

Abstract
Stem cells are crucial for the formation and maintenance of tissues and organs. The role of stem cells in the pathogenesis of mosaic skin disorders remains unclear. To study the molecular and cellular basis of mosaicism, we established a mouse model for the autosomal-dominant skin blistering disorder, epidermolytic hyperkeratosis (MIM 113800), which is caused by mutations in either keratin K1 or K10. This genetic model allows activation of a somatic K10 mutation in epidermal stem cells in a spatially and temporally controlled manner using an inducible Cre recombinase. Our results indicate that lack of selective pressure against certain mutations in epidermal stem cells leads to mosaic phenotypes. This finding has important implications for the development of new strategies for somatic gene therapy of dominant genodermatoses.
Keywords: epidermolytic hyperkeratosis, keratins, Cre recombinase, mosaic skin disease, gene therapy
Introduction
Mosaicism can be defined as a condition in which at least two genetically distinct cell populations from the same differentiation lineage exist within the same tissue. This condition is usually caused by postzygotic mutations. The genes that are mutated and the time point at which these mutations occur determine the type of tissue that will be affected and the extent of involvement. In skin, several mosaic disorders have been described (Itin and Buechner 1999) and are characterized by “islands” of abnormal skin cells that presumably correspond to a clone of “mutated” cells that arose from a single cell in which the mutation initially occurred (Happle 1993). The mechanisms that lead to clinical mosaicism are poorly understood. It remains unclear how certain genetic defects lead to a mosaic disease, whereas others never become apparent in the same tissue.
A candidate disease to analyze the mechanisms that determine how genetic mosaicism leads to a clinical phenotype is the keratin disorder epidermolytic hyperkeratosis (EHK), which is characterized by a generalized and a mosaic form (Paller et al. 1994). Keratins are members of the intermediate filament (IF) gene family and are mainly expressed in epithelial cells. They form a cytoskeletal scaffold in keratinocytes of the epidermis to provide stability, thus contributing to the mechanical integrity of the epidermis (Franke et al. 1981). Keratins are grouped into two classes, the acidic type I (K9-K20) and the neutral-basic type II (K1-K8) class (Moll et al. 1982). Keratins are obligate heteropolymers that require equimolar amounts of each type to assemble into IFs (Steinert and Liem 1990). The expression of keratin genes is highly regulated. The pair of keratins that is expressed is usually specific for the cell type and state of differentiation (Franke et al. 1981; Roop 1995). In the epidermis, IF assembly is a dynamic process where newly synthesized filaments are integrated into the existing network. In the basal cell compartment, keratins K5 and K14 form the IF cytoskeleton and defects in either keratin are associated with the autosomal-dominant blistering disease, epidermolysis bullosa simplex (EBS; Bonifas et al. 1991; Coulombe et al. 1991; Rothnagel and Roop 1995). Keratins K1 and K10 are expressed in keratinocytes that are committed to terminal differentiation and point mutations in the corresponding genes have been identified in EHK (Cheng et al. 1992; Chipev et al. 1992; Rothnagel et al. 1992). The majority of mutations in EHK are located in the same codon, affecting an evolutionary highly conserved arginine residue. This site, codon 156 of the keratin 10 gene, has consequently been found to be a “hot spot” for mutations due to CpG methylation and deamination (Rothnagel et al. 1993).
The clinical course of EHK is severe and is characterized by blistering and erythroderma (redness of the skin) at birth, and development of hyperkeratoses (thickening of the uppermost layer of the epidermis) later in life, predominantly over the flexural folds and in areas exposed to mechanical stress (Digiovanna 1999). Histologically, perinuclear vacuolar degeneration, lysis of the suprabasal keratinocytes and a thickened stratum corneum are characteristic findings (Bale et al. 1993). At the ultrastructural level, clumps of keratin filaments are seen in a perinuclear distribution in suprabasal keratinocytes; these clumps have been shown to contain keratins K1 and K10 (Ishida-Yamamoto et al. 1992).
Whereas mosaic forms have been reported for EHK where stripes of affected and unaffected skin alternate (Paller et al. 1994), this has never been reported for EBS. Why do these two keratin disorders behave so differently?
To investigate this question and to study the molecular and cellular basis of mosaic diseases, we established a genetic mouse model for EHK that allows focal activation of a mutant K10 allele using a ligand-inducible Cre recombinase (Kellendonk et al. 1996). Induction of the EHK lesions in a circumscribed area of the skin resulted in a phenotype characteristic of mosaic diseases, with patches of affected and unaffected skin. Analysis of dissected keratinocytes from lesional areas demonstrated activation of the mutant allele in epidermal stem cells. We demonstrate that mutant EHK stem cells and wild-type stem cells can coexist in the basal layer of the epidermis, which may explain the persistent “islands” of phenotypic skin. Furthermore, we show that the severity of the clinical phenotype in this mouse model correlates with the expression level of the mutant allele. These findings have important implications for gene therapy approaches for dominant diseases and these strategies can be tested in our model, as it mimics the human disease at the genetic level. Finally, our results indicate that lack of selective pressure against certain mutations in epidermal stem cells could explain how genetic mosaicism results in clinical mosaicism.
Materials and Methods
Gene Targeting and Generation of Transgenic Mice
A BAC clone containing the full-length mouse keratin 10 gene was isolated from a 129 SVJ genomic library (Incyte Genomics). Two overlapping genomic fragments, a 4-kb EcoRI and a 6-kb BamHI fragment, were fused at an EcoRI site in intron 2 to generate a 4.8-kb BglII-XhoI fragment. The point mutation (CGC→ΤGC) was introduced by PCR-mediated mutagenesis in the context of a 400-bp BstEII-NdeI fragment, which was then reinserted into the targeting construct, thus replacing the wild-type codon at position 154. A neo cassette (PGKneo) was inserted into a unique NdeI site in intron 1 and this construct was introduced into the KpnI and NotI sites of the pPNT vector which contains a herpes simplex virus thymidine kinase gene under the control of the phosphoglycerate kinase promotor (PGKtk; Tybulewicz et al. 1991). The procedures for embryonic stem (ES) cell culture, electroporation, drug selection, and Southern blot analysis of targeted clones have been described previously (Ramirez-Solis et al. 1993). In brief, the NotI-linearized targeting vector (see Fig. 1 B) was electroporated into AB2.2 ES cells, which were maintained on SNL76/7 feeder plates (Bradley et al. 1992). ES clones were selected in geneticin (G418) and FIAU (1(1-2-deoxy-2-fluoro-β-darabinofuransyl)-5-iodouracil) and screened for homologous recombination by Southern blot analysis, using probes derived from sequences not included in the targeting construct (see Fig. 1 A) and an internal neomycin probe to confirm a single integration event (data not shown). To excise the neomycin resistance gene from the genome of targeted ES clones, ES cells were electroporated with pOG231, a mammalian Cre expression vector. The recombinant ES cells were injected into C57BL/6 blastocysts, followed by transfer to pseudopregnant ICR females. Chimeric offspring were crossed with C57BL/6 mice and agouti offspring carrying the targeted alleles (+/mutneo; +/mutlox) were identified by PCR analysis of genomic DNA obtained from tail biopsies. Homozygous pups (mutneo/mutneo) were obtained from heterozygous (+/mutneo) intercrosses. Bigenic (+/mutneo. CrePR1) pups were obtained by mating heterozygous +/mutneo mice to the previously described RU486-inducible Cre transgenic line (Berton et al. 2000). RU486 (0.5 mg/ml in ethanol/DMSO) was topically applied onto the skin of newborn bigenic mice once a day for 4–5 d until blisters were visible.
Figure 1.

Targeting of the mouse keratin 10 gene. (A) Genomic organization of the mouse keratin 10 locus. Exon 1 contains codon 154 encoding arginine (R154). (B) The targeting vector contains a C to T mutation encoding cysteine (C154) which also destroys an AciI restriction site. (C) Recombinant mutneo allele after homologous recombination in ES cells. (D) Recombinant mutloxP allele after Cre-mediated excision of PGKneo. 5′ and 3′: probes used for Southern blot analysis. R, EcoRV; B, BglII; X, XhoI; S, ScaI; A, ApaI; N, NotI; K, KpnI.
Histology and Immunohistochemistry
Skin samples were processed as described and stained with hematoxylin and eosin (Wojcik et al. 2000). Samples for double-label immunofluorescence were prepared as described previously (Bickenbach et al. 1996) and sequentially incubated with primary antibodies: rabbit anti–mouse K10 and guinea pig anti–mouse K14 (Roop et al. 1984); then secondary antibodies: swine anti–rabbit conjugate with FITC (Dako) and Texas red–conjugated goat anti–guinea pig IgG (Vector Laboratories).
RNA Analysis
RNA was isolated from the epidermis of heterozygous pups with RNAzol B (Tel-Test), as described previously (Wang et al. 1995). 2 μg of RNA were reverse transcribed using Moloney murine leukemia virus (M-MLV) reverse transcriptase and random primers, according to the manufacturer's recommendation (Promega). A 280-bp fragment encompassing the point mutation was amplified from the cDNA: 5′-biotin GGATTCGGAGGAGATGGTGG-3′ and 5′-biotin CAATCTGCAGCAGCACGTTG-3′. Reverse transcription (RT)-PCR samples were digested with AciI, analyzed by gel electrophoresis, followed by transfer to a positively charged nylon membrane (Gene Screen Plus; NEN Life Science Products). As the point mutation destroys an AciI restriction site, the 280-bp PCR product is cleaved into fragments of 210 and 70 bp in DNA from the wild-type allele, but is resistant to digestion in DNA from the allele harboring the point mutation. The biotinylated DNA fragments were detected using a nonisotopic chemiluminescent detection system (BrightStar BioDetect; Ambion Inc.). Signals were visualized by autoradiography and quantitated.
Laser Capture Microdissection
Skin biopsies from previously RU486-induced and -uninduced areas were fixed in 10% formalin overnight, dehydrated through graded ethanol, and embedded in paraffin. 5-μm sections were deparaffinized in xylenes, rinsed in graded alcohols, and stained with Nuclear Fast Red (Vector Laboratories). Sections were visualized using the PixCell I laser capture microdissection (LCM) system (Arcturus Engineering). A thermoplastic polymer coating (ethylene vinyl acetate; CapSure) attached to a rigid support was placed in contact with the tissue section. The transfer film is activated by a near infrared laser pulse which melts the film onto the targeted cells, thus forming a strong focal bond. 500–700 cells from the stratum spinosum were captured per sample and DNA was extracted in a buffer containing 1 mg/ml proteinase K, 0.5% Nonidet P-40, 0.25% Tween 20, 0.2 mM EDTA pH 8.0, and 10 mM Tris-HCl pH 8.0 and analyzed by PCR with two primer pairs specific for the loxP (5′-GGGTTATTGAATATGATCGG-3′ and 5′-AGGTAAGGCGCTTCAGACTC-3′) and neomycin sequences (5′-ACAGCAAGGGGGAGGATTGG-3′ and 5′-TACTATTCCTGCAGCCGACC-3′), respectively.
Results and Discussion
We have established a mouse model for the skin disease EHK by introducing a K10 mutation, which is found in the majority of human cases of EHK (Rothnagel et al. 1993), into the mouse keratin 10 gene (Krieg et al. 1985). The knock-in/replacement strategy was designed to replace the wild-type amino acid arginine encoded by codon 154 (CGC) with a sequence coding for cysteine (TGC) in ES cells by homologous recombination (Fig. 1). Heterozygous +/mutneo and +/mutloxP mice were derived from ES clones that contain a neomycin (neo) cassette or in which neo had been deleted in vitro before injection into wild-type blastocysts, respectively. These two lines express the mutant allele at different levels, resulting in different phenotypes (see below).
For the inducible system, heterozygous +/mutneo mice were used, as the neo cassette suppresses expression of the mutant allele. Consequently, the animals develop a very mild phenotype which is mainly restricted to skin exposed to mechanical stress. Two mouse lines are required for the inducible system, the first line harbors the target sequence flanked by loxP sites (“floxed” neo cassette), and the second line expresses an inducible form of Cre recombinase (Kellendonk et al. 1996). The Cre transgene encodes a fusion protein consisting of Cre recombinase and a truncated form of a progesterone receptor (PR1), which is sequestered in the cytoplasm until activated by topical application of an inducer to the skin (e.g., RU486). Upon ligand binding, the fusion protein translocates to the nucleus, where Cre exerts its effect by excising any sequence that is flanked by loxP sites in the same orientation. In regenerating tissues like the epidermis, the excision event will only be permanent if epidermal stem cells are targeted. Therefore, the CrePR1 construct was placed under the transcriptional control of an epithelial-specific promotor that drives transgene expression in the basal layer of the epidermis, where epidermal stem cells are located (Berton et al. 2000). Bigenic mice were generated by crossing these two lines (+/mutneo, CrePR1). Topical application of RU486 to the ventral side of the trunk and extremities of newborn bigenic mice induced blistering at the site of induction after three to five treatments (Fig. 2 A). After the blisters ruptured, scaling developed around the site of the previous blisters. With the onset of hair growth, scaling diminished, leaving hyperkeratotic areas on the paws (Fig. 2 B). These hyperkeratotic areas persisted and developed into thick, brownish hyperkeratoses as seen in older EHK patients (Digiovanna 1999). Persistence of the phenotype for 6 mo after the initial RU486 application suggested that Cre-mediated excision of the neo cassette had occurred in epidermal stem cells, as the epidermis is renewed every 8–10 d in mice (Potten et al. 1987).
Figure 2.
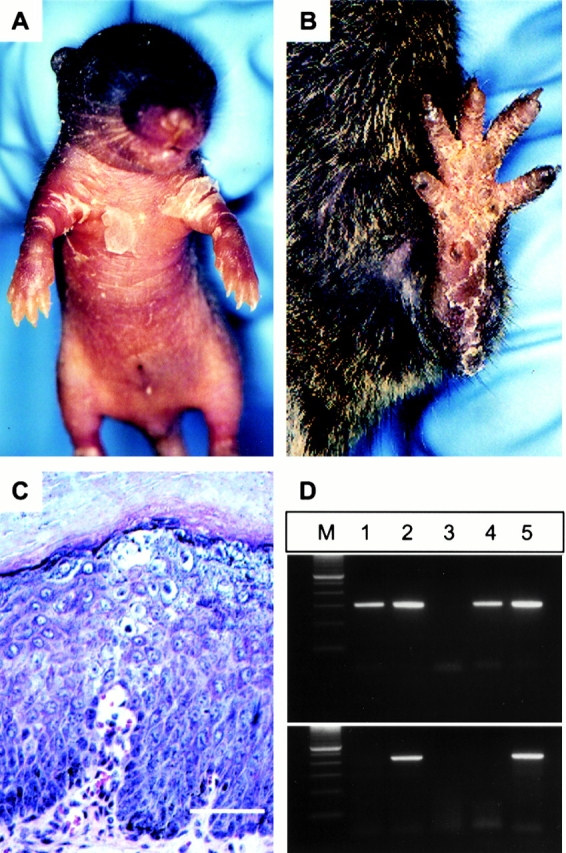
Inducible mouse model for EHK. (A) Bigenic +/mutneo. CrePR1 pup after four topical applications of RU486. Scaling around the arm folds after rupture of the blister. No blisters developed in untreated bigenic +/mutneo.CrePR1 pups or +/mutneo or CrePR1 pups treated with RU486 (data not shown). (B) Same mouse 3 mo after induction. Note the thick brownish hyperkeratoses on the paws. (C) Light micrograph of lesional skin from B. Note the thickened stratum corneum, vacuolization, and translucency in the suprabasal layer. Hematoxylin and eosin, bar = 50 μm. (D) PCR analysis of keratinocytes captured by LCM. PCR analysis to detect the loxP site (top panel) and neo cassette (bottom panel). 1, suprabasal cells from previously treated area; 2, untreated area; 3, wild-type control mouse; 4, +/mutloxP mouse; and 5, +/mutneo mouse. M, DNA size marker.
To investigate this hypothesis, LCM was performed to isolate keratinocytes from persisting hyperkeratotic lesions (Fig. 2 C). As expected, the neo cassette was absent in these cells (Fig. 2 D). This suggested that Cre-mediated excision of the neo cassette and therefore activation of the mutant allele had occurred in epidermal stem cells and that these cells persisted and gave rise to daughter cells that expressed the mutant allele in the suprabasal layers of the epidermis. Cells from surrounding uninduced, clinically normal areas contained the neo cassette. These results clearly demonstrate that keratinocytes containing the neo cassette did not migrate into lesional areas.
Persistent “islands” of phenotypic skin were also observed in chimeric mice derived from +/mutloxP ES cell clones, where the neo cassette had been excised in vitro before injection into wild-type blastocysts. These mice exhibited focal keratotic lesions on the paws at birth that developed into thick, brownish hyperkeratoses (Fig. 3A and Fig. B). The focal lesions in both the inducible model system and the chimeras are equivalent to the linear, asymmetric hyperkeratotic areas in humans in the mosaic form of EHK, which is characterized by alternating stripes of affected and unaffected skin that follow the lines of Blaschko (Happle 1987). It has been shown that the mosaic form is caused by postzygotic K10 mutations that occur during embryogenesis (Paller et al. 1994). Our analysis of the inducible mouse model clearly suggests that in EHK, mutant epidermal stem cells exist side by side with wild-type stem cells. As the mutant K10 allele is not expressed in the basal layer, there is no selection against mutant EHK epidermal stem cells. In contrast, a selection process takes place against defective epidermal stem cells in a mouse model for EBS, when the mutant allele is focally activated in the basal layer (Cao et al. 2001). This could explain why mosaic forms exist for EHK, but not for EBS.
Figure 3.

Chimeric mice derived from +/mutloxP ES cells. Thick hyperkeratoses on the paws at 5 wk (A) and 3 mo of age (B). (C) Normal paw.
An interesting aspect of the mouse models generated in this study (+/mutneo, +/mutloxP) is the correlation of a mild phenotype with reduced expression of the mutant EHK allele. Heterozygous mice that contain the point mutation and the neo cassette (+/mutneo) exhibit a very mild scaling phenotype due to suppressed expression levels of the mutant allele. RNA analysis revealed that the mRNA from the mutant allele was significantly reduced to 35–40% of the levels of the wild-type allele (data not shown). To confirm that mutant K10 mRNA was efficiently translated into protein, we mated heterozygous +/mutneo mice to obtain mice that were homozygous for the mutant allele (mutneo/mutneo). These mice showed a very severe phenotype at birth with extensive blistering and erosions, and died shortly thereafter (Fig. 4 A). Skin biopsies showed a complete disintegration of the stratum spinosum in lesional areas (Fig. 4 B). Immunofluorescence microscopy revealed abundant K10 expression in the suprabasal layers of the epidermis (Fig. 4 C). As the wild-type K10 allele is not present in this mouse, all of the K10 protein detected is expressed from the mutant alleles. Therefore, the mutant K10 mRNA is translated into protein, but the presence of wild-type K10 in heterozygous +/mutneo mice is sufficient to overcome the effects of the reduced level of mutant K10.
Figure 4.

Gross appearance and skin morphology of mutneo/mutneo mice. (A) mutneo/mutneo pup shortly after birth with severe blistering and erosions. (B) Histological analysis of a skin biopsy from A shows a split in the suprabasal layer of the epidermis. Hematoxylin and eosin, bar = 50 μm. (C) Immunolabeling shows abundant mutant K10 (yellow) throughout the disintegrating suprabasal layer against the red K14 background stain. Bars = 50 μm.
Our EHK model has important implications for gene therapy approaches. First, gene therapy approaches will be different for EHK and EBS. Whereas repopulation of a phenotypic area by corrected stem cells is predicted in the case of EBS (Cao et al. 2001), successful approaches for EHK will have to include a strategy that allows selection of genetically corrected epidermal stem cells and ablation of defective EHK stem cells. Recent studies suggest that it may be possible to achieve selection of genetically modified epidermal stem cells by topical selection with cytostatic and antimitotic compounds, such as colchicine (Pfutzner et al. 1999).
Second, in contrast to the general assumption that gene therapy approaches for dominant diseases must either correct the mutant allele or completely inhibit its expression, our data suggest that amelioration of the EHK phenotype may be achieved by partial suppression of the mutant allele or overexpression of the normal allele, thus altering the ratio of wild-type to mutant protein. As our previous transgenic studies have not revealed adverse effects from an approximate twofold overexpression of a wild-type K1 allele (Bickenbach et al. 1996), it is intriguing to speculate that overexpression of the wild-type protein two- to threefold might be sufficient to improve the phenotype in EHK patients. This gene therapy approach may also be feasible for other dominant diseases.
Acknowledgments
We are thankful to A. Bradley for ES cells, R. Behringer and Y. Mishina for the PGKneo plasmid, and J. Wang and F. DeMayo for ES cell injections. We also thank C. Allred for assistance with LCM, S. Wojcik for helpful comments, and P. Koch for help with ES cell work and valuable comments on the manuscript.
This work was supported by National Institutes of Health grant HD25479 to D.R. Roop. M.J. Arin was supported in part by the Deutsche Forschungsgemeinschaft (Ar 291/1-1).
References
- Bale S.J., Compton J.G., DiGiovanna J.J. Epidermolytic hyperkeratosis. Semin. Dermatol. 1993;12:202–209. [PubMed] [Google Scholar]
- Berton T.R., Wang X.J., Zhou Z., Kellendonk C., Schutz G., Tsai S., Roop D.R. Characterization of an inducible, epidermal-specific knockout systemdifferential expression of lacZ in different Cre reporter mouse strains. Genesis. 2000;26:160–161. [PubMed] [Google Scholar]
- Bickenbach J.R., Longley M.A., Bundman D.S., Dominey A.M., Bowden P.E., Rothnagel J.A., Roop D.R. A transgenic mouse model that recapitulates the clinical features of both neonatal and adult forms of the skin disease epidermolytic hyperkeratosis. Differentiation. 1996;61:129–139. doi: 10.1046/j.1432-0436.1996.6120129.x. [DOI] [PubMed] [Google Scholar]
- Bonifas J.M., Rothman A.L., Epstein E.H.J. Epidermolysis bullosa simplexevidence in two families for keratin gene abnormalities. Science. 1991;254:1202–1205. doi: 10.1126/science.1720261. [DOI] [PubMed] [Google Scholar]
- Bradley A., Ramirez-Solis R., Zheng H., Hasty P., Davis A. Genetic manipulation of the mouse via gene targeting in embryonic stem cells. Ciba. Found. Symp. 1992;165:256–269. doi: 10.1002/9780470514221.ch15. [DOI] [PubMed] [Google Scholar]
- Cao T., Longley M.A., Wang X.-J., Roop D.R. An inducible mouse model for epidermolysis bullosa simpleximplications for gene therapy. J. Cell Biol. 2001;152:651–656. doi: 10.1083/jcb.152.3.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J., Syder A.J., Yu Q.C., Letai A., Paller A.S., Fuchs E. The genetic basis of epidermolytic hyperkeratosisa disorder of differentiation-specific epidermal keratin genes. Cell. 1992;70:811–819. doi: 10.1016/0092-8674(92)90314-3. [DOI] [PubMed] [Google Scholar]
- Chipev C.C., Korge B.P., Markova N., Bale S.J., Digiovanna J.J., Compton J.G., Steinert P.M. A leucine—proline mutation in the H1 subdomain of keratin 1 causes epidermolytic hyperkeratosis. Cell. 1992;70:821–828. doi: 10.1016/0092-8674(92)90315-4. [DOI] [PubMed] [Google Scholar]
- Coulombe P.A., Hutton M.E., Letai A., Hebert A., Paller A.S., Fuchs E. Point mutations in human keratin 14 genes of epidermolysis bullosa simplex patientsgenetic and functional analyses. Cell. 1991;66:1301–1311. doi: 10.1016/0092-8674(91)90051-y. [DOI] [PubMed] [Google Scholar]
- Digiovanna J.J. Ichthyosiform dermatoses. In: Freedberg I.M., Eisen A.Z., Wolff K., Austen K.F., Goldsmith L.A., Katz S.I., Fitzpatrick T.B., editors. Dermatology in General Medicine. McGraw-Hill; New York: 1999. pp. 581–603. [Google Scholar]
- Franke W.W., Schiller D.L., Moll R., Winter S., Schmid E., Engelbrecht I., Denk H., Krepler R., Platzer B. Diversity of cytokeratins. Differentiation specific expression of cytokeratin polypeptides in epithelial cells and tissues. J. Mol. Biol. 1981;153:933–959. doi: 10.1016/0022-2836(81)90460-5. [DOI] [PubMed] [Google Scholar]
- Happle R. Lethal genes surviving by mosaicisma possible explanation for sporadic birth defects involving the skin. J. Am. Acad. Dermatol. 1987;16:899–906. doi: 10.1016/s0190-9622(87)80249-9. [DOI] [PubMed] [Google Scholar]
- Happle R. Mosaicism in human skin. Understanding the patterns and mechanisms. Arch. Dermatol. 1993;129:1460–1470. [PubMed] [Google Scholar]
- Ishida-Yamamoto A., McGrath J.A., Judge M.R., Leigh I.M., Lane E.B., Eady R.A. Selective involvement of keratins K1 and K10 in the cytoskeletal abnormality of epidermolytic hyperkeratosis (bullous congenital ichthyosiform erythroderma) J. Invest. Dermatol. 1992;99:19–26. doi: 10.1111/1523-1747.ep12611391. [DOI] [PubMed] [Google Scholar]
- Itin P.H., Buechner S.A. Segmental forms of autosomal dominant skin disordersthe puzzle of mosaicism. Am. J. Med. Genet. 1999;85:351–354. [PubMed] [Google Scholar]
- Kellendonk C., Tronche F., Monaghan A.P., Angrand P.O., Stewart F., Schutz G. Regulation of Cre recombinase activity by the synthetic steroid RU 486. Nucleic Acids Res. 1996;24:1404–1411. doi: 10.1093/nar/24.8.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieg T.M., Schafer M.P., Cheng C.K., Filpula D., Flaherty P., Steinert P.M., Roop D.R. Organization of a type I keratin gene. Evidence for evolution of intermediate filaments from a common ancestral gene. J. Biol. Chem. 1985;260:5867–5870. [PubMed] [Google Scholar]
- Moll R., Franke W.W., Schiller D.L., Geiger B., Krepler R. The catalog of human cytokeratinspatterns of expression in normal epithelia, tumors and cultured cells. Cell. 1982;31:11–24. doi: 10.1016/0092-8674(82)90400-7. [DOI] [PubMed] [Google Scholar]
- Paller A.S., Syder A.J., Chan Y.M., Yu Q.C., Hutton E., Tadini G., Fuchs E. Genetic and clinical mosaicism in a type of epidermal nevus. N. Engl. J. Med. 1994;331:1408–1415. doi: 10.1056/NEJM199411243312103. [DOI] [PubMed] [Google Scholar]
- Pfutzner W., Hengge U.R., Joari M.A., Foster R.A., Vogel J.C. Selection of keratinocytes transduced with the multidrug resistance gene in an in vitro skin model presents a strategy for enhancing gene expression in vivo. Hum. Gene Ther. 1999;10:2811–2821. doi: 10.1089/10430349950016546. [DOI] [PubMed] [Google Scholar]
- Potten C.S., Saffhill R., Maibach H.I. Measurement of the transit time for cells through the epidermis and stratum corneum of the mouse and guinea-pig. Cell Tissue Kinet. 1987;20:461–472. doi: 10.1111/j.1365-2184.1987.tb01355.x. [DOI] [PubMed] [Google Scholar]
- Ramirez-Solis R., Davis A.C., Bradley A. Gene targeting in embryonic stem cells. Methods Enzymol. 1993;225:855–878. doi: 10.1016/0076-6879(93)25054-6. [DOI] [PubMed] [Google Scholar]
- Roop D. Defects in the barrier. Science. 1995;267:474–475. doi: 10.1126/science.7529942. [DOI] [PubMed] [Google Scholar]
- Roop D.R., Cheng C.K., Titterington L., Meyers C.A., Stanley J.R., Steinert P.M., Yuspa S.H. Synthetic peptides corresponding to keratin subunits elicit highly specific antibodies. J. Biol. Chem. 1984;259:8037–8040. [PubMed] [Google Scholar]
- Rothnagel J.A., Dominey A. M., Dempsey L.D., Longley M.A., Greenhalgh D.A., Gagne T.A., Huber M., Frenk E., Hohl D., Roop D.R. Mutations in the rod domains of keratins 1 and 10 in epidermolytic hyperkeratosis. Science. 1992;257:1128–1130. doi: 10.1126/science.257.5073.1128. [DOI] [PubMed] [Google Scholar]
- Rothnagel J.A., Fisher M.P., Axtell S.M., Pittelkow M.R., Anton-Lamprecht I., Huber M., Hohl D., Roop D.R. A mutational hot spot in keratin 10 (KRT 10) in patients with epidermolytic hyperkeratosis. Hum. Mol. Genet. 1993;2:2147–2150. doi: 10.1093/hmg/2.12.2147. [DOI] [PubMed] [Google Scholar]
- Rothnagel J.A., Roop D.R. Analysis, diagnosis, and molecular genetics of keratin disorders. Curr. Op. Dermatol. 1995;1995:211–218. [Google Scholar]
- Steinert P.M., Liem R.K. Intermediate filament dynamics. Cell. 1990;60:521–523. doi: 10.1016/0092-8674(90)90651-t. [DOI] [PubMed] [Google Scholar]
- Tybulewicz V.L., Crawford C.E., Jackson P.K., Bronson R.T., Mulligan R.C. Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell. 1991;65:1153–1163. doi: 10.1016/0092-8674(91)90011-m. [DOI] [PubMed] [Google Scholar]
- Wang X.J., Greenhalgh D.A., Lu X.R., Bickenbach J.R., Roop D.R. TGF alpha and v-fos cooperation in transgenic mouse epidermis induces aberrant keratinocyte differentiation and stable, autonomous papillomas. Oncogene. 1995;10:279–289. [PubMed] [Google Scholar]
- Wojcik S.M., Bundman D.S., Roop D.R. Delayed wound healing in keratin 6a knockout mice. Mol. Cell. Biol. 2000;20:5248–5255. doi: 10.1128/mcb.20.14.5248-5255.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]