

Abstract
Induction of tolerance in self-reactive memory T cells is an important process in the prevention of autoimmune responses against peripheral self-antigens in autoimmune diseases. Although naive T cells can readily be tolerized, memory T cells are less susceptible to tolerance induction. Recently, we demonstrated that low avidity engagement of T cell receptor (TCR) by low densities of agonist peptides induced anergy in T cell clones. Since memory T cells are more responsive to lower antigenic stimulation, we hypothesized that a low avidity TCR engagement may induce tolerance in memory T cells. We have explored two antigenic systems in two transgenic mouse models, and have tracked specific T cells that are primed and show memory phenotype. We demonstrate that memory CD4+ T cells can be rendered anergic by presentation of low densities of agonist peptide–major histocompatibility complex complexes in vivo. We rule out other commonly accepted mechanisms for induction of T cell tolerance in vivo, such as deletion, ignorance, or immunosuppression. Anergy is the most likely mechanism because addition of interleukin 2–reversed anergy in specific T cells. Moreover, cytotoxic T lymphocyte antigen (CTLA)-4 plays a critical role in the induction of anergy because we observed that there was increased surface expression of CTLA-4 on anergized T cells, and that injection of anti–CTLA-4 blocking antibody restored anergy in vivo.
Keywords: T cell tolerance, autoimmunity, transgenic mice, CTLA-4, antigen presentation
Introduction
Induction of tolerance in self-reactive memory T cells is essential for prevention of autoimmune responses against peripheral self-antigens in autoimmune diseases. However, although naive T cells can be tolerized successfully 1 2, memory T cells are less susceptible to tolerance induction in vivo 3. Differences that distinguish naive from memory T cells may contribute to the differential susceptibility to tolerogenic signals. For example, naive and memory CD4+ T cells express different patterns of adhesion molecules, such as CD44, intercellular adhesion molecule 1, LFA-1, and CD62L, or signaling molecules such as CD45. Because of an increased expression of adhesion receptors, memory T cells require less costimulation than do naive cells, and are able to respond faster and to lower densities of antigenic challenge 3. Some differences at the levels of TCR-induced signaling pathways may also contribute to a lower activation threshold for memory cells. Specifically, stimulation through TCR/CD3 in murine naive versus memory CD4+ T cells leads to differential phosphorylation of proteins involved in signal transduction 4. These characteristics may enable memory T cells to respond positively to stimulation by all APCs, including resting B cells 5. In agreement with these findings it has been documented that lack of costimulatory signals does not lead to tolerance in memory T cells 2.
The role of CTLA-4 in maintaining peripheral tolerance is well established. Recently, Greenwald et al. 1 clearly demonstrated an essential role for CTLA-4 in regulating the induction of anergy in vivo. It is reported that CTLA-4 is produced and stored in endosomal vesicles in activated/memory, but not naive T cells 6. Thus, when needed, CTLA-4 might readily be expressed on the cell surface and act to regulate overstimulation of activated/memory T cells.
Recently, we have demonstrated that low avidity engagement of T cell receptor by low densities of agonist peptides induced anergy in T cell clones 7. Thus, we hypothesized that low avidity TCR engagement may also drive memory T cells to a state of anergy in vivo. To test this hypothesis, we designed two experimental systems using two transgenic (Tg) mouse models. In one, monoclonal 6.5 TCR Tg mice, specific for hemagglutinin (HA)110–120, and in the other, HLA-DR1 Tg mice, with a full repertoire of T cells, were tested. We observed that presentation of low densities of agonist peptides in complex with MHC class II induced tolerance in specific memory CD4+ T cells. We demonstrated that tolerance was not due to ignorance or active suppression/immunoregulation. Furthermore, by studying specific T cells tracked by specific mAb or peptide–MHC II oligomers, we established that tolerance was due to anergy but not deletion. Moreover, induction of anergy required signaling through CTLA-4.
Materials and Methods
Mice.
TCR Tg mice line 6.5 that expresses an α/β T cell receptor recognizing an I-Ed–restricted HA110–120 on a B10 background was used. NonTg B10.D2 mice at 6–8 wk of age were purchased from The Jackson Laboratory. HLA-DR1 (DR B1*0101) Tg mice (Merck) at 8–10 wk of age were studied. The chimeric HLA-DR1 molecule comprised a peptide-binding groove derived from the human DR1 sequence and a CD4-binding domain from I-Ef mice. The mice were housed in The Johns Hopkins University animal facilities under virus-free conditions. All experiments were performed in accordance with protocols approved by the Animal Care and Use Committee of the Johns Hopkins University, School of Medicine.
Peptides.
The peptides, influenza virus HA306–318 (PKYVKQNTLKLAT) and HA109–120 (SSFERFEIFPKE), were synthesized by Peptide Express. The peptides were >90% pure as analyzed by reverse-phase HPLC.
Immunizations.
To determine the immunogenicity of the HA306–318, HLA-DR1 Tg mice were immunized subcutaneously at the base of the tail with 10 nmol of HA306–318 emulsified at a 1:1 (vol/vol) in Complete Freund's Adjuvant (CFA; Sigma-Aldrich). After 2, 3, 5, 7, or 12 wk, mice were injected subcutaneously with increasing concentrations (0–50 nmol) of HA306–318 in Incomplete Freund's Adjuvant (IFA; Sigma-Aldrich). 9 d after the second injection, inguinal lymph nodes were removed and cells were used for assays.
Adoptive Transfer.
CD41 cells (2.5 × 106), positive for the clonotypic TCR, prepared from pooled lymph nodes and spleens of TCR Tg donors, were resuspended in 200 μl of sterile HBSS (Life Technologies) and injected through the tail vein of B10.D2 recipient mice. 3 d after transfer, mice were immunized (subcutaneously) with 15 nmol of HA109–120 emulsified at a 1:1 (vol/ vol) ratio with CFA. After 5 wk, recipient mice were injected with increasing concentrations of HA109–120 (0–10 nmol) mixed with IFA. 2 or 9 d after the second injection, lymph nodes were removed and cells were used for assays.
Proliferation Assay.
Cells (4 × 105) were cultured in each well of a 96-well round-bottomed plate (Becton Dickinson) with no peptide or various concentrations of HA306–318 or HA109–120 or purified protein derivative (PPD; Connaught Laboratories Ltd.), at 37°C, 5% CO2, for 72 h in RPMI 1640 (GIBCO BRL) supplemented with 10% FBS (GIBCO BRL), 2 mM l-glutamine (GIBCO BRL), 10 mM Hepes (GIBCO BRL), 50 U/ml penicillin/streptomycin (GIBCO BRL) and 50 mM 2-mercaptoethanol (Sigma-Aldrich). Each well was then pulsed with 1 μCi of [3H]thymidine (Amersham Pharmacia Biotech). 18 h later, cells were harvested with a Packard Micromate cell harvester and the incorporated radioactivity was measured by a Packard Matrix 96 Direct b Counter.
Cytokine Assays.
IL-2 release was measured with the IL-2–sensitive cell line CTLL-2 (American Type Culture Collection[ATCC]). Inguinal lymph node cells (4 × 105 cells per well) were cultured with 0, 0.1, or 1 μM HA306–318 for 24 h. Cell-free culture supernatants were then collected, stored at −70°C, and thawed once. CTLL-2 cells (4 × 105 cells per well) were incubated at 37°C, with 5% CO2 for 24 h in RPMI 1640 with supplements (10% FBS, 2 mM l-glutamine, 1.5 gram per liter sodium bicarbonate; Sigma-Aldrich), 10 mM Hepes plus 24-h culture supernatants, in culture wells of 96-well round-bottomed plates. Plates were then pulsed with 1 μCi of [3H]thymidine for an additional 18 h, harvested, and counted. The level of IL-2 was calibrated against a standard curve of rIL-2. Cell-free culture supernatants were collected after 48 h and IFN-γ was measured by ELISA for mouse IFN-γ using Quantikine cytokine ELISA kit (R&D Systems) according to the manufacturer's protocol. All assays were performed in triplicate.
Flow Cytometric Analysis.
Pooled lymph node and spleen cells (106) from various mice (adoptively transferred), 3 d after transfer of cells, 2, 3, or 5 wk after immunization were preincubated with Fc-γ receptor-blocking antibody 2.4G2 (HB-197, ATCC). Cells were washed and stained with biotinylated anticlonotypic TCR mAb (6.5, provided by H. von Boehmer, Harvard University, Boston, MA; reference 8) and avidin-cychrome (Cyc). The following mAbs were used for analysis: anti–mouse CD4-FITC; anti–mouse CD4-PE; anti–mouse CD62L-PE; anti–mouse CD45RB-PE; anti–mouse CD69-PE; anti–mouse CD25-FITC; and anti–mouse CD44-FITC. 48 h after the second peptide injection, 106 cells of recipient mice were stained with anticlonotypic TCR mAb-Cyc, anti–mouse CD4-FITC, and anti–mouse CTLA-4 PE. 9 d after the second peptide injections, 5 × 106 cells of HLA-DR1 Tg mice were cultured either with peptide (10 μM) alone or with rIL-2 (10 U/ml) for 8 d. Cells were stained with HA-DR1-SA-PE oligomers (provided by T. Cameron and L. Stern, MIT, Cambridge, MA) for 3.5 h at 37°C followed by anti–mouse CD4-FITC or anti–mouse CD4-Cyc and anti–mouse CTLA-4–FITC (Southern Biotechnology Associates, Inc.). All antibodies were purchased from BD PharMingen. The samples were analyzed on a FACScan™ by using CELLQuest™ software (Becton Dickinson). In each case, 5–10 × 104 events were collected.
In Vivo Antibody Treatments.
HLA-DR1 Tg mice received intraperitoneal injections of 85 μg of hamster anti–mouse CTLA-4 mAb from the culture supernatant of the UC10-4F10-11 cell line (ATCC), which was purified on a protein A column in our laboratory and showed >95 purity by SDS-PAGE and silver staining. Injections were done on days 0, 1, 2, 3, 5, and 7 after the tolerogenic injection of various doses of HA306–318 in IFA.
Mixing Experiments for Potential Presence of Immunoregulatory Cells in Anergic Groups.
The regulatory capacity of anergic T cells was analyzed in a culture in vitro. Anergic cells (2 × 105) either from 0.0005 nmol or 0.05 nmol groups were incubated with 4 × 105 responder cells from each nonanergized group of HLA-DR1 Tg mice together with various concentrations of HA306–318 (0, 0.1, 1, 10 μM). As positive controls, responder cells (0 and 5 nmol groups) were treated with rhTGF-β (0.1, 1, 10 ng/ml; R&D Systems) for 48 h. Proliferative responses were examined by [3H]thymidine incorporation.
Results
Acquisition of Memory Phenotype in 6.5 TCR Tg Cells.
The surface phenotype of naive T cells is relatively well characterized, and they express high levels of L-selectin (CD62L), CD45RB, and low levels of CD44. In contrast, the surface phenotype of memory T cells has remained controversial. It has been proposed that memory T cells express CD45RBlo, CD62Llo, and CD44hi. In comparison to activated cells, memory T cells are presumed to be small and to have low expression of activation markers such as CD25 (IL-2R) and CD69 9. Memory T cells migrate from blood and spleen to lymph nodes at a low rate 10 11, but reintroducing antigen can accelerate their recruitment back into lymph nodes 9 11.
To trace specific T cells and to characterize their phenotype, we used 6.5 TCR Tg mice specific for complexes of HA109–120-I-Ed 8. TCR Tg CD4+ T cells were transferred into B10.D2 recipient mice, and 3 d later (to allow for homing of transferred cells to the lymphoid organs), recipients were immunized with 15 nmol of HA109–120 in CFA. The group called “naive” did not receive any peptide. Lymph node and spleen cells were analyzed by size and three-color FACS® for expression of CD4, clonotypic TCR epitope, and levels of expression of memory markers. HA109–120–specific CD4+ T cells were stained for CD44, CD25, CD69, CD45RB, and CD62L on day 3 after transfer and 2, 3, and 5 wk after immunization. As seen in Fig. 1 A, during the 5 wk after immunization the expression level of CD44 increased, whereas levels of CD45RB, CD62L, CD25, and CD69 decreased. Moreover, the average cell size decreased during the 5-wk period, consistent with distinctive smaller size for memory versus activated T cells.
Figure 1.


(A) Surface phenotypes of clonotypic HA-specific CD4 T cells in naive and immunized mice: splenocytes and lymph node cells were pooled and triple stained with anti-CD4, anticlonotypic TCR antibody (mAb 6.5), and indicated antibodies. Cell size and levels of expression of CD25, CD44, CD45RB, CD62L, and CD69 were measured by gating on the CD4+ and mAb 6.5+ population. (B) Memory cells respond faster and more efficiently to lower peptide doses than do naive cells; lymph node cells from adoptively transferred mice at 3 d (▴) and 5 wk after immunization with HA109–120 (▵) were prepared. Cells were restimulated with various concentration of HA109–120 peptide. Proliferation assay was performed as described in Materials and Methods.
Memory T Cells Respond more efficiently to Lower Doses of Peptide.
A more reliable marker for memory T cells is their characteristic higher sensitivity to antigenic stimulation compared with naive T cells 12. For further evaluation of the generation of memory T cells 5 wk after immunization, we compared the dose-proliferative response of cells from adoptively transferred mice 3 d after transfer (naive phenotype) and 5 wk after immunization (memory phenotype). Fig. 1 B shows that memory T cells proliferated in response to 100-fold lower peptide concentrations than did naive T cells.
Induction of Tolerance in TCR Tg Cells In Vivo.
The above data strongly suggested that memory T cells developed 5 wk after immunization. Next, we examined whether low doses of agonist peptides could induce unresponsiveness in memory T cells in vivo. Induction of unresponsiveness occurred when low doses of peptide were administered 5 wk after priming (Fig. 2 A). Lack of vigorous proliferation in tolerized groups was not due to the deletion of antigen-specific cells, because we detected comparable percentages of CD4+ 6.5 TCR+ cells in all groups that ranged between 0.40–0.64% (0 nmol group, 0.40%; 0.5 nmol group, 0.44%; 10 nmol group, 0.64%).
Figure 2.


Induction of anergy in adoptively transferred mice with T cells from HA109–120-specific TCR Tg donors. 3 d after adoptive transfer mice were immunized with HA109–120 in CFA, 5 wk after they received different doses of HA109–120 in IFA. 9 d later cells were harvested and cultured with different doses of HA109–120 (A) and PPD (B) and proliferation was measured by [3H]thymidine incorporation. Representative of five independent experiments.
Specificity of Tolerance Induced by Low Doses of Peptide.
The mycobacterium debris antigens in CFA used in the first immunization could prime non-TCR Tg cells to respond to the bacterial PPD challenge in vitro. Thus, in order to control for specificity of the tolerance induced we tested the response levels of all groups to two different doses of PPD in vitro. The loss of proliferation of tolerized cells to HA109–120 was clearly antigen specific as the proliferation to PPD was not altered significantly (Fig. 2 B).
Induction of Tolerance by Low Doses of HA306–318 Peptide in HLA-DR1 Tg Mice.
The above experiments demonstrated that tolerance can be induced among memory T cells of a single clonal origin adoptively transferred to a syngenic nonTg recipient. To further establish these findings and to test whether multiple T cell clones specific for the same pair of peptide–MHC could also be tolerized by this treatment, we used Tg mice that express the human class II molecules, HLA-DR1. These Tg mice carry chimeric I-E/DR1 molecules where the peptide-binding groove is composed of HLA-DR1, but the membrane proximal domain is murine I-E to allow undisturbed interactions with murine CD4 on T cells. Importantly, these Tg mice develop a diverse T cell repertoire and can give rise to human DR–restricted immune responses after challenge with peptides that bind HLA-DR1 13. In addition, our previous results demonstrating that anergy can be induced in T cell clones by low densities of peptide–MHC had used HLA-DR1 in complex with HA306–318. Notably, interaction of HLA-DR1 and HA306–318 peptide has been well characterized biochemically 14 15 16. Thus, testing HLA-DR1 mice had multiple advantages.
To generate memory T cells, an immunogenic dose (10 nmol ∼15 μg) of HA306–318 in CFA was injected and different groups of mice received a second injection of variable doses of HA306–318 2, 3, or 5 wk later. We also tested 2 and 3 wk intervals because in vivo tracing of DR1/ HA306–318–specific T cells is not currently available 17 18. Slightly lower doses of HA306–318 were used to compensate for the higher binding affinity of HA306–318 for DR1. HA306–318-DR1 complex has a dissociation half-time of 6 d at 37°C 19. Cells from the draining nodes were removed and tested in a proliferation assay 9 d later. At short intervals between CFA priming and administration of peptide in IFA, no tolerogenic effects were observed (Fig. 3a and Fig. b), consistent with the later development of memory phenotype. When low peptide doses were administered 5 wk after the initial priming we observed tolerance. Fig. 3 C depicts proliferation of cells from mice tolerized by low doses/densities of HA306–318 (0.0005–0.05 nmol) in vivo. Cells from these groups proliferated significantly less well than cells from other groups that had received peptide doses <0.0005 nmol or higher than 0.05 nmol. The in vivo doses capable of inducing unresponsiveness spanned 3–4 logs. The inverse bell-shaped pattern of unresponsiveness to a range of peptide doses resembled T cell clones 7, where densities between 1–10 peptide–DR1 complexes per APC had the greatest inhibitory effects. Similar results were observed when the time required for induction of anergy was extended to 7 and 12 wk (see Fig. 4a and Fig. b) as a further proof for the longevity of memory T cells and their susceptibility to tolerance induction by low avidity stimulation. Although in most experiments we have used draining lymph nodes isolated 9 d after tolerogenic peptide injection, tolerance was established at time points tested as early as 2 d after second peptide injection, and persisted up to 27 d, that latest time point tested (data not shown).
Figure 3.





Low densities of agonist peptide induce anergy in memory T cells. Mice were immunized with 10 nmol of HA306–318 in CFA; mice received a second peptide injection 2 (A), 3 (B), or 5 wk later (C) with indicated doses of HA306–318 in IFA. After 9 d, draining lymph nodes were collected and restimulated with peptide in culture. Proliferative response was measured. The spectrum of induction of anergy by low densities of agonist peptide spans 3–4 logs. The experiment shown represents five independent experiments and each point represents an average of five mice. Anergized cells are unable to produce IL-2 and IFN-γ; supernatants from lymph node cell cultures stimulated with various concentration of HA306–318 were removed after 24 and 48 h of incubation and assayed by measuring proliferation of IL-2 sensitive CTLL-2 cell line (D), and ELISA for IFN-γ (E). One out of three representative experiments is shown.
Figure 4.


Memory T cells are targets for anergy. Seven (A) or 12 wk (B) after immunization with HA306–318 in CFA, mice received a second peptide injection as described in Materials and Methods. The proliferative response to in vitro peptide restimulation was determined by [3H]thymidine incorporation. The experiment shown represents five independent experiments and each point represents an average of five mice.
It has been proposed that T cell anergy is a consequence of the inability of a T cell to produce IL-2 20. To determine the effect of various doses of HA306–318 on IL-2 production, culture supernatants of draining lymph node cells were assayed for IL-2 release. Measurement of IL-2 levels showed that cells from mice exposed to 0.0005–0.05 nmol of HA306–318 in vivo had markedly diminished IL-2 levels (Fig. 3 D). As shown in Fig. 3 E, production of IFN-γ was also significantly reduced in tolerized cells. The cytokine responses were antigen specific because cells cultured without peptide contained no detectable cytokines. It has been reported that low densities of antigen 21 22 in IFA 23 may cause Th cells to differentiate into Th2 rather than Th1 cells. We examined cells for Th2 cytokines such as IL-4, IL-5, and IL-10 secretions in culture supernatants. We also analyzed intracellular IL-4 production in all groups. None of the assays used detected measurable levels of IL-4, IL-5, and IL-10 in any of the groups tested (data not shown). Furthermore, IFN-γ secreted in nontolerized groups (0, 0.5, 5 nmol) reflects their differentiation into Th1 cells (Fig. 3 E).
Tolerance Is not due to Suppressor T Cells or Deletion.
A potential mechanism responsible for induction of tolerance in mature peripheral T cells is immunoregulation. Two models have been proposed to explain the effects of immunoregulatory T cells. One involves suppressive effects of T cell–derived cytokines, such as IL-4, IL-10, and TGF-β, which inhibit the activation of IL-2–producing T cells 24 25, and the other postulates the suppressive effects based on competition with responding cells for access to APC surface antigens or costimulatory molecules 26. To determine whether tolerance was induced by suppressor T cells in our system, we mixed responder cells (nonanergized groups) with cells from two anergized groups (0.0005–0.05 nmol) in the presence of various concentrations of HA306–318, and then tested for T cell proliferation (Fig. 5a and Fig. b). To control for whether the responding cells were responsive to suppressor cytokines, responder cells (0 and 5 nmol groups) were treated with different doses of TGF-β in vitro (Fig. 5 C). Fig. 5 shows that tolerized cells were unable to inhibit proliferation of antigen-specific responder T cells, although TGF-β treatment had inhibitory effects.
Figure 5.



Anergic cells cannot suppress proliferation of responder cells. Cells from nonanergized groups (responders) plus anergized groups (A) 0.0005 nmol and (B) 0.05 nmol were cultured at 2:1 ratio with various doses of HA306–318 peptide for 72 h. (C) Responding cells (0 and 5 nmol groups) were treated with rhTGF-β (0, 0.1, 1, 10 ng/ml) for 48 h.
Apoptosis 27 and deletion 28 are well-established mechanisms for peripheral tolerance. To determine whether tolerance was caused by deletion of the specific T cells, we used HA-DR1 oligomers to trace HA306–318-specific T cells in DR1 Tg lymph node cells that had been tolerized. Similar to other preparations of oligomeric human class II 17, HA-DR1-SA-PE oligomer 18 failed to detect specific cells directly from a pool of lymphocytes. Nonetheless, specific cells were detectable after 8 d in culture. We followed the protocol established previously 18 and detected specific T cells that were stained by HA-DR1-SA-PE oligomers in all groups of mice (Fig. 6). The percentages of positive cells were comparable in anergized and nonanergized groups, ranging from 0.7–1.3%, indicating that they were not deleted. It has been demonstrated in models in vitro 20 and in vivo 29 that anergic T cells are unable to divide because they do not produce IL-2 and that addition of exogenous IL-2 can prevent or reverse anergy 7 30. Upon addition of peptide and IL-2, the percentage of positive cells in the anergized group (0.0005 nmol peptide in IFA) doubled, confirming persistence of anergic cells (Fig. 6). The background counts for unprimed naive cells were 0.09%.
Figure 6.

Anergized cells are able to expand in the presence of rIL-2. Splenocytes and lymph node cells from anergized and nonanergized mice were cultured without peptide, with peptide (10 μM) alone or with rIL-2 (10 U/ml) for 8 d. Cells were stained with HA-DR1-SA-PE oligomers and anti-CD4–FITC.
These experiments rule out suppression or deletion as an explanation for tolerance in our system, but provide clear evidence for anergy as the most likely explanation.
Upregulation of CTLA-4 Expression by Tolerogenic Doses of HA Peptides.
A critical role for CTLA-4 in negative regulation of the immune response has been established by the findings that CTLA-4–deficient (CTLA−/−) mice display a severe lymphoproliferative disorder combined with massive lymphocytic infiltration that leads to tissue destruction and death of the mice at 3–4 wk of age 31 32. T cell tolerance in vivo may arise from dominant engagement of B7 molecules by the CTLA-4 over CD28 and not from lack of costimulation 33. To test possible involvement of CTLA-4 we measured levels of CTLA-4 expression in clonotypic HA-specific CD4+ T cells (6.5+, CD4+) of draining lymph nodes 2 d after the second peptide injections 34 35, and in HA306–318–specific CD4+ T cells from HLA-DR1 mice, 8 d after in vitro culture in the presence of peptide. Interestingly, we observed a direct correlation between the levels of CTLA-4 expression and the induction of T cell unresponsiveness (Fig. 7a and Fig. b). Mice exposed to tolerogenic dose of peptide expressed an increased level of CTLA-4 on their T cells, whereas T cells from mock-immunized mice and those injected with no peptide or an immunogenic dose had comparable levels of CTLA-4. These observations suggest that tolerance induced by low densities of agonist peptide in vivo may occur through CTLA-4 signaling.
Figure 7.


Expression of CTLA-4 on cells exhibiting anergy. Surface expression of CTLA-4 on (A) clonotypic HA-specific CD4 cells from draining lymph nodes of adoptively transferred mice 48 h after the tolerogenic peptide administration and (B) HA306–318–specific CD4+ T cells from HLA-DR1 mice 8 d after in vitro culture in presence of peptide as examined by anti–CTLA-4 antibody staining. Results are from one out of two independent experiments.
Blocking CTLA-4 Prevents Induction of Tolerance by Low Density of Agonist Peptide.
To ascertain the regulatory role of CTLA-4 in induction of tolerance in this system, HLA-DR1 Tg mice were treated with anti–CTLA-4 mAb. Immunized mice were treated with multiple injections of anti–CTLA-4 mAb (intraperitoneally) on days 0, 1, 2, 3, 5, and 7 after injection of tolerogenic peptide. Draining lymph nodes were removed on day 9 and cell proliferation to in vitro peptide restimulation was tested. Fig. 8 shows that lymph node cells from mice given a tolerogenic peptide dose proliferated weakly. In contrast, cells from mice treated with anti–CTLA-4 mAb showed a strong proliferative response. Animals injected with 0 or 5 nmol of HA306–318 in the presence or absence of anti–CTLA-4 mAb showed similar levels of proliferation. Therefore blocking of CTLA-4 prevents transmission of signaling required for T cell tolerance induced by low density of agonist peptide. These results demonstrate that induction of tolerance in memory T cells by low density peptide presentation is accomplished by the expression of CTLA-4.
Figure 8.
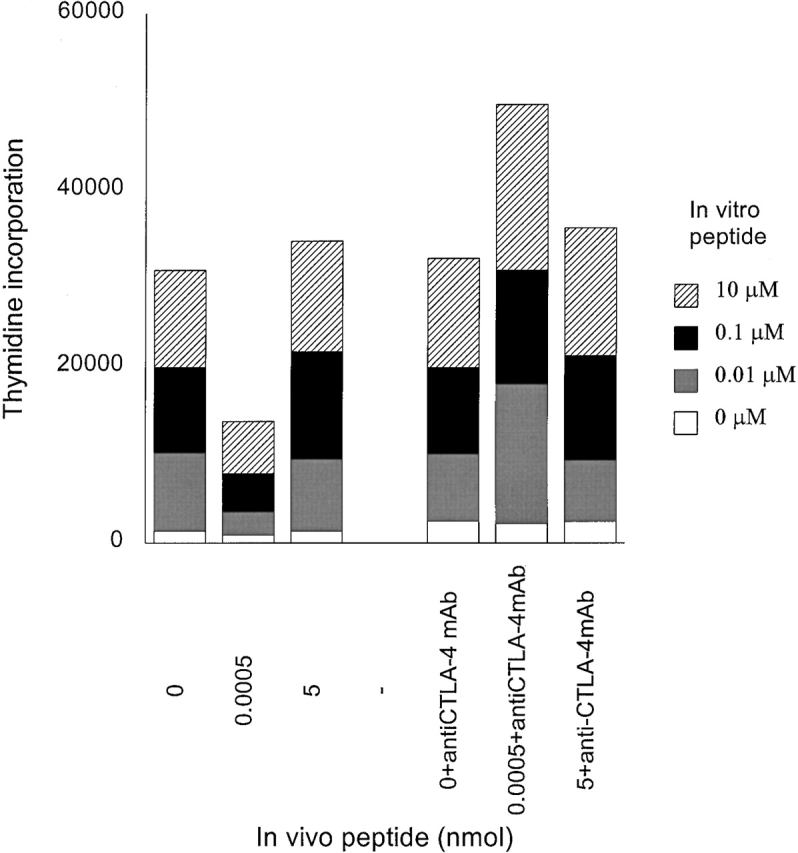
Blocking of CTLA-4 engagement prevents induction of anergy. HLA-DR1 Tg mice were immunized and injected with HA306–318 as described in Materials and Methods. One group of these mice was treated (intraperitoneally) with multiple anti–CTLA-4 mAb injections on days 0,1, 2, 3, 5, and 7 after the second peptide injection with various doses of HA306–318.
Discussion
Low Densities of Agonist Peptides Induce Anergy In Vivo.
We have recently described a novel mechanism for induction of a long-term T cell anergy by presentation of low densities of agonist peptide–MHC in CD4+ T cell clones. It was estimated that 1–10 agonist HA306–318 in complex with HLA-DR1 could internalize <1,000 TCR 7. This was in sharp contrast to altered peptide ligand effects that are generally observed upon presentation of 2–3 orders of magnitude higher densities of peptide–MHC. The anergic cells were unable to proliferate, secretion of IL-2 was inhibited, and secretion of IFN-γ was reduced. Nevertheless, no inhibitory effects were observed on downregulation of TCR or upregulation of IL-2R. Such a phenotype denotes a state of partial stimulation of T cells similar to that defined for the effects of classic antagonist peptides 36 37 or TCR engagement in the absence of signal 2 38. In this report, we address the relationship between the density of peptide–MHC complexes and the differential transduction of signals in T cells in vivo.
In this study, we provide evidence that low-avidity engagement of T cells, by low densities of agonist peptide–MHC, leads to the induction of T cell unresponsiveness in the periphery in vivo. High-avidity engagement of TCR by peptide–MHC may lead to formation of immunological synapse 39 40 and sustained signaling 41. One can assume that low-avidity engagements might interrupt cytoskeletal rearrangements and full recruitment of necessary signaling molecules to the plasma membrane, resulting in negative signaling and T cell anergy. However, the detailed mechanism of these events remains to be tested.
Memory T Cells as Targets for Anergy Induction by a Low Density of Peptide.
Although induction of anergy among naive T cells has been readily generated 42 43, induction of anergy in memory T cells in vivo has proved to be difficult 3. Qualitative changes that distinguish memory from naive T cells render memory T cells able to respond to antigenic challenge faster and more efficiently than naive T cells 9 12. Multiple reasons could account for this effect: differences in activation requirements, TCR signaling, expression of adhesion molecules 9, and dependence on costimulatory signals for activation between memory and naive T cells 3 44. In addition, memory CD8 T cells have been shown to have a distribution of Lck different from that of naive T cells. Lck seems to be associated with CD8 in memory, but evenly distributed in naive T cells 45.
Several observations in this study support the notion that low densities of peptide–MHC target memory T cells. First, and most critical, is the long generation time, a minimum of 5 wk 46 47 48, for T cells to become receptive to anergy. Second, specific memory T cells were able to proliferate at lower peptide doses than did naive cells (Fig. 1 B), consistent with the observations of Rogers et al. 12. Third, in comparison to cells from nonimmunized mice, primed cells expressed low levels of CD62L and CD45RB and increased levels of CD44 (Fig. 1 A). Finally, increased expression of CTLA-4 also correlated with induction of anergy (Fig. 7a and Fig. b), consistent with the finding that memory and not naive T cells express intracellular stores of CTLA-4 6 49.
It seems reasonable to assume that memory T cells would respond to low densities of peptide–MHC because of the specific signaling machinery that is already associated with their membranes. For naive T cells, such low densities of ligands might be ignored.
Previous studies on peripheral tolerance attributed the loss of T cell function to different mechanisms 24 50 51 52. Diminished proliferation marks the acquired state of unresponsiveness (anergy) in our system. Lack of proliferation in cells exposed to low doses of peptide was not due to cellular death for the following reasons: first, antigen-specific T cells were detectable in lymph node cells of anergized mice (Fig. 6 and Fig. 7); second, anti–CTLA-4 treatment restored vigorous proliferation of anergized cells (Fig. 8); and third, anergized HA-specific CD4+ T cells from HLA-DR1 Tg mice proliferated in the presence of rIL-2 (Fig. 6). Our conclusions are supported by reports demonstrating that CTLA-4 ligation does not induce apoptosis 53 54 and that memory T cells are more resistant to apoptosis 44 55.
Recent reports suggest a role for immunoregulatory CD25+CD4+ T cells in the control of tolerance 56. Immunoregulatory T cells are present in the subpopulation of CD4+ cells that express activation/memory markers CD25+CD45RBlow 57. Another report suggests that the CD4+CD25+ population contains a significant proportion of cells with a naive/resting phenotype 58 that seem to express CTLA-4 constitutively and to suppress the activation and proliferation of other T cells, by competing for costimulatory molecules 59 or by triggering TGF-β secretion 60. Nevertheless, cells rendered anergic by low densities of antigen did not show any suppressive function: they did not cause neither specific inhibition of responder T cell proliferation (Fig. 5a and Fig. b) nor did they induce a general suppression in response to PPD (Fig. 2 B).
Lack of proliferation and production of IL-2 and IFN-γ in anergized groups is less likely to be due to a Th2 switch. Two important observations support this notion: first, that no IL-4, IL-5, or IL-10 production was detected; and second, CTLA-4 expression, which was upregulated in the anergized group, is reported to limit Th2 differentiation 61.
Finally, ignorance has been evoked as a mechanism for T cell tolerance 62. The tolerance described here cannot be explained by ignorance because tolerized cells had to be primed and responded fully to the peptide challenge in the absence of a second low dose treatment. Thus, taken together, T cell anergy best describes our findings.
Multiple reports indicate that resting B cells may be the predominant APCs that induce tolerance. The tolerogenicity of resting B cells may be due to the absence of costimulatory molecules such as B7, which is highly expressed in DCs and activated B cells 63. It has been shown, however, that both activated and resting B cells can induce tolerance in naive but not memory T cells 2, which may be due to less stringent requirement of memory T cells for costimulatory molecules. All APCs, including resting B cells can stimulate memory T cells 5. Although a role for DCs as the most potent antigen presenters has well been documented, its role as a tolerogen is less clear. However, under the experimental conditions used here, it is likely that DCs that express high levels of MHC class II, may preferentially bind peptides offered at the tolerogenic, lower doses. At higher peptide doses all APCs might access and present antigenic peptides. Notably, some MHC class II expressed on DCs are shown to be empty 64, which provokes the thought of a new role for DCs as inducers of tolerance in peripheral T cells 65. Studies on the way address these issues.
Use of two different Tg mouse models provides clear evidence that a monoclonal 6.5+ TCR Tg mice or multiclonal T cell populations specific for HA306–318 in HLA-DR1 Tg mice, can be rendered tolerant. Other reports have also investigated differences in a similar Tg system, documenting that in the non-TCR Tg mouse model, T cell clones of different affinities for a given peptide–MHC complex can be stimulated or tolerized 66.
CTLA-4 Mediates Anergy.
Manifestation of anergy in this system correlates with the upregulation of CTLA-4 which can be inhibited by administration of anti–CTLA-4 mAb in vivo. Several groups have proposed that induction of peripheral T cell tolerance is a direct consequence of CTLA-4 engagement by B7 33 67. CTLA-4 can inhibit T cell responses extracellularly, by competing for B7 ligands 68, or intracellularly, by recruiting src homology 2 domain–containing protein tyrosin phosphatase (SHP)-2 and inhibiting tyrosine phosphorylation 69, by triggering TGF-β production 70, or by blocking cell cycle progression at the late G1 to S phase 1. Alternatively, full inhibitory function of CTLA-4 may require both extra and intracellular domains 71. We observed an increased level of CTLA-4 in cells that exhibited anergy compared with other groups of cells that received doses below or above the tolerogenic dose of peptide (Fig. 7). Strong evidence for the role of CTLA-4 in the induction of anergy is provided in Fig. 8, in which enhanced T cell proliferation of the anergized cells of mice that were treated with anti–CTLA-4 blocking antibody is observed. Lack of any measurable effect on proliferation of cells from other groups that did not express CTLA-4 indicates the specific and controlled effects of anti–CTLA-4 antibody. In most reports an increase in proliferation correlates with parallel increase in IL-2 and IFN-γ secretion 1 33 61 72 73. However, it is of interest to consider the possibility that CTLA-4 blockade may affect the sensitivity of T cells to TCR ligand and possible effects on the proportion of cytokine-producing cells in favor of specific cytokines 74. Future experiments will address these issues.
Immediate implications of T cell anergy by a low density of agonist peptide–MHC include autoimmunity and viral and tumor surveillance. Memory T cells, specific for self, might need to be kept tolerized by encountering low densities of peptide–MHC, which could be an important process in the prevention of organ-specific autoimmune diseases. Furthermore, understanding of T cell responses to presentation of low densities of agonist peptide–MHC in vivo is of great significance because of its pertinence in viral infections and in antitumor responses. Many viruses and several tumors are known to decrease expression of cell surface MHC class II 75. Alternatively, some tumor-associated peptides bind MHC poorly 76. The reduced surface expression of MHC and low affinity peptide–MHC complexes lead to presentation of low densities of specific peptide–MHC, which will induce anergy to the viral peptides or to tumor antigens through a CTLA-4–dependent mechanism. A recent report demonstrating that CTLA-4 blockade reverses CD8+ T cell tolerance to tumor by a CD4+ T cell– and IL-2–dependent mechanism supports this notion 77. Thus, our findings demand a revision in design of strategies to overcome viral infections or tumor therapy. Rather than enhancing antigen presentation, one may have to focus on the reversal of anergy.
In conclusion, our results strongly suggest that low densities of agonist peptide induce anergy in memory T cells and that CTLA-4 is the dominant regulatory mechanism.
Acknowledgments
We thank Drs. Dennis Zaller for the HLA-DR1 Tg mice, Drew Pardoll for TCR (6.5) Tg mice, Thomas Cameron and Lawrence Stern for HA-DR1-SA-PE oligomers, and Ron Germain, Drew Pardoll, Antony Rosen, Abdu Hamad, Chih-Ling Chou, and Jill Slansky for discussion and critical reading of the manuscript.
This work was supported by National Institutes of Health (NIH) grant R01GM5354 (to S. Sadegh-Nasseri), and a pilot project grant from Fujisawa to S. Sadegh-Nasseri. S. Mirshahidi thanks NIH training grants to Immunology and Pathobiology Graduate Programs.
Footnotes
Abbreviations used in this paper: CFA, Complete Freund's Adjuvant; Cyc, cychrome; HA, hemagglutinin; IFA, Incomplete Freund's Adjuvant; Tg, transgenic; PPD, purified protein derivative.
A portion of this paper was presented as a poster in Immunology 2000 in Seattle, WA on May 12–16, 2000.
References
- Greenwald R.J., Boussiotis V.A., Lorsbach R., Abbas A.K., Sharpe A.H. CTLA-4 regulates induction of anergy in vivo. Immunity. 2001;14:145–155. doi: 10.1016/s1074-7613(01)00097-8. [DOI] [PubMed] [Google Scholar]
- Fuchs E.J., Matzinger P. B cells turn off virgin but not memory T cells. Science. 1992;258:1156–1159. doi: 10.1126/science.1439825. [DOI] [PubMed] [Google Scholar]
- London C.A., Lodge M.P., Abbas A.K. Functional responses and costimulator dependence of memory CD4+ T cells. J. Immunol. 2000;164:265–272. doi: 10.4049/jimmunol.164.1.265. [DOI] [PubMed] [Google Scholar]
- Farber D.L., Acuto O., Bottomly K. Differential T cell receptor-mediated signaling in naive and memory CD4 T cells. Eur. J. Immunol. 1997;27:2094–2101. doi: 10.1002/eji.1830270838. [DOI] [PubMed] [Google Scholar]
- Croft M., Bradley L.M., Swain S.L. Naive versus memory CD4 T cell response to antigen. Memory cells are less dependent on accessory cell costimulation and can respond to many antigen-presenting cell types including resting B cells. J. Immunol. 1994;152:2675–2685. [PubMed] [Google Scholar]
- Linsley P.S., Bradshaw J., Greene J., Peach R., Bennett K.L., Mittler R.S. Intracellular trafficking of CTLA-4 and focal localization towards sites of TCR engagement. Immunity. 1996;4:535–543. doi: 10.1016/s1074-7613(00)80480-x. [DOI] [PubMed] [Google Scholar]
- Korb L.C., Mirshahidi S., Ramyar K., Sadighi Akha A.A., Sadegh-Nasseri S. Induction of T cell anergy by low numbers of agonist ligands. J. Immunol. 1999;162:6401–6409. [PubMed] [Google Scholar]
- Kirberg J., Baron A., Jakob S., Rolink A., Karjalainen K., von Boehmer H. Thymic selection of CD8+ single positive cells with a class II major histocompatibility complex-restricted receptor. J. Exp. Med. 1994;180:25–34. doi: 10.1084/jem.180.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutton R.W., Bradley L.M., Swain S.L. T cell memory. Annu. Rev. Immunol. 1998;16:201–223. doi: 10.1146/annurev.immunol.16.1.201. [DOI] [PubMed] [Google Scholar]
- Tietz W., Hamann A. The migratory behavior of murine CD4+ cells of memory phenotype. Eur. J. Immunol. 1997;27:2225–2232. doi: 10.1002/eji.1830270916. [DOI] [PubMed] [Google Scholar]
- Bradley L.M., Harbertson J., Freschi G.C., Kondrack R., Linton P.J. Regulation of development and function of memory CD4 subsets. Immunol. Res. 2000;21:149–158. doi: 10.1385/IR:36:1:149. [DOI] [PubMed] [Google Scholar]
- Rogers P.R., Dubey C., Swain S.L. Qualitative changes accompany memory T cell generationfaster, more effective responses at lower doses of antigen. J. Immunol. 2000;164:2338–2346. doi: 10.4049/jimmunol.164.5.2338. [DOI] [PubMed] [Google Scholar]
- Rosloniec E.F., Brand D.D., Myers L.K., Whittington K.B., Gumanovskaya M., Zaller D.M., Woods A., Altmann D.M., Stuart J.M., Kang A.H. An HLA-DR1 transgene confers susceptibility to collagen-induced arthritis elicited with human type II collagen. J. Exp. Med. 1997;185:1113–1122. doi: 10.1084/jem.185.6.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan S.K., Stern L.J., Sadegh-Nasseri S. Sodium dodecyl sulfate stability of HLA-DR1 complexes correlates with burial of hydrophobic residues in pocket 1. J. Immunol. 1999;162:3463–3470. [PubMed] [Google Scholar]
- Natarajan S.K., Assadi M., Sadegh-Nasseri S. Stable peptide binding to MHC class II molecule is rapid and is determined by a receptive conformation shaped by prior association with low affinity peptides. J. Immunol. 1999;162:4030–4036. [PubMed] [Google Scholar]
- Chou C.-L., Sadegh-Nasseri S. HLA-DM recognizes the flexible conformation of major histocompatibility complex class II. J. Exp. Med. 2000;192:1697–1706. doi: 10.1084/jem.192.12.1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak E.J., Liu A.W., Nepom G.T., Kwok W.W. MHC class II tetramers identify peptide-specific human CD4+ T cells proliferating in response to influenza A antigen. J. Clin. Invest. 1999;104:R63–R67. doi: 10.1172/JCI8476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron T.O., Cochran J.R., Yassine-Diab B., Sekaly R.P., Stern L.J. Cutting edgedetection of antigen-specific CD4+ T cells by HLA-DR1 oligomers is dependent on the T cell activation state. J. Immunol. 2001;166:741–745. doi: 10.4049/jimmunol.166.2.741. [DOI] [PubMed] [Google Scholar]
- Sadegh-Nasseri S., Stern L.J., Wiley D.C., Germain R.N. MHC class II function preserved by low-affinity peptide interactions preceding stable binding. Nature. 1994;370:647–650. doi: 10.1038/370647a0. [DOI] [PubMed] [Google Scholar]
- Schwartz R.H. Models of T cell anergyis there a common molecular mechanism? J. Exp. Med. 1996;184:1–8. doi: 10.1084/jem.184.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constant S., Pfeiffer C., Woodard A., Pasqualini T., Bottomly K. Extent of T cell receptor ligation can determine the functional differentiation of naive CD4+ T cells. J. Exp. Med. 1995;182:1591–1596. doi: 10.1084/jem.182.5.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer C., Stein J., Southwood S., Ketelaar H., Sette A., Bottomly K. Altered peptide ligands can control CD4 T lymphocyte differentiation in vivo. J. Exp. Med. 1995;181:1569–1574. doi: 10.1084/jem.181.4.1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yip H.C., Karulin A.Y., Tary-Lehmann M., Hesse M.D., Radeke H., Heeger P.S., Trezza R.P., Heinzel F.P., Forsthuber T., Lehmann P.V. Adjuvant-guided type-1 and type-2 immunityinfectious/noninfectious dichotomy defines the class of response. J. Immunol. 1999;162:3942–3949. [PubMed] [Google Scholar]
- Groux H., O'Garra A., Bigler M., Rouleau M., Antonenko S., de Vries J.E., Roncarolo M.G. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature. 1997;389:737–742. doi: 10.1038/39614. [DOI] [PubMed] [Google Scholar]
- Buer J., Lanoue A., Franzke A., Garcia C., von Boehmer H., Sarukhan A. Interleukin 10 secretion and impaired effector function of major histocompatibility complex class II-restricted T cells anergized in vivo. J. Exp. Med. 1998;187:177–183. doi: 10.1084/jem.187.2.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardi G., Sidhu S., Batchelor R., Lechler R. Anergic T cells as suppressor cells in vitro. Science. 1994;264:1587–1589. doi: 10.1126/science.8202711. [DOI] [PubMed] [Google Scholar]
- Lenardo M.J. Interleukin-2 programs mouse αβ T lymphocytes for apoptosis. Nature. 1991;353:858–861. doi: 10.1038/353858a0. [DOI] [PubMed] [Google Scholar]
- Rocha B., von Boehmer H. Peripheral selection of the T cell repertoire. Science. 1991;2:1225–1228. doi: 10.1126/science.1900951. [DOI] [PubMed] [Google Scholar]
- Sundstedt A., Sigvardsson M., Leanderson T., Hedlund G., Kalland T., Dohlsten M. In vivo anergized CD4+ T cells express perturbed AP-1 and NF-κB transcription factors. Proc. Natl. Acad. Sci. USA. 1996;93:979–984. doi: 10.1073/pnas.93.3.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boussiotis V.A., Freeman G.J., Berezovskaya A., Barber D.L., Nadler L.M. Maintenance of human T cell anergyblocking of IL-2 gene transcription by activated Rap1. Science. 1997;278:124–128. doi: 10.1126/science.278.5335.124. [DOI] [PubMed] [Google Scholar]
- Waterhouse P., Penninger J.M., Timms E., Wakeham A., Shahinian A., Lee K.P., Thompson C.B., Griesser H., Mak T.W. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995;270:985–988. doi: 10.1126/science.270.5238.985. [DOI] [PubMed] [Google Scholar]
- Tivol E.A., Borriello F., Schweitzer A.N., Lynch W.P., Bluestone J.A., Sharpe A.H. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3:541–547. doi: 10.1016/1074-7613(95)90125-6. [DOI] [PubMed] [Google Scholar]
- Perez V.L., Van Parijs L., Biuckians A., Zheng X.X., Strom T.B., Abbas A.K. Induction of peripheral T cell tolerance in vivo requires CTLA-4 engagement. Immunity. 1997;6:411–417. doi: 10.1016/s1074-7613(00)80284-8. [DOI] [PubMed] [Google Scholar]
- Walunas T.L., Lenschow D.J., Bakker C.Y., Linsley P.S., Freeman G.J., Green J.M., Thompson C.B., Bluestone J.A. CTLA-4 can function as a negative regulator of T cell activation. Immunity. 1994;1:405–413. doi: 10.1016/1074-7613(94)90071-x. [DOI] [PubMed] [Google Scholar]
- Metzler B., Burkhart C., Wraith D.C. Phenotypic analysis of CTLA-4 and CD28 expression during transient peptide-induced T cell activation in vivo. Int. Immunol. 1999;11:667–675. doi: 10.1093/intimm/11.5.667. [DOI] [PubMed] [Google Scholar]
- De Magistris M.T., Alexander J., Coggeshall M., Altman A., Gaeta F.C., Grey H.M., Sette A. Antigen analog-major histocompatibility complexes act as antagonists of the T cell receptor. Cell. 1992;68:625–634. doi: 10.1016/0092-8674(92)90139-4. [DOI] [PubMed] [Google Scholar]
- Sloan-Lancaster J., Evavold B.D., Allen P.M. Induction of T-cell anergy by altered T-cell-receptor ligand on live antigen-presenting cells. Nature. 1993;363:156–159. doi: 10.1038/363156a0. [DOI] [PubMed] [Google Scholar]
- Jenkins M.K., Schwartz R.H. Antigen presentation by chemically modified splenocytes induces antigen-specific T cell unresponsiveness in vitro and in vivo. J. Exp. Med. 1987;165:302–319. doi: 10.1084/jem.165.2.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monks C.R., Freiberg B.A., Kupfer H., Sciaky N., Kupfer A. Three-dimensional segregation of supramolecular activation clusters in T cells. Nature. 1998;395:82–86. doi: 10.1038/25764. [DOI] [PubMed] [Google Scholar]
- Grakoui A., Bromley S.K., Sumen C., Davis M.M., Shaw A.S., Allen P.M., Dustin M.L. The immunological synapsea molecular machine controlling T cell activation. Science. 1999;285:221–227. doi: 10.1126/science.285.5425.221. [DOI] [PubMed] [Google Scholar]
- Iezzi G., Karjalainen K., Lanzavecchia A. The duration of antigenic stimulation determines the fate of naive and effector T cells. Immunity. 1998;8:89–95. doi: 10.1016/s1074-7613(00)80461-6. [DOI] [PubMed] [Google Scholar]
- Kearney E.R., Pape K.A., Loh D.Y., Jenkins M.K. Visualization of peptide-specific T cell immunity and peripheral tolerance induction in vivo. Immunity. 1994;1:327–339. doi: 10.1016/1074-7613(94)90084-1. [DOI] [PubMed] [Google Scholar]
- Ridge J.P., Fuchs E.J., Matzinger P. Neonatal tolerance revisitedturning on newborn T cells with dendritic cells. Science. 1996;271:1723–1726. doi: 10.1126/science.271.5256.1723. [DOI] [PubMed] [Google Scholar]
- Swain S.L., Croft M., Dubey C., Haynes L., Rogers P., Zhang X., Bradley L.M. From naive to memory T cells. Immunol. Rev. 1996;150:143–167. doi: 10.1111/j.1600-065x.1996.tb00700.x. [DOI] [PubMed] [Google Scholar]
- Bachmann M.F., Gallimore A., Linkert S., Cerundolo V., Lanzavecchia A., Kopf M., Viola A. Developmental regulation of Lck targeting to the CD8 coreceptor controls signaling in naive and memory T cells. J. Exp. Med. 1999;189:1521–1530. doi: 10.1084/jem.189.10.1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opferman J.T., Ober B.T., Ashton-Rickardt P.G. Linear differentiation of cytotoxic effectors into memory T lymphocytes. Science. 1999;283:1745–1748. doi: 10.1126/science.283.5408.1745. [DOI] [PubMed] [Google Scholar]
- Jacob J., Baltimore D. Modelling T-cell memory by genetic marking of memory T cells in vivo. Nature. 1999;399:593–597. doi: 10.1038/21208. [DOI] [PubMed] [Google Scholar]
- Boursalian T.E., Bottomly K. Stability of naive and memory phenotypes on resting CD4 T cells in vivo. J. Immunol. 1999;162:9–16. [PubMed] [Google Scholar]
- Metz D.P., Farber D.L., Taylor T., Bottomly K. Differential role of CTLA-4 in regulation of resting memory versus naive CD4 T cell activation. J. Immunol. 1998;161:5855–5861. [PubMed] [Google Scholar]
- Falb D., Briner T.J., Sunshine G.H., Bourque C.R., Luqman M., Gefter M.L., Kamradt T. Peripheral tolerance in T cell receptor-transgenic miceevidence for T cell anergy. Eur. J. Immunol. 1996;26:130–135. doi: 10.1002/eji.1830260120. [DOI] [PubMed] [Google Scholar]
- Critchfield J.M., Racke M.K., Zuniga-Pflucker J.C., Cannella B., Raine C.S., Goverman J., Lenardo M.J. T cell deletion in high antigen dose therapy of autoimmune encephalomyelitis. Science. 1994;263:1139–1143. doi: 10.1126/science.7509084. [DOI] [PubMed] [Google Scholar]
- Adler A.J., Huang C.T., Yochum G.S., Marsh D.W., Pardoll D.M. In vivo CD4+ T cell tolerance induction versus priming is independent of the rate and number of cell divisions. J. Immunol. 2000;164:649–655. doi: 10.4049/jimmunol.164.2.649. [DOI] [PubMed] [Google Scholar]
- Walunas T.L., Bakker C.Y., Bluestone J.A. CTLA-4 ligation blocks CD28-dependent T cell activation. J. Exp. Med. 1996;183:2541–2550. doi: 10.1084/jem.183.6.2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krummel M.F., Allison J.P. CTLA-4 engagement inhibits IL-2 accumulation and cell cycle progression upon activation of resting T cells. J. Exp. Med. 1996;183:2533–2540. doi: 10.1084/jem.183.6.2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeths M.J., Kedl R.M., Mescher M.F. CD8+ T cells become nonresponsive (anergic) following activation in the presence of costimulation. J. Immunol. 1999;163:102–110. [PubMed] [Google Scholar]
- Salomon B., Lenschow D.J., Rhee L., Ashourian N., Singh B., Sharpe A., Bluestone J.A. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 2000;12:431–440. doi: 10.1016/s1074-7613(00)80195-8. [DOI] [PubMed] [Google Scholar]
- Powrie F., Carlino J., Leach M.W., Mauze S., Coffman R.L. A critical role for transforming growth factor-β but not interleukin 4 in the suppression of T helper type 1-mediated colitis by CD45RBlow CD4+ T cells. J. Exp. Med. 1996;183:2669–2674. doi: 10.1084/jem.183.6.2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton A.M., Shevach E.M. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J. Exp. Med. 1998;188:287–296. doi: 10.1084/jem.188.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T., Tagami T., Yamazaki S., Uede T., Shimizu J., Sakaguchi N., Mak T.W., Sakaguchi S. Immunologic self-tolerance maintained by CD25+CD4+ regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J. Exp. Med. 2000;192:303–310. doi: 10.1084/jem.192.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read S., Malmström V., Powrie F. Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25+CD4+ regulatory cells that control intestinal inflammation. J. Exp. Med. 2000;192:295–302. doi: 10.1084/jem.192.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oosterwegel M.A., Mandelbrot D.A., Boyd S.D., Lorsbach R.B., Jarrett D.Y., Abbas A.K., Sharpe A.H. The role of CTLA-4 in regulating Th2 differentiation. J. Immunol. 1999;163:2634–2639. [PubMed] [Google Scholar]
- Ohashi P.S., Oehen S., Buerki K., Pircher H., Ohashi C.T., Odermatt B., Malissen B., Zinkernagel R.M., Hengartner H. Ablation of and induction of diabetes by virus infection in viral antigen transgenic mice. Cell. 1991;65:305–317. doi: 10.1016/0092-8674(91)90164-t. [DOI] [PubMed] [Google Scholar]
- Yuschenkoff V.N., Sethna M.P., Freeman G.J., Parker D.C. Coexpression of B7-1 and antigen blocks tolerance induction to antigen presented by resting B cells. J. Immunol. 1996;157:1987–1995. [PubMed] [Google Scholar]
- Santambrogio L., Sato A.K., Fischer F.R., Dorf M.E., Stern L.J. Abundant empty class II MHC molecules on the surface of immature dendritic cells. Proc. Natl. Acad. Sci. USA. 1999;96:15050–15055. doi: 10.1073/pnas.96.26.15050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman R., Turley S., Mellman I., Inaba K. The induction of tolerance by dendritic cells that have captured apoptotic cells. J. Exp. Med. 2000;191:411–416. doi: 10.1084/jem.191.3.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R., Wang-Zhu Y., Gabaglia C.R., Kimachi K., Grey H.M. The stimulation of low-affinity, nontolerized clones by heteroclitic antigen analogues causes the breaking of tolerance established to an immunodominant T cell epitope. J. Exp. Med. 1999;190:983–994. doi: 10.1084/jem.190.7.983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bluestone J.A. Is CTLA-4 a master switch for peripheral T cell tolerance? J. Immunol. 1997;158:1989–1993. [PubMed] [Google Scholar]
- Nakaseko C., Miyatake S., Iida T., Hara S., Abe R., Ohno H., Saito Y., Saito T. Cytotoxic T lymphocyte antigen 4 (CTLA-4) engagement delivers an inhibitory signal through the membrane-proximal region in the absence of the tyrosine motif in the cytoplasmic tail. J. Exp. Med. 1999;190:765–774. doi: 10.1084/jem.190.6.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K.M., Chuang E., Griffin M., Khattri R., Hong D.K., Zhang W., Straus D., Samelson L.E., Thompson C.B., Bluestone J.A. Molecular basis of T cell inactivation by CTLA-4. Science. 1998;282:2263–2266. doi: 10.1126/science.282.5397.2263. [DOI] [PubMed] [Google Scholar]
- Chen W., Jin W., Wahl S.M. Engagement of cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) induces transforming growth factor β (TGF-β) production by murine CD4+ T cells. J. Exp. Med. 1998;188:1849–1857. doi: 10.1084/jem.188.10.1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masteller E.L., Chuang E., Mullen A.C., Reiner S.L., Thompson C.B. Structural analysis of CTLA-4 function in vivo. J. Immunol. 2000;164:5319–5327. doi: 10.4049/jimmunol.164.10.5319. [DOI] [PubMed] [Google Scholar]
- Alegre M.L., Shiels H., Thompson C.B., Gajewski T.F. Expression and function of CTLA-4 in Th1 and Th2 cells. J. Immunol. 1998;161:3347–3356. [PubMed] [Google Scholar]
- Griffin M.D., Hong D.K., Holman P.O., Lee K.M., Whitters M.J., O'Herrin S.M., Fallarino F., Collins M., Segal D.M., Gajewski T.F. Blockade of T cell activation using a surface-linked single-chain antibody to CTLA-4 (CD152) J. Immunol. 2000;164:4433–4442. doi: 10.4049/jimmunol.164.9.4433. [DOI] [PubMed] [Google Scholar]
- Kuhns M., Epshteyn V., Sobel R., Allison J. Cytotoxic T lymphocyte antigen-4 (CTLA-4) regulates the size, reactivity, and function of a primed pool of CD4+ T cells. Proc. Natl. Acad. Sci. USA. 2000;97:12711–12716. doi: 10.1073/pnas.220423597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky F.M., Lem L., Solache A., Bennett E.M. Human pathogen subversion of antigen presentation. Immunol. Rev. 1999;168:199–215. doi: 10.1111/j.1600-065x.1999.tb01294.x. [DOI] [PubMed] [Google Scholar]
- Topalian S.L., Gonzales M.I., Parkhurst M., Li Y.F., Southwood S., Sette A., Rosenberg S.A., Robbins P.F. Melanoma-specific CD4+ T cells recognize nonmutated HLA-DR-restricted tyrosinase epitopes. J. Exp. Med. 1996;183:1965–1971. doi: 10.1084/jem.183.5.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrikant P., Khoruts A., Mescher M.F. CTLA-4 blockade reverses CD8+ T cell tolerance to tumor by a CD4+ T cell- and IL-2-dependent mechanism. Immunity. 1999;11:483–493. doi: 10.1016/s1074-7613(00)80123-5. [DOI] [PubMed] [Google Scholar]