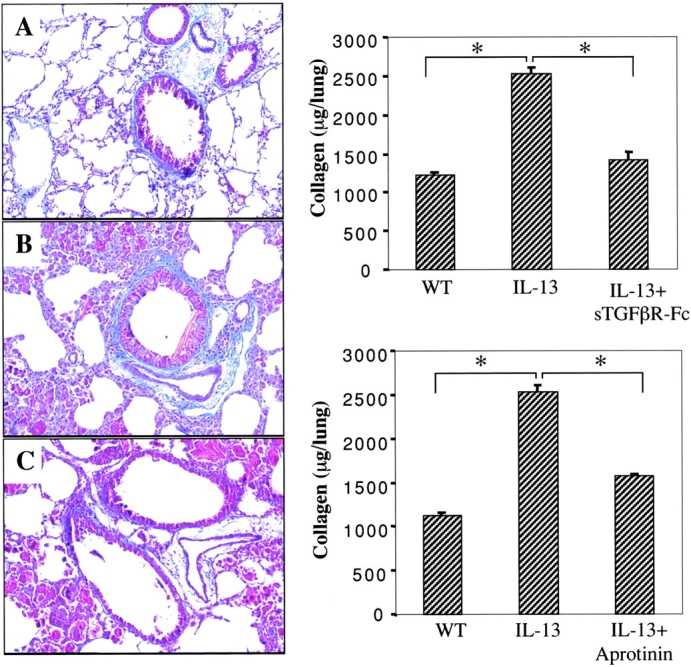
Effects of sTGFβR-Fc and aprotinin on IL-13–induced fibrosis. In the panels on the left 1 mo-old IL-13 transgene (+) mice were treated with sTGFβR-Fc or control and collagen was assessed with trichrome stains. Comparisons are made of WT littermate transgene (−) control mice (A), transgene (+) mice that received control IgG (B), and transgene (+) mice that received sTGFβR-Fc (C). In the panels on the right Sircol biochemical assays were used to characterize the effects of sTGFβR-Fc or aprotinin on lung collagen content. Comparisons are made of WT littermate transgene (−) control mice, transgene (+) mice that received appropriate controls (IL-13) and transgene (+) mice that received sTGFβR-Fc (+ sTGFβR-Fc) or aprotinin (+ aprotinin). Each bar represents the mean ± SEM of a minimum of four animals. *P < 0.01.
Discussion
To further understand the mechanism(s) by which IL-13 generates tissue fibrosis, we took advantage of a transgenic system developed in our laboratory in which the chronic pulmonary overexpression of IL-13 induces airway and parenchymal tissue fibrosis. By comparing the regulation of TGF-β cytokines and related proteins in lungs from transgene (+) mice and transgene (−) littermate controls, and comparing the fibrotic responses induced by IL-13 in animals that received and did not receive TGF-β antagonists, we were able to define the effects of IL-13 on TGF-β cytokines and the role of TGF-β cytokines in IL-13–induced fibrosis. These studies demonstrate that IL-13 is a potent stimulator of TGF-β1. They also demonstrate that IL-13 activates TGF-β1 in vivo and that this activation is mediated by a serine protease/plasmin– and MMP-9–dependent mechanism. Lastly, these studies demonstrate that the fibrotic effects of IL-13 are mediated, at least in part, by this TGF-β1 induction and activation pathway. When viewed in combination, these studies highlight a novel relationship between IL-13 and TGF-β1 which is likely to play a key role in the pathogenesis of Th2- and IL-13–induced tissue remodeling responses.
Although there are >30 members of the TGF-β supergene family, three moieties (TGF-β1, TGF-β2, and TGF-β3) are the major moieties involved in mammalian regulatory systems 45 52. These cytokines are products of separate genes that are differentially regulated and expressed 53. As a result, studies were undertaken to determine if IL-13 stimulated these TGF-β moieties in a selective fashion and define the sites of TGF-β1 production and localization after this induction. These studies demonstrated that TGF-β1 is the major moiety induced by IL-13 in our animals and that TGF-β1 mRNA and protein can be found in large quantities in macrophages and lesser quantities in bronchiolar epithelial cells, alveolar type II cells, and eosinophils from these animals. These findings are in accord with studies that demonstrate that TGF-β1 plays a central role in the pathogenesis of a variety of fibrotic disorders and models of these diseases 35 45 54 55 and studies that demonstrate that macrophages, alveolar epithelial cells, alveolar type II epithelial cells, and eosinophils are important sites of TGF-β1 production and/or localization in murine models of bleomycin-induced pulmonary fibrosis, human IPF, and human asthma 49 54 55 56. It is important to point out that these studies do not rule out a role for TGF-β2 and/or TGF-β3 in the pathogenesis of the phenotype of our transgenic mice or in other IL-13–associated fibrotic disorders because TGF-β2 and TGF-β3 are expressed in a ubiquitous fashion in normal pulmonary tissues 55.
TGF-β moieties are elaborated by a wide array of hematopoietic and structural cells 53 57. In these cells, TGF-β1 is produced as a propeptide which is proteolytically processed into its propeptide fragment (LAP) and a COOH-terminal peptide which forms a disulfide-linked homodimer 43 44 45 50 58. In virtually all cases, TGF-β1 is elaborated as a biologically inactive latent complex which is formed by the covalent bonding of LAP and the TGF-β dimer and the linking to LTBP-1 43 44 45 50 58. The amino terminal of LTBP-1 is critical for tissue transglutaminase cross-linking into matrix. Activation of TGF-β1 is required for TGF-β effector functions to be appreciated. This is accomplished via multiple complex mechanisms that release TGF-β1 from the matrix and cleave and/or induce conformational changes that allow TGF-β1 to bind to its receptor 41 43 44 45 50 58. Decreases in the levels of LTBP-1 50, plasmin activation 43 46 48 49 50, and pathways involving CD-36 and tissue thrombospondin-1 46 47, the αVβ6 integrin 43 and MMP-9 expressed on cell surface CD44 molecules 51 can all play important roles in TGF-β1 activation. In contrast to the majority of models of pulmonary fibrosis in which only latent TGF-β1 can be detected in BAL fluids, our studies demonstrate that a significant amount of the TGF-β1 in the BAL fluids from CC10-IL-13 transgenic mice is spontaneously bioactive. They also demonstrate that the levels of mRNA encoding LTBP-1 are markedly diminished and the levels encoding uPA (which activates plasminogen to bioactive plasmin), MMP-9, and CD44 are increased in IL-13 transgenic mice. This suggests that the decrease in LTBP-1 and the increase in plasmin and MMP-9 play important roles in this activation. Studies using aprotinin corroborated this speculation since this well-known plasmin inhibitor 48 51 59 totally abrogated the ability of IL-13 to spontaneously activate TGF-β1 in the lungs from our transgenic animals. The importance of MMP-9 was also confirmed by breeding the CC10-IL-13 transgene (+) mice with mice with a null mutation of MMP-9, since the levels of spontaneously bioactive TGF-β1 in BAL fluids from IL-13 transgene (+) mice with a null MMP-9 locus were significantly lower than the levels in BAL fluids from IL-13 transgene (+) mice with a WT MMP-9 locus. Interestingly, the effects on TGF-β1 activation of aprotinin exceeded the effects of the MMP-9 null mutation in our transgenic system. uPA-activated plasmin activates MMP-9, both directly and via an indirect pathway that involves MMP-3 60. TGF-β1 is also known to stimulate uPA production 61. Thus, the ability of uPA/plasmin to active TGF-β1 in lungs from IL-13 transgene (+) mice may be mediated, in part, via the ability of plasmin to activate MMP-9. In addition, it is tempting to hypothesize that the LTBP-1, uPA, plasmin, IL-13, and TGF-β1, in our transgenic animals, are involved in an amplification loop that drives tissue fibrosis. In this schema, IL-13 stimulates TGF-β1 and MMP-9 production and inhibits LTBP-1. IL-13 also stimulates uPA either directly or via TGF-β1. The decrease in LTBP-1, increase in uPA-induced plasmin, and increase in MMP-9 all feedback to activate additional TGF-β1. The TGF-β1, in turn stimulates the elaboration of additional uPA and activates additional MMP-9 which activates more TGF-β1. The increased levels of active TGF-β1 then generate additional tissue fibrosis.
An interesting relationship between the hyaluronan receptor CD44, MMP-9, and TGF-β cytokines has been documented in in vitro studies using tumor cells and normal keratinocytes. In these studies CD44 was shown to provide a docking receptor that localized bioactive MMP-9 to the cell surface, and the CD44-MMP-9 complex was shown to proteolytically activate latent TGF-β moieties 51. To determine if similar mechanisms were operative in vivo, we compared the levels of spontaneously bioactive TGF-β1 in BAL fluids from IL-13 transgene (+), IL-13+/MMP-9−/−, and IL-13+/CD44−/− mice. These studies demonstrated that MMP-9 plays an essential role in IL-13–induced TGF-β activation since significantly decreased levels of spontaneously bioactive TGF-β1 were noted in BAL fluids from mice with the null MMP-9 mutation. Surprisingly, similar levels of spontaneous TGF-β1 bioactivity were found in BAL fluids from IL-13 transgene (+) mice with WT (+/+) and null CD44 loci. This demonstrates that MMP-9 does not require CD44 to activate TGF-β1 in vivo in the lung. This CD44 independence may reflect the ability of other molecules to present active MMP-9 on the surface of cells in this complex in vivo system. Alternatively, MMP-9 may only require CD44 to activate specific TGF-β moieties since, in vitro, TGF-β2 and TGF-β3 are more susceptible to activation via the CD44/MMP-9 complex than TGF-β1 51.
In early studies, IL-13 was noted to have effector properties that are relevant to Th2 inflammation including the ability to stimulate IgE production, endothelial VCAM-1 expression, and activate B cells 62 63 64. More recently, IL-13 has been shown to be associated with and induce tissue fibrosis 12 13 25. This effect was assumed to be due to direct effects of IL-13 on fibroblasts since IL-13 stimulates fibroblast proliferation and collagen production in vitro 12 29 30. Our studies add to our understanding of this response by demonstrating that IL-13 also stimulates the production and activates TGF-β in vivo. They also demonstrate that IL-13 induces tissue fibrosis, in great extent, via this TGF-β1 induction and activation pathway. These findings have important implications for diseases such as asthma, IPF, PPS, and hepatic fibrosis which are characterized by IL-13 production, increased TGF-β production, and tissue fibrosis. They suggest that IL-13–stimulated TGF-β1 production and activation may be a major contributor to the pathogenesis of the tissue fibrosis seen in these diseases. They also suggest that IL-13 stimulation and activation of TGF-β1 may represent a healing response in the host designed to control and repair IL-13–induced tissue injury. Lastly, they allow for the speculation that interventions that decrease IL-13–induced TGF-β1 production, TGF-β1 activation, and/or TGF-β1 effector functions will have beneficial effects in these and other IL-13–mediated fibrotic diseases. The latter may be especially important since our present therapeutic approaches do not control the tissue fibrosis that is a major contributor to the morbidity and mortality of these disorders.
Acknowledgments
The authors wish to thank the investigators and institutions that provided the reagents that were employed and Susan Ardito and Kathleen Bertier for their excellent secretarial and administrative assistance.
This work was supported by grants HL-56389, HL-61904, and HL-64242 (to J.A. Elias), HL-60539 (to P. Noble), HL-47328 (to R.M. Senior), and the Alan and Edith L. Wolf Charitable Trust (to R.M. Senior).
Footnotes
Abbreviations used in this paper: BAL, bronchoalveolar lavage; IPF, idiopathic pulmonary fibrosis; ISH, in situ hybridization; LAP, latency-associated peptide; LTBP, latent TGF-β–binding protein; MMP, matrix metalloproteinase; PAI, plasminogen activator inhibitor; RT, reverse transcription; s, soluble; uPA, urokinase type plasminogen activator; WT, wild-type.
References
- Bradding P., Redington A.E., Holgate S.T. Airway wall remodelling in the pathogenesis of asthmacytokine expression in the airways Stewart A.G. Airway Wall Remodelling in Asthma 1997. 29 63 CRC Press, Inc; Boca Raton, FL: pp [Google Scholar]
- Ray A., Cohn L. Th2 cells and GATA-3 in asthmanew insights into the regulation of airway inflammation. J. Clin. Invest. 1999;104:1001–1006. doi: 10.1172/JCI8204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias J.A., Zhu Z., Chupp G., Homer R.J. Airway remodeling in asthma. J. Clin. Invest. 1999;104:1001–1006. doi: 10.1172/JCI8124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa M., Fujimoto M., Kikuchi K., Takehara K. Elevated serum levels of interleukin 4 (IL-4), IL-10, and IL-13 in patients with systemic sclerosis. J. Rheumatol. 1997;24:328–332. [PubMed] [Google Scholar]
- Majumdar S., Li D., Ansari T., Pantelidis P., Black C.M., Gizycki M., du Bois R.M., Jeffery P.K. Different cytokine profiles in cryptogenic fibrosing alveolitis and fibrosing alveolitis associated with systemic sclerosis. Eur. Respir. J. 1999;14:251–257. doi: 10.1034/j.1399-3003.1999.14b03.x. [DOI] [PubMed] [Google Scholar]
- Romagnani S. Th1/Th2 cells. Inflamm. Bowel Dis. 1999;5:285–294. doi: 10.1097/00054725-199911000-00009. [DOI] [PubMed] [Google Scholar]
- Wallace W.A., Ramage E.A., Lamb D., Howie S.D. A type 2 (Th2-like) pattern of immune response predominates in the pulmonary interstitium of patients with cryptogenic fibrosing alveolitis (CFA) Clin. Exp. Immunol. 1995;101:436–441. doi: 10.1111/j.1365-2249.1995.tb03131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziesche R., Hofbauer E., Wittmann K., Petkov V., Block L.H. A preliminary study of long term treatment with interferon γ-1β and low dose prednisolone in patients with idiopathic pulmonary fibrosis. New Engl. J. Med. 1999;341:1264–1269. doi: 10.1056/NEJM199910213411703. [DOI] [PubMed] [Google Scholar]
- Westermann W., Schobl R., Rieber E.P., Frank K.H. Th2 cells as effectors in postirradiation pulmonary damage preceding fibrosis in the rat. Int. J. Radiat. Biol. 1999;75:629–638. doi: 10.1080/095530099140276. [DOI] [PubMed] [Google Scholar]
- Shirwan H. Chronic allograft rejection. Do the Th2 cells preferentially induced by indirect alloantigen recognition play a dominant role? Transplantation. 1999;68:715–726. doi: 10.1097/00007890-199909270-00001. [DOI] [PubMed] [Google Scholar]
- Gharaee-Kermani M., Phan S. Lung interleukin-5 expression in murine bleomycin-induced pulmonary fibrosis. Am. J. Resp. Cell. Mol. Biol. 1997;16:438–447. doi: 10.1165/ajrcmb.16.4.9115755. [DOI] [PubMed] [Google Scholar]
- Chiramonte M.G., Donaldson D.D., Cheever A.W., Wynn T.A. An IL-13 inhibitor blocks the development of hepatic fibrosis during a T-helper type 2-dominated inflammatory response. J. Clin. Invest. 1999;104:777–785. doi: 10.1172/JCI7325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallon P.G., Richardson E.J., McKenzie G.J., McKenzie A.N. Schistosome infection of transgenic mice defines distinct and contrasting pathogenic roles for IL-4 and IL-13IL-13 is a profibrotic agent. J. Immunol. 2000;164:2585–2591. doi: 10.4049/jimmunol.164.5.2585. [DOI] [PubMed] [Google Scholar]
- Hoffmann K.F., Cheever A.W., Wynn T.A. IL-10 and the dangers of immune polarizationexcessive type 1 and type 2 cytokine responses induce distinct forms of lethal immunopathology in murine schistosomiasis. J. Immunol. 2000;164:6406–6416. doi: 10.4049/jimmunol.164.12.6406. [DOI] [PubMed] [Google Scholar]
- Shi Z., Wakil A.E., Rockey D.C. Strain-specific differences in mouse hepatic wound healing are mediated by divergent T helper cytokine responses. Cell Biol. 1997;94:10663–10668. doi: 10.1073/pnas.94.20.10663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wangoo A., Sparer T., Brown I.N., Snewin V.A., Janssen R., Thole J., Cook H.T., Shaw R.J., Young D.B. Contribution of Th1 and Th2 cells to protection and pathology in experimental models of granulomatous lung disease. J. Immunol. 2001;166:3432–3439. doi: 10.4049/jimmunol.166.5.3432. [DOI] [PubMed] [Google Scholar]
- Gurujeyalakshmi G., Giri S.N. Molecular mechanisms of antifibrotic effect of interferon γ in bleomycin-mouse model of lung fibrosisdownregulation of TGF-β and procollagen I and III gene expression. Exp. Lung Res. 1995;21:791–808. doi: 10.3109/01902149509050842. [DOI] [PubMed] [Google Scholar]
- Oldroyd S.D., Thomas G.L., Gabiani G., El Nahas A.M. Interferon-γ inhibits experimental renal fibrosis. Kidney Int. 1999;56:2116–2127. doi: 10.1046/j.1523-1755.1999.00775.x. [DOI] [PubMed] [Google Scholar]
- Sime P.J., O'Reily K.M.A. Fibrosis in the lung and other tissuesnew concepts in pathogenesis and treatment. Clin. Immunol. 2001;99:308–319. doi: 10.1006/clim.2001.5008. [DOI] [PubMed] [Google Scholar]
- de Vries J.E. The role of IL-13 and its receptor in allergy and inflammatory responses. J. Allergy Clin. Immunol. 1998;102:165–169. doi: 10.1016/s0091-6749(98)70080-6. [DOI] [PubMed] [Google Scholar]
- Minty A., Asselin S., Bensussan A., Shire D., Vita N., Vyakarnam A., Wijdenes J., Ferrara P., Caput D. The related cytokines interleukin-13 and interleukin-4 are distinguished by differential production and differential effects on T lymphocytes. Eur. Cytokine Netw. 1997;8:203–213. [PubMed] [Google Scholar]
- Grünig G., Warnock M., Wakil A.E., Venkayya R., Brombacher F., Rennick D.M., Sheppard D., Mohrs M., Donaldson D.D., Locksley R.M., Corry D.B. Requirement for IL-13 independently of IL-4 in experimental asthma. Science. 1998;282:2261–2263. doi: 10.1126/science.282.5397.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotsimbos T.C., Ernst P., Hamid Q.A. Interleukin-13 and interleukin-4 are coexpressed in atopic asthma. Proc. Assoc. Am. Physicians. 1996;108:368–373. [PubMed] [Google Scholar]
- Wills-Karp M., Luyimbazi J., Xu X., Schofield B., Neben T.Y., Karp C.L., Donaldson D.D. Interleukin-13central mediator of allergic asthma. Science. 1998;282:2258–2260. doi: 10.1126/science.282.5397.2258. [DOI] [PubMed] [Google Scholar]
- Zhu Z., Homer R.J., Wang Z., Chen Q., Geba G.P., Wang J., Zhang Y., Elias J.A. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities and eotaxin production. J. Clin. Invest. 1999;103:779–788. doi: 10.1172/JCI5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiaramonte M.G., Schopf L.R., Neben T.Y., Cheever A.W., Donaldson D.D., Wynn T.A. IL-13 is a key regulatory cytokine for Th2 cell-mediated pulmonary granuloma formation and IgE responses induced by Schistosoma mansoni eggs. J. Immunol. 1999;162:920–930. [PubMed] [Google Scholar]
- Hancock A., Armstrong L., Gama R., Millar A. Production of interleukin 13 by alveolar macrophages from normal and fibrotic lung. Am. J. Respir. Cell Mol. Biol. 1998;18:60–65. doi: 10.1165/ajrcmb.18.1.2627. [DOI] [PubMed] [Google Scholar]
- Ohshima K., Akaiwa M., Umeshita R., Suzumiya J., Izuhara K., Kikuchi M. Interleukin-13 and interleukin-13 receptor in Hodgkin's diseasepossible autocrine mechanism and involvement in fibrosis. Hostopathology. 2001;38:368–375. doi: 10.1046/j.1365-2559.2001.01083.x. [DOI] [PubMed] [Google Scholar]
- Doucet C., Brouty-Boye D., Pottin-Clemenceau C., Canonica G.W., Jasmin C., Azzarone B. Interleukin (IL)-4 and IL-13 act on human lung fibroblasts. J. Clin. Invest. 1998;101:2129–2139. doi: 10.1172/JCI741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oriente A., Fedarko N.S., Pacocha S.E., Huang S.K., Lichtenstein L.M., Essayan D.M. Interleukin-13 modulates collagen homeostasis in human skin and keloid fibroblasts. J. Pharmacol. Exp. Ther. 2000;292:988–994. [PubMed] [Google Scholar]
- Vu T.H., Shipley J.M., Bergers G., Berger J.E., Helms J.A., Hanahan D., Shapiro S.D., Senior R.M., Werb Z. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell. 1998;93:411–422. doi: 10.1016/s0092-8674(00)81169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmits R., Filmus J., Gerwin N., Senaldi G., Kiefer F., Kundig T., Wakeham A., Shahinian A., Catzavelos C., Rak J. CD44 regulates hematopoietic progenitor distribution, granuloma formation, and tumorigenicity. Blood. 1997;90:2217–2233. [PubMed] [Google Scholar]
- Zheng T., Zhu Z., Wang Z., Homer R.J., Ma B., Riese R., Chapman H., Shapiro S.D., Elias J.A. Inducible targeting of IL-13 to the adult lung causes matrix metalloproteinase- and cathepsin-dependent emphysema. J. Clin. Invest. 2000;106:1081–1093. doi: 10.1172/JCI10458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W., Geba G.P., Zheng T., Ray P., Homer R., Kuhn C., Favell R.A., Elias J.A. Targeted expression of IL-11 in the murine airway causes airways obstruction, bronchial remodeling and lymphocytic inflammation. J. Clin. Invest. 1996;98:2845–2853. doi: 10.1172/JCI119113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George J., Roulot D., Koteliansky V.E., Bissell D.M. In vivo inhibition of rat stellate cell activation by soluble transforming growth factor β type II receptora potential new therapy for hepatic fibrosis. Proc. Natl. Acad. Sci. USA. 1999;96:12719–12724. doi: 10.1073/pnas.96.22.12719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J.D., Bryant S.R., Couper L.L., Vary C.P., Gotwals P.J., Koteliansky V.E., Lindner V. Soluble transforming growth factor-β type II receptor inhibits negative remodeling, fibroblast transdifferentiation, and intimal lesion formation but not endothelial growth. Circ. Res. 1999;84:1212–1222. doi: 10.1161/01.res.84.10.1212. [DOI] [PubMed] [Google Scholar]
- Wang J.M., Homer R.J., Hong L., Cohn L., Lee C.G., Elias J.A. IL-11 selectively inhibits aeroallergen-induced pulmonary eosinophilia and Th2 cytokine production. J. Immunol. 2000;165:2222–2231. doi: 10.4049/jimmunol.165.4.2222. [DOI] [PubMed] [Google Scholar]
- Bosch J.J., Zyderhoudt K.S., Houtkooper F.M., van Gool J. Histophotometric estimation of volume density of collagen as an indication of fibrosis in rat liver. Histochemistry. 1986;85:129–133. doi: 10.1007/BF00491759. [DOI] [PubMed] [Google Scholar]
- Ashcroft G.S., Lei K., Jin W., Longnecker G., Kulkarnt A.B., Greenwell-Wild T., Hale-Donze H., McGrady G., Song X.-Y., Wahl S.M. Secretory leukocyte protease inhibitor mediates non-redundant functions necessary for wound healing. Nat. Med. 2000;6:1147–1153. doi: 10.1038/80489. [DOI] [PubMed] [Google Scholar]
- Junqueira L.C., Bignolas G., Brentani R.R. Picrosirius staining plus polarization microscopy, a specific method for collagen detection in tissue sections. Histochem. J. 1979;11:447–455. doi: 10.1007/BF01002772. [DOI] [PubMed] [Google Scholar]
- Khalil N. TGF-βfrom latent to active. Microbes. Infect. 1999;1:1255–1263. doi: 10.1016/s1286-4579(99)00259-2. [DOI] [PubMed] [Google Scholar]
- Khalil N. Post translational activation of latent transforming growth factor β (L-TGF-β)Clinical implications. Histol. Histopathol. 2001;16:541–551. doi: 10.14670/HH-16.541. [DOI] [PubMed] [Google Scholar]
- Munger J.S., Harpel J.G., Gleizes P.E., Mazzieri R., Nunes I., Rifkin D.B. Latent transforming growth factor-βstructural features and mechanisms of activation. Kidney Intl. 1997;51:1376–1382. doi: 10.1038/ki.1997.188. [DOI] [PubMed] [Google Scholar]
- Munger J.S., Huang X., Kawakatsu H., Griffiths M.J., Dalton S.L., Wu J., Pittet J.F., Kaminski N., Garat C., Matthay M.A. The integrin αvβ6 binds and activates latent TGF β1a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96:319–328. doi: 10.1016/s0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- Clark D.A., Coker R. Transforming growth factor-β (TGF-β) Int. J. Biochem. Cell Biol. 1998;30:293–298. doi: 10.1016/s1357-2725(97)00128-3. [DOI] [PubMed] [Google Scholar]
- Yehualaeshet T., O'Connor R., Green-Johnson J., Mai S., Silverstein R., Murphy-Ullrich J.E., Khalil N. Activation of rat alveolar macrophage-derived latent transforming growth factor β-1 by plasmin requires interaction with thrombospondin-1 and its cell surface receptor, CD36. Am. J. Pathol. 1999;155:841–851. doi: 10.1016/s0002-9440(10)65183-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yehualaeshet T., O'Connor R., Begleiter A., Murphy-Ullrich J.E., Silverstein R., Khalil N. A CD36 synthetic peptide inhibits bleomycin-induced pulmonary inflammation and connective tissue synthesis in the rat. Am. J. Respir. Cell Mol. Biol. 2000;23:204–212. doi: 10.1165/ajrcmb.23.2.4089. [DOI] [PubMed] [Google Scholar]
- Herbert J.M., Carmeliet P. Involvement of u-PA in the antiapoptotic activity of TGF-beta for vascular smooth muscle cells. FEBS Lett. 1997;413:401–404. doi: 10.1016/s0014-5793(97)00915-0. [DOI] [PubMed] [Google Scholar]
- Khalil N., Corne S., Whitman C., Yacyshyn H. Plasmin regulates the activation of cell-associated latent TGF-β1 secreted by rat alveolar macrophages after in vivo bleomycin injury. Am. J. Respir. Cell Mol. Biol. 1996;15:252–259. doi: 10.1165/ajrcmb.15.2.8703482. [DOI] [PubMed] [Google Scholar]
- Rifkin D.B., Mazzieri R., Munger J.S., Noguera I., Sung J. Proteolytic control of growth factor availability. APMIS. 1999;107:80–85. doi: 10.1111/j.1699-0463.1999.tb01529.x. [DOI] [PubMed] [Google Scholar]
- Yu Q., Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-β and promotes tumor invasion and angiogenesis. Genes and Develop. 2000;14:163–176. [PMC free article] [PubMed] [Google Scholar]
- Shah M., Revis D., Herrick S., Baillie R., Thorgeirson S., Ferguson M., Roberts A. Role of elevated plasma transforming growth factor-β1 levels in wound healing. Am. J. Pathol. 1999;154:1115–1124. doi: 10.1016/s0002-9440(10)65364-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letterio J.L., Roberts A.B. Regulation of immune responses by TGF-β. Annu. Rev. Immunol. 1998;16:137–161. doi: 10.1146/annurev.immunol.16.1.137. [DOI] [PubMed] [Google Scholar]
- Khalil N., O'Connor R.N., Unruh H.W., Warren P.W., Flanders K.C., Kemp A., Bereznay O.H., Greenberg A.H. Increased production and immunohistochemical localization of transforming growth factor-β in idiopathic pulmonary fibrosis. Am. J. Respir.Cell Mol. Biol. 1991;5:155–162. doi: 10.1165/ajrcmb/5.2.155. [DOI] [PubMed] [Google Scholar]
- Khalil N., O'Connor R.N., Flanders K.C., Unruh H. TGF-β1, but not TGF-β2 or TGF-β3, is differentially present in epithelial cells of advanced pulmonary fibrosisan immunohistochemical study. Am. J. Respir. Cell Mol. Biol. 1996;14:131–138. doi: 10.1165/ajrcmb.14.2.8630262. [DOI] [PubMed] [Google Scholar]
- Minshall E.M., Leung D.Y., Martin R.J., Song Y.L., Cameron L., Ernst P., Hamid Q. Eosinophil-associated TGF-β1 mRNA expression and airways fibrosis in bronchial asthma. Am. J. Respir. Cell Mol. Biol. 1997;17:326–333. doi: 10.1165/ajrcmb.17.3.2733. [DOI] [PubMed] [Google Scholar]
- Letterio J.J., Roberts A.B. Molecule of the month. TGF-βa critical modulator of immune cell function. Clin. Immunol. Immunopath. 1997;84:244–250. doi: 10.1006/clin.1997.4409. [DOI] [PubMed] [Google Scholar]
- Macatonia S.E., Hosken N.A., Litton M., Vieira P., Hsieh C., Culpepper J.A., Wysocka M., Trinchieri G., Murphy K.M., O'Garra A. Dendritic cells produce IL-12 and direct the development of Th1 cells from naive CD4+ T cells. J. Immunol. 1995;154:5071–5079. [PubMed] [Google Scholar]
- Davis G.E., Pintar Allen K.A., Salazar R., Maxwell S.A. Matrix metalloproteinase-1 and -9 activation by plasmin regulates a novel endothelial cell-mediated mechanism of collagen gel contraction and capillary tube regression in three dimensional collagen matrices. J. Cell Sci. 2001;114:917–930. doi: 10.1242/jcs.114.5.917. [DOI] [PubMed] [Google Scholar]
- Ramos-DeSimone N., Hahn-Datona E., Sipley J., Nagase H., French D.L., Quigley J.P. Activation of matrix metalloproteinase-9 (MMP-9) via a converging plasmin/stromolysin-1 cascade enhances tumor cell invasion. J. Biol. Chem. 1999;274:13066–13076. doi: 10.1074/jbc.274.19.13066. [DOI] [PubMed] [Google Scholar]
- Santibanez J.F., Iglesias M., Frontello P., Martinez J., Quintinilla M. Involvement of the Ras/MAPK signaling pathway in the modulation of urokinase production and cellular invasiveness by transforming growth factor-β1 in transformed keratinocytes. Biochem. Biophys. Res. Comm. 2000;273:521–527. doi: 10.1006/bbrc.2000.2946. [DOI] [PubMed] [Google Scholar]
- Emson C.L., Bell S.E., Jones A., Wisden W., McKenzie A.N.J. Interleukin (IL)-4-independent induction of immunoglobulin (Ig)E, and perturbation of T cell development in transgenic mice expressing IL-13. J. Exp. Med. 1998;188:399–404. doi: 10.1084/jem.188.2.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zurawski G., de Vries J.E. Interleukin-13, an interleukin-4-like cytokine that acts on monocytes and B cells, but not on T cells. Immunol. Today. 1994;15:19–26. doi: 10.1016/0167-5699(94)90021-3. [DOI] [PubMed] [Google Scholar]
- Bochner B.S., Klunk D.A., Sterbinsky S.A., Coffman R.L., Schleimer R.P. IL-13 selectively induces vascular cell adhesion molecule-1 expression in human endothelial cells. J. Immunol. 1995;154:799–803. [PubMed] [Google Scholar]