

Abstract
Interstitial fluid is constantly drained into lymph nodes (LNs) via afferent lymph vessels. This conduit enables monocyte-derived macrophages and dendritic cells to access LNs from peripheral tissues. We show that during inflammation in the skin, a second recruitment pathway is evoked that recruits large numbers of blood-borne monocytes to LNs via high endothelial venules (HEVs). Inhibition of monocyte chemoattractant protein (MCP)-1 blocked this inflammation-induced monocyte homing to LNs. MCP-1 mRNA in inflamed skin was over 100-fold upregulated and paralleled MCP-1 protein levels, whereas in draining LNs MCP-1 mRNA induction was much weaker and occurred only after a pronounced rise in MCP-1 protein. Thus, MCP-1 in draining LNs was primarily derived from inflamed skin. In MCP-1−/− mice, intracutaneously injected MCP-1 accumulated rapidly in the draining LNs where it enhanced monocyte recruitment. Intravital microscopy showed that skin-derived MCP-1 was transported via the lymph to the luminal surface of HEVs where it triggered integrin-dependent arrest of rolling monocytes. These findings demonstrate that inflamed peripheral tissues project their local chemokine profile to HEVs in draining LNs and thereby exert “remote control” over the composition of leukocyte populations that home to these organs from the blood.
Keywords: homing, MCP-1, high endothelial venules, phagocytes, lymphatic system
Introduction
Blood-borne monocytes possess developmental plasticity allowing them to differentiate into macrophages or subsets of dendritic cells (DCs)* upon entering tissues. These two monocyte-derived cell types perform crucial immune functions, particularly in peripheral LNs (PLNs). How monocytes and their descendants enter PLNs is incompletely understood. Some monocytes exit the blood at peripheral sites of inflammation, load themselves with antigen, and become mature antigen-presenting DCs during their transit to draining PLNs via afferent lymphatics (1). Tissue resident macrophages, dermal DCs, and Langerhans cells enter PLNs by the same route (2, 3). Some mechanisms by which monocyte-derived cells enter PLNs via the lymph have been uncovered recently (1, 4, 5). In addition, monocytes may enter PLNs directly from the blood through high endothelial venules (HEVs), the route taken by naive lymphocytes and subsets of memory cells (6, 7). Classic electron microscopy studies have suggested that monocyte homing via HEVs may be induced by unknown signals in PLNs that drain inflamed tissues. In resting PLNs, macrophages reside mostly in subcapsular and medullary sinuses distant from HEVs, but in inflamed PLNs monocytes/macrophages were detected within and around HEVs (8). Consistent with this concept, the monocyte cell line WEHI78/24 was found to adhere in an integrin-dependent fashion to HEVs in frozen sections of inflamed, but not resting PLNs (9). However, the magnitude, kinetics, and mechanisms by which monocytes home to inflamed PLNs via HEVs have not been explored in vivo.
Since monocyte-expressed integrins are likely to play a role (9) and since these molecules must be activated for efficient binding to endothelial cells, we hypothesized that inflamed HEVs display chemoattractants that trigger integrin activation on intravascular monocytes. One candidate molecule is the inflammatory CC chemokine monocyte chemotactic protein (MCP)-1 (CCL2), which regulates monocyte recruitment from blood into many nonlymphoid tissues (10). In vitro, MCP-1 stimulates arrest of rolling human monocytes under flow (11) and also promotes transendothelial migration (12). In addition, MCP-1 has been implicated in angiogenesis (13, 14), maintenance of Th2-lymphocyte populations (15), and modulation of HIV-1 infection of mononuclear cells (16).
Chemokines are commonly thought to act locally, directing leukocyte migration into and within the tissue in which they are generated. However, previous observations suggest a mechanism by which chemokines may act far away from the site of their origin (17, 18). In this paradigm, chemokines use the afferent lymphatics, and the fibroblastic reticular cell (FRC) network within draining PLNs, as conduits for transport from peripheral tissues to HEVs (17, 18). However, these earlier studies have examined the fate of relatively large quantities of recombinant chemokines after bolus injection into the skin. The question whether endogenous chemokines are transported from peripheral tissues to draining LNs and how this would affect the nodal environment has not been addressed.
Here we show that a remotely generated chemokine plays a profound role in PLNs that drain inflamed skin. Circulating monocytes are recruited into such PLNs by MCP-1, which is rapidly released in inflamed skin, transported via the lymph into PLNs, and translocated to the luminal surface of HEVs. Intravascular MCP-1 then activates CCR2 on rolling monocytes, which triggers integrin-mediated arrest. These observations suggest a mechanism by which multistep adhesion cascades in HEVs can be modulated remotely, thus enabling peripheral tissues to control the composition and function of leukocytes in the draining LNs.
Materials and Methods
Animals
Mice were used in accordance with NIH guidelines, in experiments approved by the Institutional Review Committees of Harvard Medical School and the Center for Blood Research. MCP-1−/− mice on C57BL/6 background (19) and CX3CR1+/GFP mice on C57 × 129Sv background (20) were bred in a specific pathogen-free, viral antibody-free (SPF/VAF) barrier facility. Wild-type C57BL/6 mice were from The Jackson Laboratory. All mice tested negative for specific pathogens and were kept on sterile bedding with unrestricted access to autoclaved water and lab chow.
Induction of Cutaneous Inflammation
Equal volumes of complete Freund's adjuvant (CFA) and sterile PBS containing keyhole limpet hemocyanin (KLH; final concentration 2.5 mg/ml) were emulsified (CFA/KLH). Mice were anaesthetized with ketamine and xylazine, and 50 μl of the CFA/KLH emulsion was injected intracutaneously over one or both flanks or the upper right lateral thorax. At various time points thereafter the injection site and draining PLNs were removed for histology or analysis of cellular composition, MCP-1 protein, and mRNA levels. In some experiments draining subiliac PLNs were prepared for observation by intravital microscopy (21).
For enumeration of the number of CD11b+F4/80+ leukocytes, PLNs were removed, digested in type II collagenase (0.5% wt/vol in HBSS, 37°C, 40 min; Worthington Biochemical) and stained with anti–CD45-PE (Ly-5; BD PharMingen), anti–CD11b-FITC (M1/70; BD PharMingen), and biotinylated mAb F4/80 (Serotec), followed by streptavidin-CyChrome, and analyzed on a flow cytometer (FACscan™; Becton Dickinson). To determine the total number of leukocytes, a known number of PKH26-labeled microspheres (Sigma-Aldrich) were added to each sample.
Adoptive Transfer Homing Assays Using CX3CR1+/GFP Leukocytes
For each experiment, blood was obtained from ∼50 donor CX3CR1+/GFP mice by cardiac puncture and PBMCs were isolated using Histopaque 1083 (Sigma-Aldrich). For phenotypic analysis aliquots of PBMCs were stained with mAb as described previously, and with rabbit polyclonal anti-CCR2 Ab. 5 × 106 CX3CR1+/GFP PBMCs, which contained ∼10% cells that expressed detectable levels of green fluorescent protein (GFP), were warmed to 37°C and then injected intravenously into recipient animals 5–7 d after inducing unilateral cutaneous inflammation. Recipient mice were pretreated with anti–MCP-1 (2H5; BD PharMingen) or isotype control mAb (G235–2356; BD PharMingen; 25 μg intraperitoneally at −2 h and 25 μg intravenously at −20 min). To compare the magnitude of monocyte recruitment to a well-characterized LN-tropic reference population, five mice with 5-d-old skin inflammation received 5 × 106 LN cells (containing ∼75% GFP+ naive T cells) from T-GFP mice (17). 4 h after injection of donor CX3CR+/GFP PBMCs or TGFP LN cells, mice were killed, a blood sample was taken by cardiac puncture, and the spleen, draining, and contralateral PLNs were removed and digested in collagenase. Samples were stained with anti–NK1.1-PE (NKR-P1C; BD PharMingen) and anti–CD45-CyChrome and GFP+CD45+NK1.1− cells were counted by flow cytometry.
Analysis of CX3CR1+/GFP Leukocyte Homing To Inflamed PLNs by Confocal Microscopy
5 d after intracutaneous injection of CFA/KLH in the flank skin of six wild-type mice, three animals received an intravenous injection of 5 × 106 CX3CR1+/GFP PBMCs (∼5 × 105 GFP+ cells), while the other three mice were injected into the skin granuloma with CX3CR1+/GFP PBMCs containing 2.5 × 105 GFP+ cells. The draining subiliac LNs were harvested after 1 h, frozen in OCT, and 20-μm thick sections were stained with anti-PNAd mAb MECA-79 followed by Cy5-anti–rat IgM. GFP+ leukocytes were classified according to their position relative to the nearest MECA-79+ vessel using confocal microscopy (Olympus BX50WI; BioRad Radiance 2000 scan-head).
Reconstitution of CX3CR1+/GFP Leukocyte Homing in MCP-1–deficient Mice
MCP-1−/− mice were injected intracutaneously with CFA/KLH over both flanks and the upper right lateral thorax. On day 5, animals were injected into the inflamed skin with recombinant murine MCP-1 (25 μg in 50 μl PBS) over the left subiliac LN and with vehicle alone (50 μl PBS) over the right subiliac and brachial LNs. 10 min later, all animals received 1.5 × 107 CX3CR1+/GFP PBMCs intravenously. Both subiliac LNs and the right brachial LN were harvested 90 min after cell injection for quantification of GFP+ cells by flow cytometry. To determine the distribution of intracutaneously injected MCP-1 in draining and contralateral PLNs, three additional MCP-1−/− mice were injected with CFA/KLH and, on day 5, with 25 μg MCP-1 and vehicle as described previously. One control animal received CFA/KLH, but no chemokine. Both subiliac LNs and the inflamed brachial LNs were removed 90 min after injection and prepared for ELISA. Readings from the control animal were used to subtract unspecific background signals for each organ.
Murine MCP-1 ELISA
For studies on endogenous MCP-1 protein concentration in inflamed tissues, CFA/KLH cutaneous injection sites and draining subiliac PLNs were removed from killed wild-type mice, weighed, homogenized in lysis buffer (PBS with 1 mM PMSF, 0.01 mg/ml aprotonin, and 0.01 mg/ml leupeptin), sonicated, and centrifuged (14,000× g at 4°C for 20 min). The supernatant was assayed for MCP-1 immunoreactivity by ELISA (Quantikine; R&D Systems). The same approach was taken to assess MCP-1 concentration in PLN of MCP-1−/− mice after intracutaneous injection of chemokine or PBS.
Intravital Microscopy of the Subiliac PLNs
The left or right subiliac PLN was prepared for viewing by intravital microscopy as described previously (21). In some experiments, PLNs were inflamed by intracutaneous injection of CFA/KLH into the flank 3–7 d before observation.
Observation of WEHI78/24 Cells in the Subiliac PLN Microvasculature
Calcein-labeled WEHI78/24 cells were injected through a femoral artery catheter and visualized in the downstream PLN microvasculature as described previously (21). PLN venules were classified based on their branching order ranging from I-V, where the order I venule is the large hilar collecting venule that drains blood out of the PLN and higher order venules are successively smaller branches upstream (21). Within each venule, WEHI78/24 cells were classified as rolling (cells obviously interacting with the vessel wall, with a slower velocity than the blood stream) or noninteracting. The rolling fraction was calculated as the percentage of rolling cells in the total number of cells entering the venule. Cells that arrested on the vessel wall for at least 30 s were classified as sticking cells. The sticking fraction was calculated as the percentage of arrested cells in the total number of rolling cells. In some experiments where observation times were limited by the number of cells that could be injected (to enable multiple injections of differently treated cells), only higher order (IV-V) vessels were analyzed, because homing occurs primarily in these subcortical branches (21).
Observation of Intracutaneously Injected MCP-1 in the Draining PLN
Human MCP-1 was synthesized with an Alexa565-conjugated 6×Histidine tag at the COOH terminus (Alexa-conjugated human MCP-1 [hMCP-1ALEXA]). hMCP-1ALEXA (0.5 μg/injection site) was injected intracutaneously 40 min before preparation of the draining PLN for epifluorescence intravital microscopy. At the end of the observation period the PLN vasculature was visualized with intravenous FITC-dextran (mol. wt. 150 KD; Sigma-Aldrich). In some experiments, 125I-labeled human MCP-1 (250 μCi/μg protein, 1.3 ng/injection site) was injected intracutaneously into recipient mice. 1 h later, the draining PLNs were removed and prepared for autoradiography and immunohistology as described previously (22).
Analysis of Murine MCP-1 mRNA Using Real-Time PCR (TaqMan®)
The CFA/KLH injection site (50–100 mg) and draining PLNs were flash frozen in liquid N2. Total RNA was extracted with the ULTRASPEC-II RNA Isolation System (BioTecx Laboratories) and mRNA from 100 μg total RNA was purified using a QIAGEN Oligotex mRNA kit (QIAGEN). cDNA was synthesized using mRNA equivalent to ∼30 μg total RNA. Multiplex Real Time Quantitative mRNA analyses were performed in an ABI Prism 7700 Sequence Detection System using FAM-labeled MCP-1 and VIC-labeled GAPDH probes and appropriate primers (PE Applied Biosystems). ΔCT values (MCP-1 CT minus GAPDH CT) for each triplicate sample were averaged and ΔΔCT was calculated as follows: ΔCT values from the control group (t = 0 h) were averaged and subtracted from ΔCT values of individual mice. mRNA amplification was determined by the formula 2-ΔΔCT.
Statistical Analysis
Data are presented as mean ± SEM. The Student's t test was used for comparison of two groups. One-way ANOVA followed by Dunnett's test was used for comparison of multiple test groups with a control group. P < 0.05 was considered significant.
Results
Cutaneous Inflammation Stimulates Monocyte/Macrophage Accumulation in Draining PLNs
To examine monocyte recruitment to reactive PLNs and to dissect the underlying molecular mechanisms we injected CFA/KLH into the flank skin, an approach previously shown to induce a strong local inflammatory response that alters the physiology of draining PLNs (23, 24). We analyzed the number of mononuclear phagocytes (CD11b+F4/80+, reference 25) in the inflamed (i.e., ipsilateral) and the contralateral subiliac LNs (also called superficial inguinal LNs) at various times thereafter (Fig. 1 A). A small, but statistically significant increase in CD11b+F4/80+ leukocytes was seen in inflamed PLNs as early as 24 h after CFA/KLH injection. A much more dramatic rise was observed on days 3 and 7. In contrast, monocyte numbers remained low in the contralateral PLNs. This indicates that intracutaneous injection of CFA/KLH did not alter systemic monocyte behavior, but exerted its effects only at the injection site and in anatomically connected PLNs. It is possible that some of the injected CFA/KLH became lymph-borne and exerted local responses in the draining PLNs. However, as will be shown below, the dramatic effect of CFA/KLH on monocyte accumulation in draining PLNs was mainly due to continuous release of secondary mediators from inflamed skin into the lymph.
Figure 1.
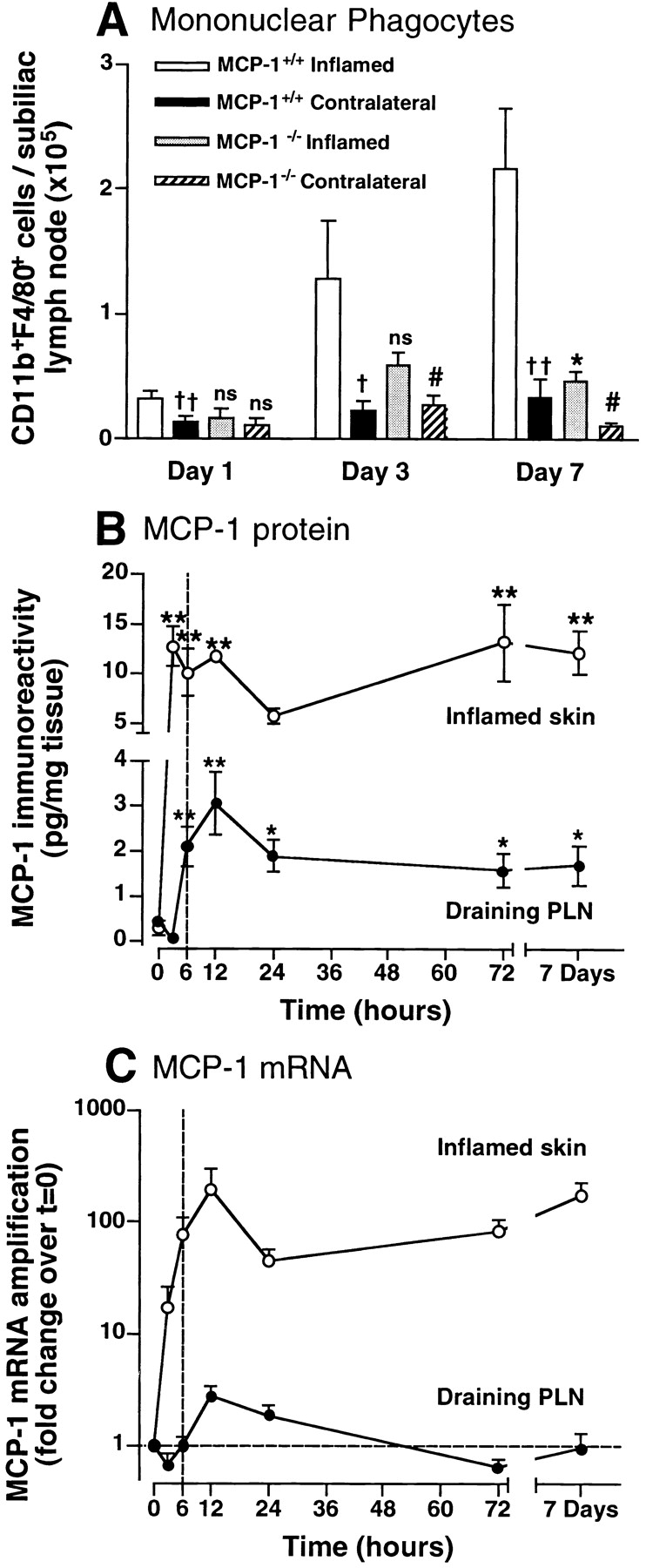
Rapid, sustained induction of MCP-1 in inflamed skin precipitates monocyte/macrophage accumulation in draining PLNs. (A) Time course of monocyte/macrophage recruitment to PLNs of wild-type and MCP-1−/− mice. CD11b+F4/80+ leukocytes were counted in subiliac PLNs at different times after intracutaneous injection of CFA/KLH into the ipsilateral flank. Monocyte numbers in the contralateral, noninflamed subiliac PLNs are shown for comparison. † P < 0.05; †† P < 0.01 vs. wild-type inflamed; *P < 0.01 MCP-1−/− vs. wild-type; # P < 0.05 vs. MCP-1−/− inflamed. n = 3–6 mice per group. (B) Time course of MCP-1 immunoreactivity in lysates of cutaneous sites of CFA/KLH injection (○) and the draining subiliac PLNs (•) measured by ELISA. *P < 0.05; **P < 0.01 vs. noninflamed tissue. n = 6. (C) Time course of MCP-1 mRNA expression in the CFA/KLH-inflamed subiliac PLN (•) and skin injection site (○). Fold change in MCP-1 mRNA levels over preinjection expression was measured in triplicate samples by real-time RT-PCR (TaqMan®) as described in Materials and Methods. Note that at 6 h (broken line in B and C), MCP-1 protein levels in draining PLNs were significantly increased above baseline, whereas mRNA levels were not. n = 3–6 mice per group. Symbols and bars represent mean ± SEM.
MCP-1 Mediates Mononuclear Phagocyte Recruitment to Inflamed PLNs
Since several studies have shown that MCP-1 (CCL2) is a key regulator of monocyte recruitment to sites of inflammation (19, 26), we studied the role of this chemokine in our model. Injection of CFA/KLH into the skin of MCP-1−/− mice resulted in markedly diminished recruitment of CD11b+F4/80+ leukocytes in draining PLNs (Fig. 1 A). Unlike in wild-type PLNs, monocyte numbers in ipsi and contralateral subiliac LNs of MCP-1−/− mice did not differ significantly at day 1. When compared with wild-type PLNs, monocyte numbers in draining MCP-1−/− PLNs were reduced on days 1 and 3 by 48 and 54%, respectively, but this difference did not reach statistical significance. At day 7, 78% fewer monocytes were found than in wild-type PLNs (P < 0.01). Similar results were obtained using mice deficient in the MCP-1 receptor, CCR2 (data not shown).
The Site of Inflammation, not the Draining PLN Is the Major Source of MCP-1
Having determined that MCP-1 is a crucial mediator of monocyte accumulation in PLNs draining inflamed skin, we set out to localize the source(s) of this chemokine. MCP-1 protein was measured by ELISA, and MCP-1 mRNA levels were quantified using real-time RT-PCR (TaqMan®; Applied Biosystems) in tissue homogenates from the injection site and the draining subiliac LN. Very low MCP-1 protein levels were detected in normal tissues (Fig. 1 B). Upon injection of CFA/KLH, MCP-1 protein levels in the skin increased to a maximum that was 43-fold higher than baseline levels as early as 3 h after injection, and they remained markedly elevated for the 7-d duration of the experiment. In contrast, in the draining PLN MCP-1 protein levels did not change significantly until 6 h after CFA/KLH injection, when a fivefold increase over preinjection levels was measured. A peak sevenfold increase was observed in draining PLNs at 12 h, and MCP-1 protein levels plateaued thereafter.
MCP-1 mRNA in the injection site increased 17-fold over preinjection levels within 3 h after CFA/KLH injection, reached a maximal 200-fold increase at 12 h, and remained elevated throughout the 1 wk experiment (Fig. 1 C). Interestingly, although MCP-1 protein was increased in the draining PLN at 6 h, MCP-1 mRNA expression did not exceed baseline levels in that organ until 12 h after CFA/KLH injection. At this time, a modest threefold increase was measured, which steadily declined during the following days. Thus, at the site of inflammation a dramatic rise in MCP-1 protein was paralleled by an increase in MCP-1 mRNA, whereas in draining PLNs the increase in MCP-1 protein preceded the induction of MCP-1 mRNA, which was subtle and transient. These results suggest that the inflamed skin was the predominant source of MCP-1, and that MCP-1 produced at the site of inflammation may be transported to the draining PLN.
MCP-1 Mediates Monocyte Homing to Inflamed PLNs
Blood-borne monocytes may access PLNs either via afferent lymph after leaving the circulation in inflamed peripheral tissues (1) or directly via HEVs (27). Since MCP-1 was expressed in both locations after CFA/KLH injection, we investigated which route was taken by monocytes in our model. To this end, we isolated PBMCs from heterozygous CX3CR1+/GFP knockin mice in which a cDNA for GFP was targeted to the CX3CR1 locus. Among CX3CR1+/GFP PBMCs, only monocytes and a small subset of NK cells express GFP (20). A further analysis of CX3CR1+/GFP PBMC (which constitute ∼1–3% of circulating leukocytes in CX3CR+/GFP mice) showed that 86% of GFP+ leukocytes expressed F4/80, and 96% were CD11bHIGH (Fig. 2 A). Two subsets of CD11bHIGHGFP+ leukocytes were apparent based on their differential expression of CCR2 and L-selectin (CD62L), which were only found on GFPMED cells, while most GFPHIGH cells were CCR2− and L-selectin−. Accordingly, only the GFPMED leukocytes migrated toward MCP-1 in chemotaxis assays (data not shown). Thus, there are at least two F4/80+CD11b+ mononuclear subsets in murine blood. Considering their surface phenotype, it seems likely that the CCR2+CD62L+F4/80+CD11b+ GFPMED population represents bona fide monocytes. Of note, these cells did not respond to the CCR7 agonist secondary lymphoid tissue chemokine (SLC; also called TCA-4, 6-C-kine, exodus 2, CCL21), which is constitutively expressed in HEVs and mediates T cell homing to PLNs (references 17 and 28, and data not shown).
Figure 2.

Short-term homing of adoptively transferred L-selectin+CCR2+ monocytes to inflamed PLNs is mediated by MCP-1. (A) Flow cytometry of PBMCs from CX3CR1+/GFP knockin mice stained for the monocyte-macrophage markers F4/80 or CD11b (Mac-1) reveals two populations of GFP+ leukocytes based upon differential expression of GFP, L-selectin (CD62L), and CCR2. The F4/80+GFP− cells were identified as eosinophils by their high side scatter (data not shown). (B) Flow cytometric analysis of leukocyte populations in homing assays. CX3CR1+/GFP PBMCs were injected intravenously into wild-type recipients 5–7 d after induction of skin inflammation by CFA/KLH injection. 4 h later, GFP+ donor populations in recipient blood and inflamed PLNs were compared with input PBMCs. To avoid counting of contaminating GFP+ NK cells, all samples were stained with anti-NK1.1. Numbers in dot plots indicate the percentage of GFP+ cells in the GFPMED and GFPHIGH gates in one representative experiment (out of 6). (C) GFPMED cells (white bars), but not GFPHIGH cells (black bars) were preferentially recruited to inflamed PLNs. Data presented as mean ± SEM, n = 6 mice per group. **P < 0.01 vs. frequency of input GFPMED CX3CR1+/GFP PBMCs in the input. ## P < 0.01 vs. input frequency of GFPHIGH CX3CR1+/GFP PBMCs. (D) Homing of all GFP+ and GFPMED CX3CR1+/GFP NK1.1− leukocytes to inflamed subiliac PLNs (black bars) and to contralateral noninflamed PLNs (white bars). The inflammation-induced increased homing of the GFPMED subset was completely blocked by anti–MCP-1. Anti–MCP-1 treatment did not affect homing of GFPHIGH cells and did not alter CX3CR1+/GFP NK1.1− cell numbers in recipient blood or spleen (data not shown). Data presented as mean ± SEM, n = 5 mice per group. *P < 0.05 control mAb inflamed versus contralateral PLNs. # P < 0.05 vs. control mAb-treated inflamed PLNs.
Initially, we set out to determine whether GFP expression is retained in monocytes/macrophages that accumulate in inflamed PLNs of CX3CR+/GFP mice. 5 d after induction of skin inflammation in three animals, 88 ± 3% of the F4/80+CD11b+ cells in inflamed PLNs expressed GFP, and nearly all of these were GFPMED (data not shown). This suggested that CX3CR1+/GFP PBMCs may be a useful tool to study whether and by what route monocytes home to inflamed PLNs. Thus, short-term homing experiments were performed by intravenous injection of CX3CR1+/GFP PBMCs into wild-type recipient mice 5–7 d after intracutaneous injection of CFA/KLH. 4 h later, a marked increase in homed NK1.1–GFP+ monocytes was observed in inflamed PLNs compared with contralateral PLNs in animals receiving a control mAb. The GFPMED monocytes constituted the majority (83%) of homed GFP+ cells in PLNs, while their mean frequency in the recipients' blood (45%) was significantly lower than in the input (62%) indicating that the CD62L+CD11b+CCR2+ subset is preferentially recruited to inflamed PLNs (Fig. 2 B and C). Consistent with the idea that CCR2/MCP-1 is required for inflammation-induced homing, concomitant injection of anti–MCP-1 significantly reduced monocyte recruitment to inflamed PLNs (Fig. 2 D). This effect of MCP-1 inhibition was limited to the GFPMED subset, whereas anti–MCP-1 did not affect the small number of GFPHIGH cells that were recovered from inflamed PLNs. Furthermore, anti–MCP-1 only affected monocyte recruitment to inflamed PLNs; it did not alter the frequency of GFP+ cells in resting PLNs, peripheral blood, or the spleen nor did it affect the overall cellularity of inflamed PLNs (data not shown).
It should be emphasized that the small number of homed monocytes recovered from recipient PLNs reflects the limited availability of donor monocytes and not inefficient trafficking (although PBMCs from as many as 50 donors were pooled for every experiment, each of the two PBMC recipients received only ∼500,000 GFP+ cells). The mean hourly rate of recruitment to inflamed PLNs was 164 GFPMED monocytes for every million GFPMED cells in the input (Table I). To assess the significance of this result relative to a cell population with known PLN-tropism, we also measured the recruitment rate of naive T cells to PLNs using the same protocol. The hourly rate of T cell recruitment to inflamed PLNs was 7.3 times higher than GFPMED monocytes. However, adoptively transferred T cells circulated in the recipients' blood stream at a 2.2-fold higher concentration than GFPMED monocytes (Table I). When the rate of recruitment of each subset was normalized to reflect equivalent blood levels, naive T cells homed 3.3 times more efficiently to inflamed PLNs than monocytes. It must be considered that every recruited monocyte has the potential to exert biological activity upon entering PLNs, whereas only a minute fraction of homed T cells is likely to encounter a cognate antigen and become activated. Thus, the observed rate of monocyte recruitment to inflamed PLNs is not only quantitatively, but probably also immunologically highly significant.
Table I.
Comparison of Monocyte and Naive T Cell Migration to Resting and Inflamed PLNs
| Cell type | Monocytes
|
Naive T cells
|
||
|---|---|---|---|---|
| PLNs | Contralateral | Inflamed | Contralateral | Inflamed |
| Total no. of cells/PLN (× 106) | 3.43 ± 1.21 | 10.58 ± 2.10 | 0.89 ± 0.17 | 10.31 ± 2.27 |
| Total no. of homed GFPMED cells/PLN | 37 ± 18 | 173 ± 48 | 1,712 ± 229 | 19,228 ± 3,837 |
| Recruitment rate (× h−1 × 10−6 cells injected) |
28.9 ± 12.9 | 164 ± 53 | 107 ± 14 | 1,202 ± 240 |
| GFPMED cells/μl blood | 4.4 ± 0.7 | 9.7 ± 2.1 | ||
Comparison of T cell and monocyte recruitment efficiency to resting and inflamed PLNs. Recipient mice with unilateral flank skin inflammation (5–7 d) were injected with 5 × 106 donor cells from either CX3CR+/GFP mice (PBMCs containing ∼10% GFP+ cells, half of which were GFPMED monocytes) or T-GFP mice (LN cells containing ∼80% GFP+ naive T cells). Peripheral blood and the subiliac LNs contralateral and ipsilateral to the inflamed tissue were harvested 4 h later and analyzed for the presence of GFP+ cells and the rate of cell recruitment was calculated. In animals that received CX3CR+/GFP cells the frequency and rate of recruitment were also determined for the GFPMED subset (shown in parenthesis). Data are shown as mean ± SEM of five experiments in each group.
The low blood concentration of adoptively transferred monocytes also makes it unlikely that many GFP+ cells were passively trapped in PLN microvessels when tissues were harvested. Based on previous measurements of microvascular dimensions and blood flow (21), and the mean passage time through PLNs of a bolus of 150-kD FITC-dextran (3.5 ± 1.2 s; n = 5), the intranodal blood volume in a normal subiliac LN can be estimated as ∼60 nl (unpublished data). Even if one assumes a substantial increase in the blood content of the enlarged PLNs that drain inflamed skin, at a mean blood concentration of 4.4 GFPMED cells/μl it is unlikely that more than two noninteracting monocytes were present in the intranodal blood stream at the time of tissue harvest. Based on these considerations, we conclude that circulating monocytes are rapidly and efficiently recruited into PLNs that drain sites of inflammation, and MCP-1 is an important mediator of this process.
Adoptively Transferred Monocytes Home to Inflamed PLNs via HEVs
To determine the route by which monocytes entered PLNs, we removed inflamed subiliac PLNs from recipients 60 min after injecting CX3CR1+/GFP PBMC either intravenously or directly into the inflamed flank skin. Cryosections were stained with anti-PNAd mAb MECA-79 to identify HEVs, and the position of GFP+ monocytes within the PLN was analyzed by confocal microscopy (Fig. 3 A). When CX3CR+/GFP cells were injected intracutaneously 1 h before tissue harvest, no GFP+ cells could be found in multiple sections of draining PLNs suggesting that, during this relatively short time interval, monocytes do not enter afferent lymphatics in substantial numbers (data not shown). In contrast, after intravenous injection, numerous GFP+ cells were found in inflamed PLNs. The vast majority of these cells was associated with HEVs, with 31% of GFP+ leukocytes found either within the lumen or the vessel wall (Fig. 3 B) and 51% located in the T cell area within 5 cell diameters (<40 μm) of the closest HEV. Only 3% of all GFP+ leukocytes were found in the subcapsular sinus, the site of entry for leukocytes in afferent lymph. This HEV-centric distribution of monocytes in PLNs was remarkably similar to that of naive T cells (unpublished data), which are known to access PLNs exclusively via HEVs (6–8). We conclude that at least within the first hour after adoptive transfer monocytes enter inflamed PLNs preferentially via HEVs and not via afferent lymph vessels.
Figure 3.


Monocytes home to inflamed PLNs via HEVs. Adoptive transfer of CX3CR1+/GFP PBMCs into wild-type recipients was performed 5 d after induction of skin inflammation as described in Fig. 2. Draining PLNs were removed after 1 h and prepared for confocal microscopy using appropriate filters for detection of GFP+ monocytes (green) and HEVs, which were visualized using anti-PNAd mAb MECA-79 and Cy5-labeled anti–rat IgM (red). (A) Representative confocal micrographs of homed GFP+ monocytes in close proximity to MECA-79+ HEVs. (B) The frequency distribution of homed GFP+ monocytes relative to MECA-79+ HEVs and the subcapsular sinus was determined. Data are expressed as mean ± SEM of the percentage of GFP+ cells in each class. n = 90 random sections of five inflamed PLNs from two independent experiments.
Intracutaneously Injected MCP-1 Is Transported to PLN HEVs
Previous studies have shown that IL-8, RANTES, MIP-1α, and SLC can accumulate in draining PLN HEVs after intracutaneous injection (17, 18). To test whether skin-derived MCP-1 has a similar fate, we injected recombinant murine MCP-1 (1 μg ≈ 100 pmol) into the flank skin of MCP-1−/ − mice. 40 min later, MCP-1 protein was detectable by ELISA in the ipsilateral subiliac LN (45.5 ± 11.5 pg/mg tissue; mean ± SEM, n = 4), with limited spillover into the contralateral subiliac LN (5.5 ± 0.3 pg/mg tissue).
While these results established that MCP-1, like other chemokines, is transported from the periphery to draining PLNs, it remained to be determined whether MCP-1 is targeted to HEVs. Two approaches were taken to address this question: first, using autoradiography and immunohistochemistry of draining PLNs, we observed that intracutaneously injected 125I-labeled human MCP-1 was highly concentrated in MECA-79+ HEV (Fig. 4 A); second, using intravital fluorescence microscopy of subiliac PLNs in mice that were injected 40 min earlier with fluorescent hMCP-1ALEXA, we detected hMCP-1ALEXA exclusively in HEVs, but not in arterioles or capillaries (Fig. 4 B and C).
Figure 4.
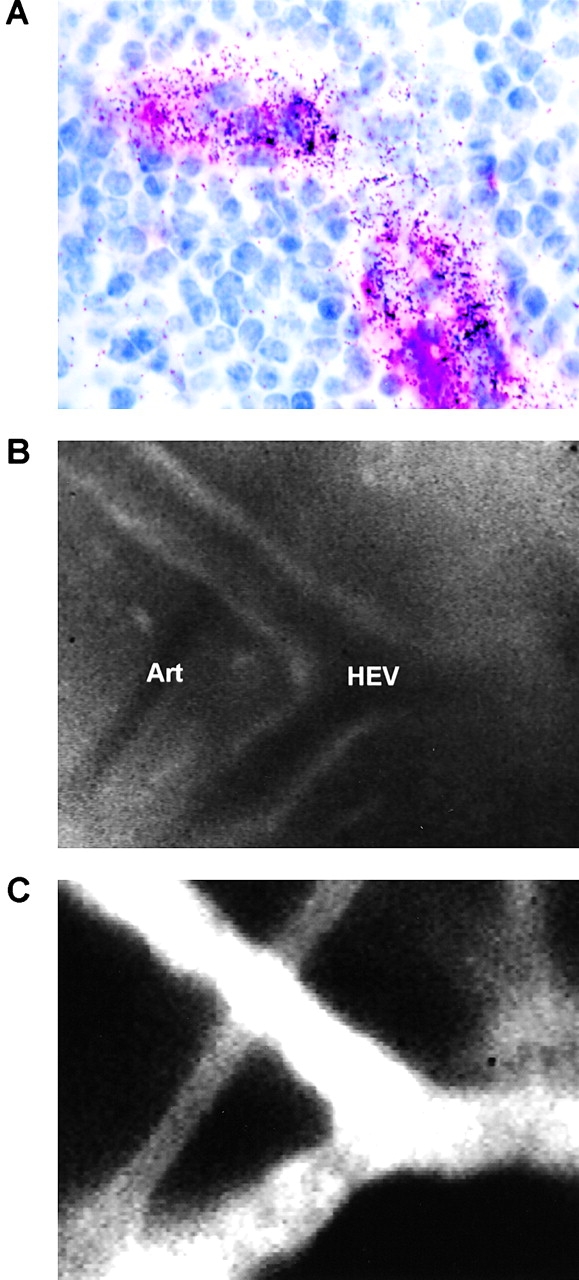
MCP-1 accumulates in HEVs after intracutaneous injection. (A) Draining PLNs were harvested from a mouse 1 h after intracutaneous injection of 125I-MCP-1. Sections were stained with anti-PNAd mAb MECA-79 (red) and hematoxylin (blue) and used to generate autoradiographs to localize radiolabeled chemokine (visible as dark grains). (B and C) Intravital micrographs of a draining PLN in an anesthetized mouse ∼1 h after intracutaneous injection of hMCP-1ALEXA. (B) Fluorescent MCP-1 was observed only in HEVs, but not in arterioles (art), whereas (C) the plasma marker FITC-dextran (150 kD) filled the entire microvasculature.
Intracutaneous Injection of MCP-1 Reconstitutes Monocyte Homing to Inflamed PLNs in MCP-1−/− Mice
To determine whether skin-derived MCP-1 is sufficient to stimulate monocyte homing to PLNs upon presentation in HEVs, CX3CR1+/GFP PBMCs were injected into MCP-1−/− mice 5 d after induction of skin inflammation in both flanks and the right lateral chest region. 10 min before adoptive transfer, the left inflamed flank skin was injected with 25 μg MCP-1, while the two inflamed contralateral regions were injected with vehicle (PBS). Homed GFP+ cells were quantitated in each PLN 90 min later. In three experiments thus performed, the frequency of GFPMED cells in the left subiliac PLN that received MCP-1 via afferent lymph was 47 and 193% higher than in the contralateral subiliac and brachial PLNs, respectively (Fig. 5 A and B). In contrast, the mean frequency of GFPHIGH cells, which do not respond to MCP-1, was similar in all PLNs. This finding and the fact that GFPHIGH cells occurred at equivalent frequency in the recipients' blood as GFPMED cells (Fig. 5 A) indicates that the observed difference in the accumulation of GFPMED cells was a direct consequence of MCP-1 injection and cannot be explained by differential hemodynamics or tissue contamination with blood.
After injection of 25 μg recombinant MCP-1 in the left flank some intracutaneously injected chemokine was found in the contralateral subiliac LN and, to a lesser degree, the brachial LN (Fig. 5 C). When tissues were harvested from MCP-1−/− mice 90 min after intracutaneous injection, optical density measurements in our ELISA assay were at least 10-fold higher in PLNs of MCP-1 recipients than in PLNs of a MCP-1−/− mouse that was not injected with chemokine (data not shown). This chemokine “spillover” is consistent with earlier experiments with SLC (17) and the experiments described previously using a lower dose (1 μg) of MCP-1. Thus, it seems plausible that some monocytes that homed to the contralateral subiliac and brachial PLNs were recruited by MCP-1 that was transported there from the injection site. Despite this, the number of monocytes in the right brachial LN was significantly smaller than in the MCP-1 draining left subiliac LN (P < 0.03), whereas intermediate monocyte numbers were present in the contralateral subiliac LN (P > 0.05 vs. right brachial and left subiliac LNs). Thus, homing of GFPMED (but not GFPHIGH) cells to recipient PLNs correlated with the presence of MCP-1 indicating that remotely-administered MCP-1 stimulates monocyte recruitment into draining PLNs of MCP-1−/− mice.
Figure 5.

Intracutaneous injection of recombinant MCP-1 into MCP-1−/− mice enhances monocyte homing in draining PLNs. MCP-1−/− mice with cutaneous inflammation in both flanks and the right lateral chest were injected intracutaneously with recombinant murine MCP-1 (25 μg) over the left flank and vehicle (50 μl PBS) elsewhere, as described in Materials and Methods. 10 min later, 1.5 × 107 CX3CR1+/GFP donor PBMCs (containing ∼106 GFP+ cells) were injected intravenously. (A) FACS® plots show the frequency of GFP+NK1.1− monocytes after 90 min in recipient blood, the PLN draining MCP-1–injected skin (subiliac LN plus MCP-1), the contralateral subiliac LNs (subiliac LN plus vehicle), and the inflamed brachial LN (brachial LN plus vehicle) from the same mouse. 6 × 105 events are plotted in each histogram. (B) GFPMED, but not GFPHIGH CX3CR+/GFP cells accumulate preferentially in the subiliac LN that drained the MCP-1 injection site. Mean ± SEM of three separate experiments are shown. (C) MCP-1 protein concentration in inflamed left and right subiliac LNs and right brachial LNs 90 min after intracutaneous injection of recombinant murine MCP-1 or vehicle. Each curve reflects MCP-1 concentration in three PLNs measured in the same mouse.
MCP-1 Induces Arrest of Rolling Monocytes in Inflamed PLN HEVs
As shown above, intracutaneously injected MCP-1 is quickly translocated to HEVs, which we identified as a port of MCP-1–dependent monocyte entry from the blood. Thus, it seemed likely that MCP-1 acted directly on monocytes within HEVs. To test this hypothesis, we performed intravital microscopy in subiliac PLNs (21). We used the murine monocytoid WEHI78/24 cell line, which has previously been shown to adhere to inflamed but not resting PLN HEVs in a Stamper–Woodruff assay (9). WEHI78/24 cells are L-selectin+ and respond avidly to MCP-1 (EC50 < 1 nM), but not to SLC in chemotaxis assays (data not shown).
Rolling fractions of WEHI78/24 monocytes were similar in inflamed and resting PLNs of both wild-type and MCP-1− /− mice (Fig. 6 A and B). However, rolling WEHI78/24 cells arrested significantly more frequently in inflamed PLNs of wild-type mice compared with noninflamed wild-type PLNs or inflamed PLNs in MCP-1−/− mice (Fig. 6 C and D). Both rolling and sticking was more frequent in paracortical high order branches of the venular tree (Fig. 6 A and C), as shown previously for lymphocytes (21, 29). Importantly, injection of MCP-1 into inflamed skin of MCP-1−/− mice restored the ability of rolling monocytes to stick in high order HEVs of the draining PLN.
Figure 6.

MCP-1 triggers the arrest of rolling monocytes in PLN HEVs. Adhesive behavior of WEHI78/24 cells in venular trees of subiliac PLNs was analyzed by intravital microscopy. Experiments were performed in wild-type mice draining either normal (white bars) or inflamed skin (black bars) and in inflamed PLNs of MCP-1−/− mice that were either untreated (hatched bars) or injected intracutaneously with MCP-1 (cross-hatched bars). Rolling fractions (percentage of total cells passing through a given venule which interact with the vessel wall) and sticking fractions (percentage of rolling cells which arrest for ≥30 s) are shown in each branching order of the venular tree (A and C) and as the mean for all venules (B and D). Mean ± SEM; total number of animals/venules analyzed are shown in B and D. (E) Desensitization of WEHI78/24 cells to MCP-1, but not MIP-1α inhibits arrest in inflamed PLN HEVs. WEHI78/24 cells were incubated for 40 min with MIP-1α (500 nM) or MCP-1 (500 nM). Desensitized WEHI78/24 cells were injected into MCP-1−/− mice, which had been pretreated 7 d and 40 min earlier with intracutaneous injections of CFA/KLH and 100 pmol MCP-1, respectively. Mean rolling and sticking fractions in 21 HEVs (13 order IV and 8 order V) in three mice are shown for each group. **P < 0.01.
While these findings are consistent with a scenario where luminal MCP-1 in HEVs triggers integrin-mediated sticking by signaling through CCR2 on rolling monocytes, these data did not exclude the possibility that MCP-1 acted indirectly by triggering the release of other integrin activating mediator(s). Therefore, we investigated the effect of CCR2 inhibition on WEHI78/24 cell behavior in inflamed PLNs. Since neutralizing reagents to murine CCR2 were not available, we incubated WEHI78/24 cells for 40 min with 500 nM MCP-1 because chemokine receptors, including CCR2, can be effectively desensitized by prolonged exposure to high concentrations of their specific ligand (references 17 and 30, and data not shown). When MCP-1 desensitized WEHI78/24 cells were analyzed in inflamed PLNs of MCP-1−/− mice injected intracutaneously with MCP-1, they rolled normally, but displayed a significantly reduced ability to arrest (Fig. 6 E). In contrast, rolling and sticking were not affected by desensitization to MIP-1α, an agonist on CCR1 and CCR5 to which WEHI78/24 cells are highly responsive in chemotaxis assays (data not shown). These results strongly support the concept that MCP-1 acts directly on rolling monocytes to stimulate arrest, most likely by signaling through CCR2.
Discussion
Here we show that inflamed cutaneous tissues discharge MCP-1 into the lymph, which is crucial for the recruitment of monocytes/macrophages in HEVs of draining PLNs. Using an experimental model of CFA/KLH-induced inflammation, we found that long-term accumulation of endogenous mononuclear phagocytes and short-term homing of adoptively transferred blood monocytes in draining PLNs was impaired in MCP-1−/− mice and in wild-type mice treated with anti–MCP-1. MCP-1 transcription occurred primarily in inflamed skin from where MCP-1 protein was rapidly transported to draining PLNs and accumulated in HEVs. Upon presentation in the HEV lumen, skin-derived MCP-1 triggered the arrest of rolling monocytes. These observations establish a novel mode of leukocyte trafficking by “remote control” where chemokines exert effects in an organ that is distant to the tissue of their production.
Data from our homing experiments with CX3CR+/GFP PBMCs suggest that monocyte recruitment via HEVs was remarkably efficient (Table I): the concentration of the GFPMED monocyte population in recipient blood was ∼4.4 cells/μl; total blood flow in subiliac LNs has been estimated as ≤1 μl/min (21); on average, 173 homed GFPMED cells were recovered from inflamed PLNs 4 h after transfer (Fig. 2 C); this suggests that about one-sixth of all monocytes that entered PLN HEVs were successfully recruited. If one assumes that murine blood contains ∼100 endogenous monocytes per μl (31), this rate of recruitment would result in a daily influx of ∼23,000 cells into each inflamed PLN. Considering the number of CD11b+F4/80+ cells recovered from inflamed subiliac LNs (Fig. 1 A), it seems likely that a substantial fraction of these cells entered the PLNs via HEVs, while the remainder may have arrived via afferent lymphatics and/or proliferated locally.
This mechanism may have evolved to support two major physiologic functions of PLNs: (a) the collection and presentation of peripheral antigens to lymphocytes; and (b) the filtration and clearance of lymph fluid from peripheral tissues. While the former has long been the subject of intense study, the latter has received relatively little attention. However, some pathogens that penetrate the skin can exploit the lymphatics as a major conduit for systemic dissemination. A point in case is Yersinia pestis, arguably the most devastating bacterial pathogen in human history (32). During bubonic plague, Y. pestis is deposited in the skin, typically by a flea bite, enters afferent lymph vessels and proliferates rapidly in draining PLNs. Interestingly, Y. pestis infection is associated with minimal recruitment of myeloid phagocytes in afflicted PLNs, whereas less virulent Yersinia strains elicit a more potent inflammatory response (33). In addition to other documented effects of Y. pestis on phagocyte function (34), it is tempting to speculate that the unprecedented success of this mammalian pathogen was due in part to interference with phagocyte recruitment to PLNs possibly by subverting the production or transportation of chemokines such as MCP-1 and/or by disabling phagocyte responsiveness to them.
While neutralization of microorganisms that enter PLNs from the periphery is a likely purpose of newly recruited monocytes, their ultimate fate and function are undetermined. Monocytes may further differentiate depending on the cytokine milieu (which may also be modulated by lymph-borne mediators). After the key diapedesis step (35), exposure to IL-4, GM-CSF, and other signals drives DC differentiation (3, 36), whereas IL-6 and M-CSF stimulate macrophage generation (37). Macrophages fulfill multiple functions in PLNs: they scavenge particulates from afferent lymph (38); maintain the phenotype of HEV (2, 39); produce angiogenic factors (40); and generate lymphotropic cytokines and chemokines. Indeed, it has been proposed that impaired monocyte/macrophage migration is responsible for the reduced Th1 cytokine production in PLNs of immunized CCR2−/− mice (41, 42).
The possibility that newly recruited monocytes in PLNs differentiate into DCs is particularly appealing. Most previous work has focused on afferent lymph as the primary route of DCs entry into PLNs (1, 3–5). Lymph-borne DCs that enter PLNs from inflamed tissues acquire a mature phenotype en route and lose their endocytic ability, preventing them from acquiring more antigen should they encounter it in draining PLNs (3). We provide evidence that monocytes, the precursors of myeloid DCs, can enter PLNs directly from the blood. These cells should be fully capable of antigen acquisition, processing, and differentiation into DCs given the appropriate cytokine stimuli.
Hendricks et. al. have reported that ligation of afferent lymphatic vessels blocks macrophage accumulation in inflamed PLNs (2). This was assumed to be caused by obstructed macrophage traffic through afferent lymphatics. Our homing and confocal microscopy experiments show that monocytes can be directly recruited via HEVs. Remotely administered MCP-1 stimulated circulating monocyte homing to draining PLNs of MCP-1−/− mice. Thus, in addition to blocking macrophage transport from peripheral tissues, ligation of afferent lymphatics might indirectly block monocyte recruitment mediated by remotely generated chemoattractants in HEVs. Occlusion of afferent lymphatics also leads to loss of luminal expression of the L-selectin ligand, PNAd (39), which might further disrupt monocyte recruitment via HEVs.
PNAd is highly expressed in resting and inflamed PLN HEVs (43) and mediates tethering and rolling of L-selectin+ leukocytes (21, 29). L-selectin binding to PNAd is the essential first step for lymphocyte homing to PLNs (44, 45). This pathway mediates also monocyte adhesion to PLN HEVs in vitro (9). Accordingly, in our homing experiments with CX3CR1+/GFP PBMCs, only the L-selectin+ GFPMED subset was efficiently recruited to PLNs, consistent with a key role for L-selectin in this setting. The requirement for L-selectin can explain why effector T cells, which are typically L-selectin−, home poorly to PLNs, even though a sizeable fraction of these cells can respond to MCP-1 and other inflammatory chemokines that may be presented in inflamed HEVs (7, 46, 47). However, while leukocyte adhesion in PLN HEVs must be initiated by L-selectin, this pathway mediates only rolling which must be followed by an activating stimulus that triggers integrin mediated arrest. Rolling T cells respond to the CCR7 agonist SLC in PLN HEVs to engage the β2 integrin LFA-1 (17), but blood monocytes do not respond to CCR7 agonists (48, 49). Thus, another activating stimulus is necessary for monocyte arrest in HEVs.
Our intravital microscopy analysis has uncovered several lines of evidence that MCP-1 provides this stimulus: (a) both monocyte sticking and the concentration of MCP-1 were increased in inflamed versus resting PLNs; (b) arrest of rolling monocytes was impaired in inflamed PLNs of MCP-1−/− mice; (c) sticking in MCP-1−/− PLNs was restored after intracutaneous injection of MCP-1; and (d) monocyte desensitization to MCP-1–inhibited sticking. MCP-1 probably exerted its effects via CCR2 signaling leading to integrin activation. In vitro studies have shown that MCP-1 can activate the α4β1 integrin on human monocytes and T cells (50). WEHI78/24 cells adhered to HEVs, in part, via LFA-1 and Mac-1 in tissue sections of inflamed, but not resting PLNs (9). On the other hand, inhibition of α4 and β2 integrins had little effect on MCP-1–mediated monocyte arrest on virally transduced endothelium in a flow chamber (11), and MCP-1 stimulated T cell adhesion only to extracellular matrix molecules, but not to the purified integrin ligands intracellular adhesion molecule 1 or vascular cell adhesion molecule 1 (51). Thus, it remains to be determined which adhesion molecules are responsible for MCP-1–induced monocyte arrest in HEVs.
Even though the participation of MCP-1 in the three-step adhesion cascade in HEVs is clearly a major mechanism for monocyte recruitment to inflamed PLNs, our data do not rule out other functions for MCP-1, e.g., during transendothelial or interstitial migration. It is also possible that additional endothelial conditioning by inflammatory signals is necessary for monocyte recruitment that may be independent of or indirectly induced by the action of MCP-1. Furthermore, although nearly all GFP+ cells in frozen sections were found in the vicinity of HEVs in our confocal microscopy analysis (Fig. 3), this does not exclude that MCP-1 is also involved in monocyte recruitment into inflamed skin from where cells may reach draining PLNs after some delay. Whatever additional effects MCP-1 may have, this study demonstrates that changes in the local chemokine milieu in inflamed peripheral tissues are readily projected onto the microvasculature of draining PLNs. By using autoradiography and intravital microscopy, we observed that radiolabeled and fluorescent MCP-1 injected into the skin was rapidly and selectively transported to HEVs in draining PLNs, with very little protein detected in other PLNs. Even though our ELISA data of tissue lysates showed that the concentration of MCP-1 protein in draining PLNs was lower than in inflamed skin, this difference may not be reflected at the level of the endothelium because MCP-1 was highly concentrated in HEVs, with minimal signal in extravascular sites.
It has recently been shown how lymph-borne chemokines gain access to HEVs in draining PLNs (18). Upon entering the subcapsular sinus, chemokines are channeled along collagen fibers in the FRC network, which forms a conduit from the subcapsular sinus to HEVs (52). To induce the arrest of rolling monocytes, MCP-1 must reach the luminal surface of HEVs. This is possibly achieved by transcytosis as described in skin postcapillary venules for IL-8 and RANTES (53). There also appears to be a physical barrier preventing lymph-borne chemokines from diffusing into the T and B cell area, thus spatially restricting chemokine action and preventing disruption of the PLN microenvironment (18). The architecture of the FRC conduit and the physical limitations on chemokine diffusion can explain how intracutaneously injected chemokines are selectively targeted to HEVs, and not to arterioles or capillaries (reference 17, and this study). This anatomical arrangement might also channel immunogens, microbial products, and cytokines to HEVs. The resulting juxtaposition of emigrating monocytes, soluble antigens, and lymph-borne cytokines would provide a mechanism by which antigen can be rapidly acquired and processed.
In conclusion, we show that during cutaneous inflammation monocyte homing into draining PLNs is mediated by skin-derived MCP-1. This effect may have evolved as a host response to cutaneous infection to allow rapid stocking of draining PLNs with leukocytes innately possessing a high phagocytic activity and plasticity for further differentiation. As macrophages, monocytes limit the spread of infections, and as DCs they have the ability to process and present antigen to stimulate a vigorous adaptive immune response. To what extent homed monocytes assume such vital functions in inflamed PLNs will require further investigation. The present data suggest that inflamed peripheral tissues can remotely control the recruitment of monocytes and possibly their further differentiation in draining PLNs by releasing chemokines into the lymph. We propose that similar mechanisms may also shape the composition of other leukocyte populations that get recruited to PLNs. Thus, afferent lymphatics are not only a pipeline for drainage of interstitial fluid and the delivery of antigen from the periphery, but also a one-way communication link by which peripheral tissues may shape immune responses in draining PLNs.
Acknowledgments
We thank Zhao Min, Thomas Felbinger, Marion Zsak, and Erni Schwarzinger for technical assistance, Rodrigo Bravo for the gift of anti–murine CCR2 Ab, Manfred Auer for Alexa-MCP-1, and Joe Moore for editorial assistance.
R.T. Palframan was supported by the Wellcome Trust. S. Jung was supported by a Special Fellowship of the Leukemia & Lymphoma Society of America. U.H. von Andrian was supported by NIH grants HL54936, HL62524, HL56949, and AR42689.
The data presented in this manuscript are original and have not been submitted for publication elsewhere. This study complies with National Institutes of Health (NIH) guidelines for the humane use of laboratory animals.
Footnotes
Abbreviations used in this paper: CFA, complete Freund's adjuvant; DC, dendritic cell; FRC, fibroblastic reticular cell; GFP, green fluorescent protein; HEV, high endothelial venule; hMCP-1ALEXA, Alexa-conjugated human MCP-1; KLH, keyhole limpet hemocyanin; MCP, monocyte chemotactic protein; PLN, peripheral LN; SLC, secondary lymphoid tissue chemokine.
References
- 1.Randolph, G.J., K. Inaba, D.F. Robbiani, R.M. Steinman, and W.A. Muller. 1999. Differentiation of phagocytic monocytes into lymph node dendritic cells in vivo. Immunity. 11:753–761. [DOI] [PubMed] [Google Scholar]
- 2.Hendriks, H.R., I.L. Eestermans, and E.C. Hoefsmit. 1980. Depletion of macrophages and disappearance of postcapillary high endothelial venules in lymph nodes deprived of afferent lymphatic vessels. Cell Tissue Res. 211:375–389. [DOI] [PubMed] [Google Scholar]
- 3.Banchereau, J., and R.M. Steinman. 1998. Dendritic cells and the control of immunity. Nature. 392:245–252. [DOI] [PubMed] [Google Scholar]
- 4.Gunn, M.D., S. Kyuwa, C. Tam, T. Kakiuchi, A. Matsuzawa, L.T. Williams, and H. Nakano. 1999. Mice lacking expression of secondary lymphoid organ chemokine have defects in lymphocyte homing and dendritic cell localization. J. Exp. Med. 189:451–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Robbiani, D.F., R.A. Finch, D. Jager, W.A. Muller, A.C. Sartorelli, and G.J. Randolph. 2000. The leukotriene C(4) transporter MRP1 regulates CCL19 (MIP-3β, ELC)-dependent mobilization of dendritic cells to lymph nodes. Cell. 103:757–768. [DOI] [PubMed] [Google Scholar]
- 6.Butcher, E.C., and L.J. Picker. 1996. Lymphocyte homing and homeostasis. Science. 272:60–66. [DOI] [PubMed] [Google Scholar]
- 7.von Andrian, U.H., and C.R. Mackay. 2000. T-cell function and migration. Two sides of the same coin. N. Engl. J. Med. 343:1020–1034. [DOI] [PubMed] [Google Scholar]
- 8.Marchesi, V.T., and J.L. Gowans. 1964. The migration of lymphocytes through the endothelium of venules in lymph nodes: an electron microscope study. Proc. R. Soc. B. 159:283–290. [DOI] [PubMed] [Google Scholar]
- 9.McEvoy, L.M., M.A. Jutila, P.S. Tsao, J.P. Cooke, and E.C. Butcher. 1997. Anti-CD43 inhibits monocyte-endothelial adhesion in inflammation and atherogenesis. Blood. 90:3587–3594. [PubMed] [Google Scholar]
- 10.Gu, L., S.C. Tseng, and B.J. Rollins. 1999. Monocyte chemoattractant protein-1. Chem. Immunol. 72:7–29. [DOI] [PubMed] [Google Scholar]
- 11.Gerszten, R.E., E.A. Garcia-Zepeda, Y.C. Lim, M. Yoshida, H.A. Ding, M.A. Gimbrone, Jr., A.D. Luster, F.W. Luscinskas, and A. Rosenzweig. 1999. MCP-1 and IL-8 trigger firm adhesion of monocytes to vascular endothelium under flow conditions. Nature. 398:718–723. [DOI] [PubMed] [Google Scholar]
- 12.Randolph, G.J., and M.B. Furie. 1995. A soluble gradient of endogenous monocyte chemoattractant protein-1 promotes the transendothelial migration of monocytes in vitro. J. Immunol. 155:3610–3618. [PubMed] [Google Scholar]
- 13.Bendeck, M.P. 2000. Mining the myocardium with macrophage drills: a novel mechanism for revascularization. Circ. Res. 87:341–343. [DOI] [PubMed] [Google Scholar]
- 14.Salcedo, R., M.L. Ponce, H.A. Young, K. Wasserman, J.M. Ward, H.K. Kleinman, J.J. Oppenheim, and W.J. Murphy. 2000. Human endothelial cells express CCR2 and respond to MCP-1: direct role of MCP-1 in angiogenesis and tumor progression. Blood. 96:34–40. [PubMed] [Google Scholar]
- 15.Gu, L., S. Tseng, R.M. Horner, C. Tam, M. Loda, and B.J. Rollins. 2000. Control of TH2 polarization by the chemokine monocyte chemoattractant protein-1. Nature. 404:407–411. [DOI] [PubMed] [Google Scholar]
- 16.Frade, J.M.R., M. Llorente, M. Mellado, J. Alcami, J.C. Gutierrez-Ramos, A. Zaballos, G. Real, and A.C. Martinez. 1997. The amino-terminal domain of the CCR2 chemokine receptor acts as coreceptor for HIV-1 infection. J. Clin. Invest. 100:497–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stein, J.V., A. Rot, Y. Luo, M. Narasimhaswamy, H. Nakano, M.D. Gunn, A. Matsuzawa, E.J. Quackenbush, M.E. Dorf, and U.H. von Andrian. 2000. The CC chemokine thymus-derived chemotactic agent 4 (TCA-4, secondary lymphoid tissue chemokine, 6Ckine, exodus-2) triggers lymphocyte function-associated antigen 1-mediated arrest of rolling T lymphocytes in peripheral lymph node high endothelial venules. J. Exp. Med. 191:61–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gretz, J.E., C.C. Norbury, A.O. Anderson, A.E. Proudfoot, and S. Shaw. 2000. Lymph-borne chemokines and other low molecular weight molecules reach high endothelial venules via specialized conduits while a functional barrier limits access to the lymphocyte microenvironments in lymph node cortex. J. Exp. Med. 192:1425–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu, B., B.J. Rutledge, L. Gu, J. Fiorillo, N.W. Lukacs, S.L. Kunkel, R. North, C. Gerard, and B.J. Rollins. 1998. Abnormalities in monocyte recruitment and cytokine expression in monocyte chemoattractant protein 1-deficient mice. J. Exp. Med. 187:601–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jung, S., J. Aliberti, P. Graemmel, M.J. Sunshine, G.W. Kreutzberg, A. Sher, and D.R. Littman. 2000. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol. Cell. Biol. 20:4106–4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.von Andrian, U.H. 1996. Intravital microscopy of the peripheral lymph node microcirculation in mice. Microcirc. 3:287–300. [DOI] [PubMed] [Google Scholar]
- 22.Hub, E., and A. Rot. 1996. Methods for study of chemokine receptors in the tissues. Methods. 10:119–125. [DOI] [PubMed] [Google Scholar]
- 23.Bogen, S.A., D.S. Weinberg, and A.K. Abbas. 1991. Histologic analysis of T lymphocyte activation in reactive lymph nodes. J. Immunol. 147:1537–1541. [PubMed] [Google Scholar]
- 24.Hillman, B.J., P.G. Herman, and W.M. Baldwin. 1979. Microvascular alterations in the lymph node during the BCG-induced immune response. Lymphology. 12:241–246. [PubMed] [Google Scholar]
- 25.Hume, D.A., A.P. Robinson, G.G. MacPherson, and S. Gordon. 1983. The mononuclear phagocyte system of the mouse defined by immunohistochemical localization of antigen F4/80. Relationship between macrophages, Langerhans cells, reticular cells, and dendritic cells in lymphoid and hematopoietic organs. J. Exp. Med. 158:1522–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ajuebor, M.N., R.J. Flower, R. Hannon, M. Christie, K. Bowers, A. Verity, and M. Perretti. 1998. Endogenous monocyte chemoattractant protein-1 recruits monocytes in the zymosan peritonitis model. J. Leukoc. Biol. 63:108–116. [DOI] [PubMed] [Google Scholar]
- 27.Marchesi, V.T. 1961. The site of leucocyte emigration during inflammation. Qu. J. Exp. Physiology. 46:115–133. [DOI] [PubMed] [Google Scholar]
- 28.Gunn, M.D., K. Tangemann, C. Tam, J.G. Cyster, S.D. Rosen, and L.T. Williams. 1998. A chemokine expressed in lymphoid high endothelial venules promotes the adhesion and chemotaxis of naive T lymphocytes. Proc. Natl. Acad. Sci. USA. 95:258–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Warnock, R.A., S. Askari, E.C. Butcher, and U.H. von Andrian. 1998. Molecular mechanisms of lymphocyte homing to peripheral lymph nodes. J. Exp. Med. 187:205–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aragay, A.M., M. Mellado, J.M. Frade, A.M. Martin, M.C. Jimenez-Sainz, A.C. Martinez, and F. Mayor, Jr. 1998. Monocyte chemoattractant protein-1-induced CCR2B receptor desensitization mediated by the G protein-coupled receptor kinase 2. Proc. Natl. Acad. Sci. USA. 95:2985–2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Furth, R., and W. Sluiter. 1986. Distribution of blood monocytes between a marginating and a circulating pool. J. Exp. Med. 163:474–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Titball, R.W., and S.E. Leary. 1998. Plague. Br. Med. Bull. 54:625–633. [DOI] [PubMed] [Google Scholar]
- 33.Smith, J., and B. Reisner. 1997. Plague. Pathology of Infectious Diseases. D.H. Connor, editor. Appleton and Lange, Stamford, CT. 1:729-738.
- 34.Cornelis, G.R. 2000. Molecular and cell biology aspects of plague. Proc. Natl. Acad. Sci. USA. 97:8778–8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Randolph, G.J., S. Beaulieu, S. Lebecque, R.M. Steinman, and W.A. Muller. 1998. Differentiation of monocytes into dendritic cells in a model of transendothelial trafficking. Science. 2828:480–483. [DOI] [PubMed] [Google Scholar]
- 36.Sallusto, F., and A. Lanzavecchia. 1994. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by GM-CSF plus IL-4 and downregulated by TNF-α. J. Exp. Med. 179:1109–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mitani, H., N. Katayama, H. Araki, K. Ohishi, K. Kobayashi, H. Suzuki, K. Nishii, M. Masuya, K. Yasukawa, N. Minami, and H. Shiku. 2000. Activity of interleukin 6 in the differentiation of monocytes to macrophages and dendritic cells. Br. J. Haematol. 109:288–295. [DOI] [PubMed] [Google Scholar]
- 38.Delemarre, F.G., N. Kors, and N. van Rooijen. 1990. Elimination of spleen and of lymph node macrophages and its difference in the effect on the immune response to particulate antigens. Immunobiology. 182:70–78. [DOI] [PubMed] [Google Scholar]
- 39.Mebius, R.E., P.R. Streeter, J. Breve, A.M. Duijvestijn, and G. Kraal. 1991. The influence of afferent lymphatic vessel interruption on vascular addressin expression. J. Cell Biol. 115:85–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sunderkotter, C., K. Steinbrink, M. Goebeler, R. Bhardwaj, and C. Sorg. 1994. Macrophages and angiogenesis. J. Leukoc. Biol. 55:410–422. [DOI] [PubMed] [Google Scholar]
- 41.Boring, L., J. Gosling, S.W. Chensue, S.L. Kunkel, R.V. Farese, Jr., H.E. Broxmeyer, and I.F. Charo. 1997. Impaired monocyte migration and reduced type 1 (Th1) cytokine responses in C-C chemokine receptor 2 knockout mice. J. Clin. Invest. 100:2552–2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peters, W., M. Dupuis, and I.F. Charo. 2000. A mechanism for the impaired IFN-γ production in C-C chemokine receptor 2 (CCR2) knockout mice: role of CCR2 in linking the innate and adaptive immune responses. J. Immunol. 165:7072–7077. [DOI] [PubMed] [Google Scholar]
- 43.Streeter, P.R., B.T.N. Rouse, and E.C. Butcher. 1988. Immunohistologic and functional characterization of a vascular addressin involved in lymphocyte homing into peripheral lymph nodes. J. Cell Biol. 107:1853–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gallatin, W.M., I.L. Weissman, and E.C. Butcher. 1983. A cell-surface molecule involved in organ-specific homing of lymphocytes. Nature. 304:30–34. [DOI] [PubMed] [Google Scholar]
- 45.Arbones, M.L., D.C. Ord, K. Ley, H. Ratech, C. Maynard-Curry, G. Otten, D.J. Capon, and T.F. Tedder. 1994. Lymphocyte homing and leukocyte rolling and migration are impaired in L-selectin-deficient mice. Immunity. 1:247–260. [DOI] [PubMed] [Google Scholar]
- 46.Butcher, E.C. 1991. Leukocyte-endothelial cell recognition: three (or more) steps to specificity and diversity. Cell. 67:1033–1036. [DOI] [PubMed] [Google Scholar]
- 47.Springer, T.A. 1994. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multi-step paradigm. Cell. 76:301–314. [DOI] [PubMed] [Google Scholar]
- 48.Kim, C.H., L.M. Pelus, J.R. White, E. Applebaum, K. Johanson, and H.E. Broxmeyer. 1998. CKβ-11/macrophage inflammatory protein-3β/EBI1-ligand chemokine is an efficacious chemoattractant for T and B cells. J. Immunol. 160:2418–2424. [PubMed] [Google Scholar]
- 49.Nagira, M., T. Imai, K. Hieshima, J. Kusuda, M. Ridanpaa, S. Takagi, M. Nishimura, M. Kakizaki, H. Nomiyama, and O. Yoshie. 1997. Molecular cloning of a novel human CC chemokine secondary lymphoid-tissue chemokine that is a potent chemoattractant for lymphocytes and mapped to chromosome 9p13. J. Biol. Chem. 272:19518–19524. [DOI] [PubMed] [Google Scholar]
- 50.Weber, C., R. Alon, B. Moser, and T.A. Springer. 1996. Sequential regulation of α4β1 and α5β1 integrin avidity by CC chemokines in monocytes: implications for transendothelial chemotaxis. J. Cell Biol. 134:1063–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Carr, M.W., R. Alon, and T.A. Springer. 1996. The C-C chemokine MCP-1 modulates the avidity of β1 but not β2 integrins on T lymphocytes. Immunity. 4:179–187. [DOI] [PubMed] [Google Scholar]
- 52.Gretz, J.E., A.O. Anderson, and S. Shaw. 1997. Cords, channels, corridors and conduits: critical artichitectural elements facilitating cell interactions in the lymph node cortex. Immunol. Rev. 156:11–24. [DOI] [PubMed] [Google Scholar]
- 53.Middleton, J., S. Neil, J. Wintle, I. Clark-Lewis, H. Moore, C. Lam, M. Auer, E. Hub, and A. Rot. 1997. Transcytosis and surface presentation of IL-8 by venular endothelial cells. Cell. 91:1001–1011. [DOI] [PubMed] [Google Scholar]