

Abstract
Survival of endothelial cells is critical for cellular processes such as angiogenesis. Cell attachment to extracellular matrix inhibits apoptosis in endothelial cells both in vitro and in vivo, but the molecular mechanisms underlying matrix-induced survival signals or detachment-induced apoptotic signals are unknown. We demonstrate here that matrix attachment is an efficient regulator of Fas-mediated apoptosis in endothelial cells. Thus, matrix attachment protects cells from Fas-induced apoptosis, whereas matrix detachment results in susceptibility to Fas-mediated cell death. Matrix attachment modulates Fas-mediated apoptosis at two different levels: by regulating the expression level of Fas, and by regulating the expression level of c-Flip, an endogenous antagonist of caspase-8. The extracellular signal–regulated kinase (Erk) cascade functions as a survival pathway in adherent cells by regulating c-Flip expression. We further show that detachment-induced cell death, or anoikis, itself results from activation of the Fas pathway by its ligand, Fas-L. Fas-L/Fas interaction, Fas–FADD complex formation, and caspase-8 activation precede the bulk of anoikis in endothelial cells, and inhibition of any of these events blocks anoikis. These studies identify matrix attachment as a survival factor against death receptor–mediated apoptosis and provide a molecular mechanism for anoikis and previously observed Fas resistance in endothelial cells.
Keywords: apoptosis, endothelium, extracellular matrix, integrin, signaling
Introduction
Endothelium is a critical site for the control of apoptosis during processes such as inflammation, vascular remodeling, and angiogenesis (Karsan and Harlan 1996). On one hand, suppression of apoptosis in endothelial cells is required for the maintenance of blood vessel integrity and for angiogenesis. On the other hand, programmed cell death has been suggested to contribute to pathological tissue destruction during vascular injury, inflammation, atherosclerosis, and allograft arteriopathy. It has been proposed that Fas-mediated apoptosis has a central role in physiological and pathological turnover in the vasculature. Fas, which belongs to the tumor necrosis factor receptor family, induces a death signal when bound to its ligand, Fas-L (Nagata and Golstein 1995). Ligation of Fas receptor induces receptor aggregation, which in turn results in the recruitment of the adaptor protein Fas-associated death domain (FADD) and caspase-8 and in the activation of downstream caspases that lead to irreversible cell damage (Krammer 1996). Expression of both Fas and Fas-L has been detected in endothelial cells. It has been demonstrated that one function of the endothelial Fas-L is to inhibit leukocyte extravasation by inducing apoptosis in Fas-expressing mononuclear cells invading the vessel wall in the absence of normal inflammatory stimuli (Sata and Walsh 1998c). Endothelial cells themselves are resistant to Fas-mediated apoptosis under normal conditions, despite the fact that they express detectable Fas on their cell surface (Sata and Walsh 1998c). These cells can become dramatically sensitized to Fas-mediated apoptosis in response to specific stimuli or injuries, and it has been suggested that the increased sensitization to Fas-mediated apoptosis may play an important role in regulation of pathological tissue destruction in the vessel wall (Sata and Walsh 1998b). The mechanisms by which stimuli sensitize cells to Fas-mediated apoptosis to date have remained largely unelucidated.
Previous studies have indicated that endothelial cells are dependent on adhesion to the extracellular matrix (ECM) for their continued survival. Thus, disruption of appropriate cell–matrix contacts both in vitro and in vivo is sufficient to initiate caspase-dependent apoptosis in endothelium, culminating with rapid involution of vascular structures (Meredith et al. 1993; Brooks et al. 1994; Re et al. 1994). As a corollary, agents that block appropriate cell–matrix interactions are known to have an important therapeutic utility in inhibiting tumor-induced neovascularization and tumor progression in animal models (Brooks et al. 1994). It is known that ligand binding of integrins, which are the main receptors for ECM proteins, protects cells against detachment-induced apoptosis or anoikis (for review see Frisch and Ruoslahti 1997). More recently, integrin-mediated cell attachment has been shown to mediate survival signaling against various apoptotic stimuli, such as serum withdrawal (Zhang et al. 1995; O'Brien et al. 1996), chemotherapeutic agents (Sethi et al. 1999), and activation-induced cell death (AICD) in T lymphocytes (Aoudjit and Vuori 2000). Focal adhesion kinase (FAK), which becomes activated upon integrin ligand binding, mediates survival signaling downstream of integrins and suppresses anoikis, serum withdrawal–induced apoptosis, and AICD (Frisch et al. 1996; Hungerford et al. 1996; Ilic et al. 1998; Almeida et al. 2000; Aoudjit and Vuori 2000; Gilmore et al. 2000). In addition to FAK, pathways involving phosphatidylinositol 3′-kinase (PI 3′-kinase) and the serine/threonine kinase Akt, the mitogen-activated protein kinase (MAPK)/Erk cascade, NF-κB, and the Bcl-2 family of proteins have been implicated in integrin-mediated protection against various apoptotic stimuli (Zhang et al. 1995; Scatena et al. 1998; Gilmore et al. 2000; Le Gall et al. 2000; Rosen et al. 2000). Despite these findings, which have been mainly made in epithelial cells and fibroblasts, the detailed molecular mechanism as to how integrin signaling leads to cell survival remains to be determined.
In this study, we hypothesized that the important survival role that matrix attachment has in endothelial cells might be due to its capability to regulate Fas-mediated killing in these cells. We therefore investigated whether matrix attachment provides protection against Fas-induced apoptosis and whether detachment-induced apoptosis, or anoikis, in turn might be caused by activation of the Fas pathway in endothelial cells. As detailed below, our studies identify matrix attachment as a survival factor against death receptor–mediated apoptosis and provide a molecular mechanism for Fas resistance and anoikis in endothelial cells.
Materials and Methods
Cells, Antibodies, and Other Reagents
Human umbilical vein endothelial cells (HUVECs; Clonetics Corp.) were cultured in EGM medium (2% FCS, 10 ng/ml hEGF, and 1 μg/ml hydrocortisone; Clonetics Corp.), supplemented with 12 μg/ml of bovine brain extract, 2 mM l-glutamine, 50 μg/ml streptomycin, and 50 U/ml penicillin (Irvine Scientific). This medium is termed as “complete growth medium” below. Anti–Fas-L antibodies, NOK-1, NOK-2, and G247-4 were from BD PharMingen, and anti–Fas-L antibody clone 33 was from Transduction Laboratories. Anti-Fas antibodies, ZB4, UB2, and CH11, were from Kamiya Biomedicals, and anti-Fas antibody clone 13 and anti-FADD antibody clone 1 were from Transduction Laboratories. Rabbit polyclonal anti–c-Flip antibody, which recognizes both FlipL and FlipS isoforms, was obtained from Dr. Donald Nicholson (Merck Frosst, Quebec, Canada). Anti–α-tubulin and anti–caspase-8 (Ab-1; this antibody recognizes the p18 fragment of caspase-8) antibodies were from Oncogene Research Products. The anti–phospho-p44/42 MAPK antibody E10, which recognizes the active form of Erk, was from New England Biolabs, Inc., and the anti-Erk2 antibody C14 was from Santa Cruz Biotechnology, Inc. The anti–caspase-8 antibody (clone 5F7; this antibody recognizes the proform of caspase-8), the caspase-8 and -3 inhibitors (z-IETD-fmk and z-DEVD-fmk, respectively), and the broad caspase inhibitor z-VAD-fmk were from Kamiya Biomedicals. The PI 3′-kinase inhibitor Ly294002 and the MAPk kinase (MEK) inhibitor PD 98059 were from Calbiochem.
Plasmids and Transient Transfections
HUVECs were transfected by using the Superfect reagent from QIAGEN according to the manufacturer's instructions. Cells were grown to 80% confluency in 6-well plates, and transfected using 5 μg of total DNA. 24 h later, the cells were washed with PBS and used for subsequent experiments. The plasmids encoding activated and dominant negative forms of PI 3′-kinase and Akt were obtained from Drs. Jerrold Olefsky (University of California at San Diego, La Jolla, CA), and Phil Tsichlis (Thomas Jefferson University, Philadelphia, PA), respectively. The plasmids encoding human c-FlipL, procaspase-8, and dominant negative (dn) FADD cDNAs were from Drs. John Reed, Guy Salvesen, and Steven Frisch, respectively (The Burnham Institute). The plasmids encoding Fas and the activated form of Raf-1 were from Drs. Taka-Aki Sato (Columbia University, New York, NY) and John Hancock (University of Queensland Medical School, Brisbane, Australia), respectively.
Apoptosis Assay
HUVECs grown into 80% confluency were detached by trypsinization, and anoikis was induced by either keeping cells in complete growth medium (see above) in suspension at 37°C at a concentration of 105 cells/ml, or by plating the cells on polyhema at 37°C for the indicated time periods. Apoptosis was monitored by trypan blue exclusion, by using a cell death detection ELISA kit (Boehringer), or by staining cells with propidium iodide or Hoechst dye, as indicated. For anti-Fas induced apoptosis, the attached cells were incubated for 24 h and the detached cells for 12 h with 1 μg/ml of anti-Fas antibody CH11. Apoptosis was monitored as above.
Flow Cytometric Analysis
3 × 105 cells in 200 μl of PBS containing 1% FCS and 0.2% sodium azide (FACS buffer) were incubated with 10 μg/ml of anti-Fas antibody UB2, anti–Fas-L antibody NOK-1, or isotype-matched control antibodies for 30 min at 4°C. The cells were washed three times in PBS and incubated with phycoerythrin-conjugated goat anti–mouse IgG secondary antibodies at a dilution of 1:100 (Jackson ImmunoResearch Laboratories) in 200 μl of FACS buffer for another 30 min. The cells were washed and immediately analyzed with a FACScan™ (Becton Dickinson).
RNA Isolation and Analysis
Total cellular RNA was isolated from HUVECs using the Trizol reagent (Life Technologies). The first strand cDNA was prepared using the Superscript Amplification System kit (Life Technologies) according to the manufacturer's instructions. After reverse transcription, the FlipL and FlipS isoforms were amplified by using specific primers described previously (Perlman et al. 1999). As a control, glyceraldehyde phosphate dehydrogenase (GAPDH) was amplified by using the following primers: forward, 5′-TTCATTGACCTCAACTACATGG-3′; and reverse, 5′-GTGGCAGTGATGGCATGGAC-3′. The PCR reaction was carried out with 1 U Taq polymerase (PerkinElmer) in a total volume of 50 μl. Amplification was performed for 28 cycles (30 s denaturing at 94°C, 45 s annealing at 50°C, and 90 s extension at 72°C) in a PerkinElmer thermal cycler. Fas was amplified by using specific primers described previously (Moulian et al. 1999). Amplification was performed for 35 cycles (1 min denaturing at 94°C, 1 min annealing at 53°C, and 2 min extension at 72°C). The PCR products were analyzed by agarose gel electrophoresis.
Immunoprecipitation and Immunoblotting
The cell lysates for the immunoprecipitations and immunoblotting studies were prepared as described previously (Aoudjit and Vuori 2000). To analyze Fas/Fas-L complex formation and death-inducing signaling complex formation (DISC), cell lysates prepared from attached and detached cells were subjected to immunoprecipitations with anti–Fas-L (clone 33) and anti-FADD antibodies, respectively. Immunoprecipitates and cytosolic proteins (20 μg of total protein) were analyzed by immunoblotting with anti-Fas antibodies (clone 13). The blots were stripped and reprobed with anti–Fas-L (clone 33) and anti-FADD antibodies to assess the amount of immunoprecipitated protein. In some of the studies, anti–Fas-L antibody G247-4 was used, and similar results were obtained. Protein expression levels of Fas, Fas-L, and c-Flip were determined by an immunoblot analysis of total cytosolic proteins (20 μg of total protein) by using the respective antibodies. The blots were stripped and reprobed with anti–α-tubulin antibody to confirm equal loading. Caspase-8 expression was determined by immunoblotting (20 μg of total protein) by using the 5F7 antibody which recognizes the proform of caspase-8. Caspase-8 activity was determined by an immunoblot analysis (50 μg of total protein) by using an antibody (Ab-1) that specifically recognizes the active forms of caspase-8. In all experiments, immunoblots were visualized by using an HRP-conjugated secondary antibody followed by enhanced chemiluminescence detection (Pierce Chemical Co.).
Results
Matrix Attachment Modulates Fas-mediated Apoptosis in HUVECs: Anoikis Is Induced by Activation of the Fas/Fas-L Pathway
Previous studies have indicated that HUVECs undergo anoikis when denied matrix attachment (Meredith et al. 1993; Re et al. 1994), and we found the same here. As shown in Fig. 1 A, endothelial cell viability decreases by 75% over a 48-h time period after cell detachment in the presence of serum, and a clear induction (25%) in cell death is observed 12 h after detachment of the cells. The loss of cell viability is a result of apoptosis as attested by the significant induction in DNA fragmentation, a hallmark of apoptotic death, after cell detachment (Fig. 1 B). To examine whether anoikis in HUVECs involves activation of the Fas/Fas-L pathway, we used inhibitory antibodies against Fas and Fas-L that have been shown previously to suppress the Fas/Fas-L pathway and inhibit Fas-mediated apoptosis (Kayagaki et al. 1995; Srivastava et al. 1999). As shown in Fig. 1 C, treatment of detached HUVECs with anti–Fas-L antibodies inhibits anoikis in these cells by ∼60%. Inhibitory anti-Fas antibodies, alone or in combination with the anti–Fas-L antibodies, also significantly block anoikis in HUVECs (Fig. 1 C). Several irrelevant isotype–matched control antibodies failed to have any effect on cell survival or anoikis in HUVECs (Fig. 1 C). AICD in T lymphocytes is known to take place via the Fas/Fas-L pathway, and as a control, we found that AICD in Jurkat cells can similarly be blocked by ∼70% with anti–Fas-L or anti-Fas antibodies (not shown; Aoudjit and Vuori 2000). These antibodies did not have any effect on serum withdrawal–induced apoptosis in endothelial cells, which occurs in a Fas-independent manner (not shown). Thus, these results suggest that activation of Fas by its ligand, Fas-L, has a significant functional role in the induction of anoikis in HUVECs.
Figure 1.
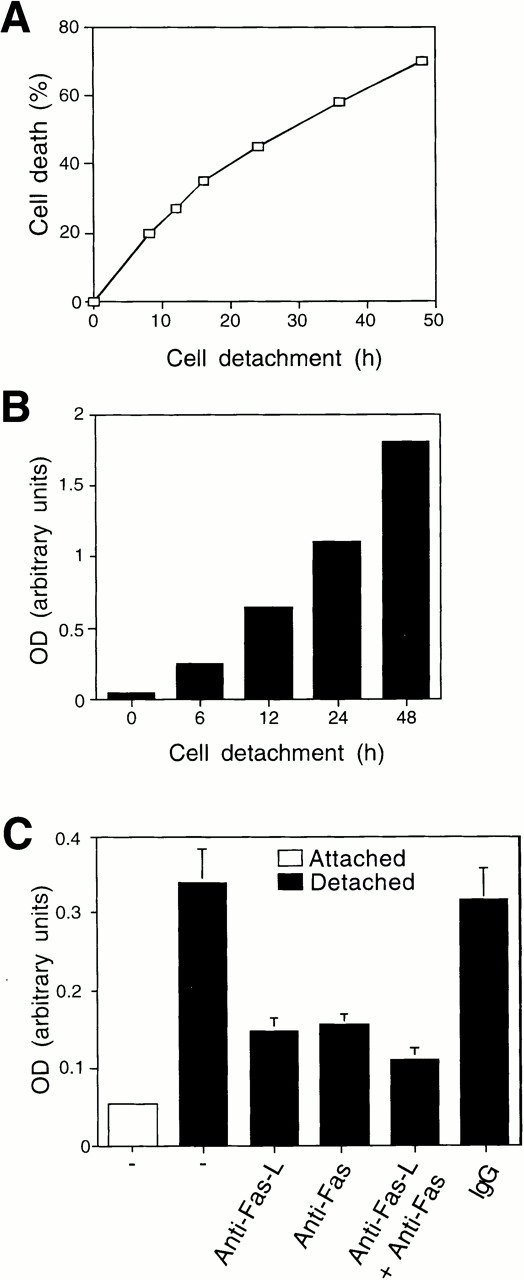
Detachment-induced apoptosis in HUVECs is Fas/Fas-L dependent. HUVECs were detached and either kept in suspension or on polyhema-coated dishes in complete growth medium (EGM medium containing 2% FCS, 10 ng/ml hEGF, and 1 μg/ml hydrocortisone, supplemented with 12 μg/ml of bovine brain extract, 2 mM l-glutamine, 50 μg/ml streptomycin, and 50 U/ml penicillin) for the indicated time periods, and cell death was determined by (A) trypan blue exclusion or (B) DNA fragmentation analysis. Identical results were obtained under the two experimental conditions; results shown are for cells kept in suspension, and these experimental conditions were used in all subsequent experiments involving detachment-induced cell death. (C) HUVECs were kept adherent or in suspension for 12 h in the presence or absence of 5 μg/ml of inhibitory anti–Fas-L antibodies (NOK-2), inhibitory anti-Fas antibodies (ZB4), or with control antibodies (IgG). Apoptosis was determined by DNA fragmentation analysis. In A and B, results are shown for representative experiments independently carried out three times. In C, bars indicate SD in a representative experiment done in triplicate.
To examine whether matrix attachment in turn might protect HUVECs against Fas-mediated apoptosis, adherent HUVECs were treated with an agonistic Fas-antibody, CH11, to induce activation of Fas signaling. We found that adherent HUVECs are highly resistant to Fas-mediated killing (Fig. 2). Upon cell detachment, HUVECs undergo programmed cell death, as expected, as a result of anoikis. However, addition of CH11 to detached cells further enhances cell death, suggesting that cell detachment sensitizes HUVECs to Fas-mediated apoptosis (Fig. 2). We used the inhibitory anti–Fas-L antibody to block Fas-L–dependent induction of Fas signaling in detached cells, and studied the effect of CH11 under these conditions. As shown in Fig. 2, a significant amount of apoptosis takes place in detached, CH11-treated HUVECs, in which Fas-L–induced apoptosis has been eliminated by the inhibitory anti–Fas-L antibody. Thus, cell detachment not only induces activation of the Fas pathway by Fas-L, but also sensitizes endothelial cells to activation of Fas-mediated apoptosis by exogenously added Fas agonists.
Figure 2.

Cell detachment sensitizes HUVECs to Fas-mediated apoptosis. The ability of agonistic anti-Fas antibody (CH11) to induce apoptosis was analyzed in attached and detached HUVECs. The cells were incubated with or without 1 μg/ml of CH11 for 12 h in the presence or absence of blocking anti–Fas-L antibody (NOK-2), and apoptosis was determined by DNA fragmentation analysis. Bars indicate SD in a representative experiment done in triplicate.
Cell Detachment Induces Fas/Fas-L Interaction, DISC Formation, and Caspase-8 Activation in HUVECs
To further examine the role of the Fas/Fas-L pathway in anoikis, we undertook several biochemical approaches to study Fas signaling in detached endothelial cells. First, we examined whether cell detachment induces Fas/Fas-L interaction in HUVECs. As shown in Fig. 3 A, Fas becomes associated with Fas-L after detachment of HUVECs, and this association precedes induction of the bulk of anoikis in these cells (see Fig. 1 A). Thus, Fas/Fas-L association can be clearly detected 6 h after cell detachment, the interaction peaking 12 h after detachment of the cells. Importantly, the molecular mass of Fas detected in the Fas-L immunoprecipitates is 110 kD, which corresponds to the oligomerized form of Fas that is induced after binding of Fas-L to Fas (Kamitani et al. 1997).
Figure 3.




Cell detachment induces Fas/Fas-L interaction, DISC formation, and caspase-8 activation in HUVECs. Inhibition of anoikis by dnFADD and caspase-8 inhibitors. (A) Cell lysates were prepared from adherent HUVECs and from HUVECs that had been kept in suspension for the indicated times, and the lysates were subjected to immunoprecipitation analysis by anti–Fas-L antibodies (clone 33). Immunoprecipitates (IP) were analyzed by immunoblotting with anti-Fas antibodies (clone 13; top). The membranes were stripped and reprobed with anti–Fas-L antibodies (clone 33) to confirm equal amounts of Fas-L in the precipitates (bottom). Similar results were obtained when anti–Fas-L antibody G247-4 was used (data not shown). (B) The cell lysates were subjected to immunoprecipitations by anti-FADD antibodies. The immunoprecipitates were analyzed by immunoblotting with anti-Fas antibodies (clone 13; top), and the membranes were stripped and reprobed with anti-FADD antibodies to confirm equal amounts of FADD in the precipitates (bottom). C, Control cell lysate prepared from Jurkat T cells that had been activated by T cell receptor stimulation. (C) HUVECs were transiently transfected with an empty vector (control) or with an expression plasmid encoding dnFADD, together with plasmid coding for the green fluorescent protein (GFP). 24 h after transfection, the cells were either kept adherent or kept in suspension for the indicated time periods. The cells were then washed and propidium iodide was added for 20 min on ice. Apoptosis analysis by FACS® was carried out in the double positive cell population for propidium iodide and fluorescent GFP. In a separate experiment, we monitored apoptosis by staining the cells with Hoechst dye and found that results obtained with propidium iodide and Hoechst were indistinguishable from each other (not shown). Thus, the results obtained with the Hoechst dye confirm that cell death observed under our experimental conditions results from apoptosis, rather than necrosis. (D) HUVECs were left adherent or kept in suspension for 12 h in the presence or absence of the indicated concentrations of caspase-8 inhibitor z-IETD-fmk. In the left panel, apoptosis was determined by DNA fragmentation analysis; bars indicate SD in a representative experiment done in triplicate. In the right panel, cell lysates were prepared and analyzed by immunoblotting with an anti–caspase-8 antibody (Ab-1) that detects the p18 active form of the enzyme.
Fas engagement and trimerization of Fas lead to the recruitment of FADD to form the DISC. FADD in turn recruits procaspase-8, which is cleaved and activated at the DISC. As shown in Fig. 3 B, HUVEC detachment induces DISC formation, as attested by the coimmunoprecipitation of oligomerized Fas and FADD. This interaction peaks 12 h after cell detachment, and decreases 24 h after detachment of the cells. To confirm that FADD has a functional role in anoikis in HUVECs, the cells were transiently transfected with a control vector or with an expression vector coding for dnFADD. As shown in Fig. 3 C, expression of dnFADD significantly blocks anoikis at all time points examined. In a control experiment, we found that AICD in Jurkat cells is also blocked by dnFADD (not shown).
We next determined whether cell detachment results in activation of caspase-8, and if so, whether activation of caspase-8 is necessary for anoikis in HUVECs. After Fas activation, caspase-8 undergoes several processing steps to generate the active caspase-8 heterodimer composed of p18 and p10/12 (Salvesen 1999). As shown in Fig. 3 D, detachment of HUVECs results in the generation of the p18 active form of caspase-8, which peaks 12 h after cell detachment. Activation and processing of caspase-8 is significantly blocked by a specific inhibitor of caspase-8, z-IETD-fmk (Fig. 3 D). Further, activation of caspase-8 was found to be necessary for anoikis to take place in HUVECs, as z-IETD-fmk also significantly blocked detachment-induced apoptosis in these cells. As a control, we found that z-IETD-fmk completely blocks AICD in Jurkat cells, whereas it has no effect on serum withdrawal–induced apoptosis in endothelial cells (data not shown). Thus, several lines of biochemical evidence support the notion that the Fas/Fas-L signaling pathway is activated in detached endothelial cells and contributes to the induction of anoikis.
Matrix Attachment Regulates Expression Levels of Fas and Fas-L in HUVECs
One possible explanation as to how matrix attachment could control Fas-mediated apoptosis is by regulating the expression levels of key components of Fas signaling, such as Fas-L and Fas. Similar to what has been reported previously with respect to HUVECs cultured as a monolayer, we found that adherent HUVECs express significant levels of Fas-L (not shown). Cell detachment did result, however, in a 1.5-fold increase in the cell surface levels of Fas-L (not shown). However, the fact that high amounts of Fas-L are expressed in attached cells and that Fas-L expressed on HUVECs is functional and capable of inducing Fas-mediated killing in target cells (data not shown; Sata and Walsh 1998c) led us to conclude that adhesion-mediated regulation of Fas-L cell surface levels is unlikely to be a rate-limiting step in Fas-mediated apoptosis and anoikis in HUVECs.
We then examined the expression levels of Fas in attached and detached HUVECs. As reported previously, HUVECs cultured as a monolayer express detectable levels of Fas (Fig. 4 A). Detachment of HUVECs results in a threefold increase in the cell surface levels of Fas by 12 h after detachment (Fig. 4 A). The detachment-induced increase in Fas expression can be detected also at the mRNA level. A semiquantitative reverse transcriptase (RT)-PCR analysis of Fas mRNA expression indicates that cell detachment induces an increase in Fas mRNA levels as early as in 3 h, with a peak in the induction of Fas mRNA levels occurring 6 h after cell detachment (Fig. 4 B). To examine whether matrix attachment protects endothelial cells from Fas-mediated apoptosis by downregulating the cell surface levels of Fas, attached HUVECs were transiently transfected with a plasmid coding for Fas. In several independent experiments, transient transfection resulted, on average, in a two- to threefold increase in the cell surface levels of Fas (not shown). However, ectopic expression of Fas did not result in an induction of apoptosis in attached HUVECs, and neither did it sensitize the cells to CH11-mediated killing (see Fig. 6). Thus, although upregulation of Fas may be necessary for anoikis and Fas-mediated apoptosis to take place in detached cells, enhanced Fas expression alone is not sufficient to sensitize adherent HUVECs for Fas-mediated killing. Upon examination of DISC formation and caspase-8 activation in Fas-transfected, CH11-treated HUVECs, we observed Fas–FADD interaction but no caspase-8 activation (not shown). Taken together, these results suggest that matrix attachment regulates (at least) one additional step in Fas signaling, which takes place downstream of DISC formation but upstream or at the level of caspase-8 activation.
Figure 4.

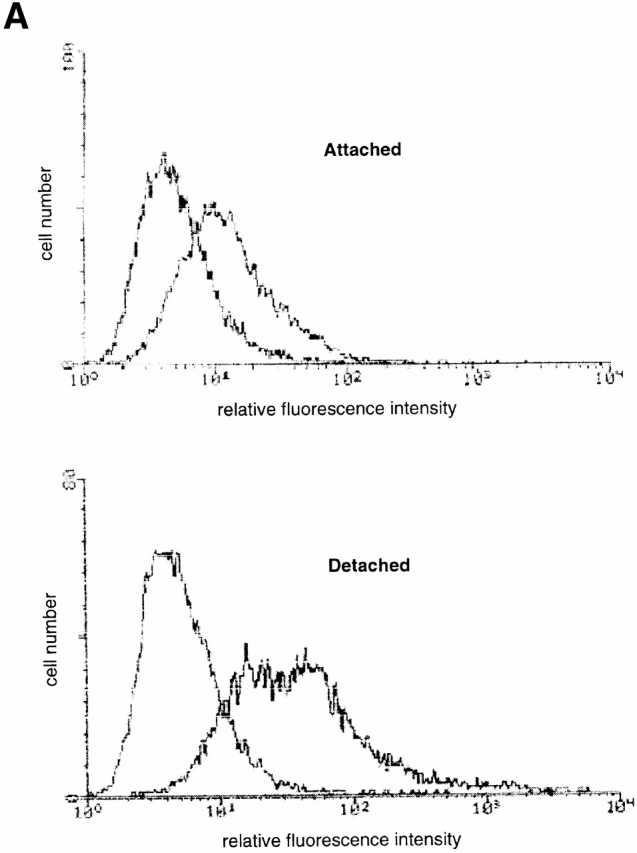
Cell detachment upregulates Fas expression in HUVECs. (A) Detachment induces cell surface levels of Fas. HUVECs were kept adherent or in suspension for 12 h. FACS® analysis with anti-Fas antibody (UB2) was carried out as described in Materials and Methods. (B) Cell detachment increases the mRNA levels of Fas. HUVECs were kept adherent or in suspension for the indicated times, and Fas mRNA levels were determined by an RT-PCR analysis as described in Materials and Methods.
Figure 6.

Exogenous expression of both Fas and caspase-8 is required to render attached HUVECs sensitive to Fas-mediated apoptosis. HUVECs were transfected with an empty vector (control), or with plasmids encoding Fas and procaspase-8, together with plasmid coding for the GFP. 24 h after transfection, the cells were stimulated or not for an additional 24 h with 1 μg/ml of anti-Fas antibody CH11. Apoptosis analysis by FACS® was carried out in the double positive cell population for propidium iodide and fluorescent GFP as described above. Bars indicate SD in a representative experiment done in triplicate.
Role of c-Flip in Adhesion-dependent Regulation of Fas Signaling
Previous reports have demonstrated that Fas-mediated apoptosis can be modulated at the level of caspase-8 activation by a mechanism that involves c-Flip, an endogenous caspase-8 inhibitor (Irmler et al. 1997), and it has been suggested that the balance between the expression levels of c-Flip and caspase-8 may control the sensitivity of a given cell to Fas-mediated killing (Scaffidi et al. 1999). Two different isoforms of c-Flip, namely Flip long (FlipL) and Flip short (FlipS), have been detected in various cell types and both isoforms are known to interfere with caspase-8 activation (Irmler et al. 1997; Scaffidi et al. 1999). As shown in Fig. 5 A, both FlipL and FlipS are expressed at relatively high levels compared with caspase-8 in adherent HUVECs. However, in HUVECs that have been detached for 8 h, the c-Flip protein levels are significantly reduced compared with caspase-8 levels (the apparent disappearance of the proform of caspase-8 protein is due to the processing of the protein that takes place during caspase-8 activation). To study the levels of c-Flip in detached HUVECs in detail, a time course analysis of the protein (Fig. 5 B, top) and mRNA levels (as measured by semiquantitative RT-PCR; Fig. 5 B, bottom) of FlipL and FlipS were carried out. Our findings indicate that HUVEC detachment induces a rapid reduction of FlipL and, more notably, FlipS at both mRNA and protein levels. A clear decrease in Flip protein can be detected as early as 6 h after cell detachment, demonstrating that the reduction in the c-Flip protein levels precedes the bulk of caspase-8 activation and anoikis observed in detached HUVECs. Thus, matrix attachment and detachment may control Fas-mediated apoptosis and caspase-8 activation by regulating the levels of the caspase-8 antagonist, c-Flip.
Figure 5.
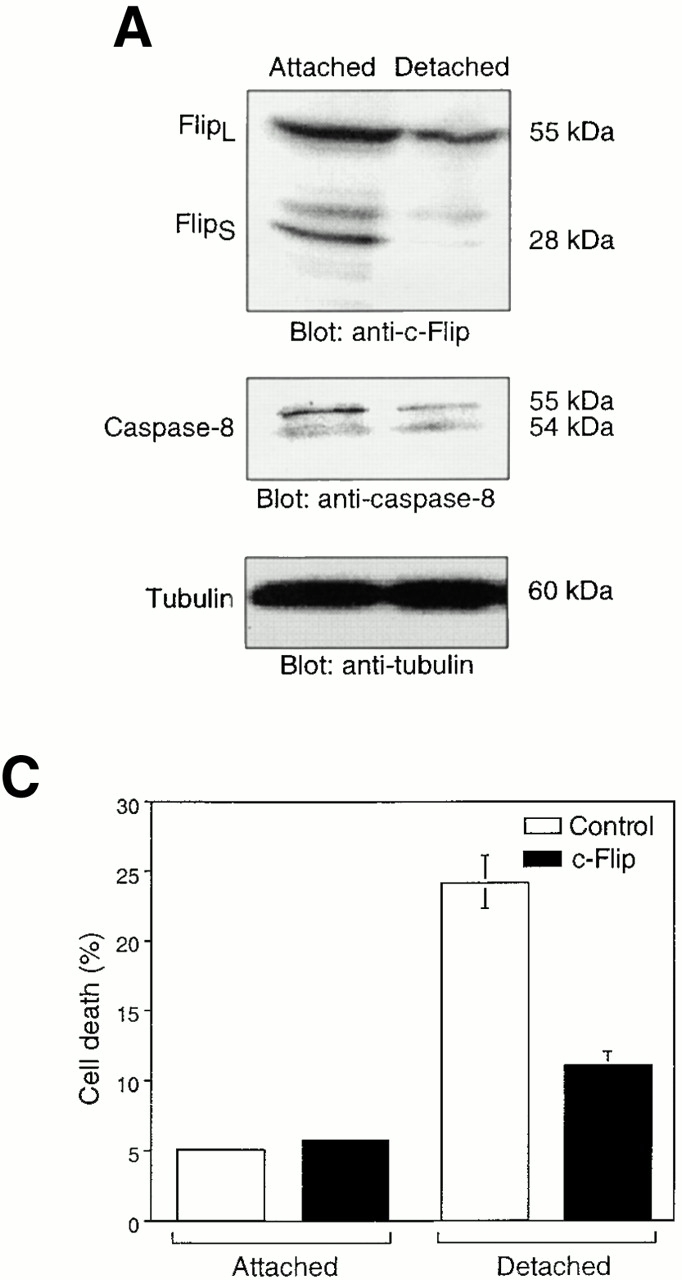
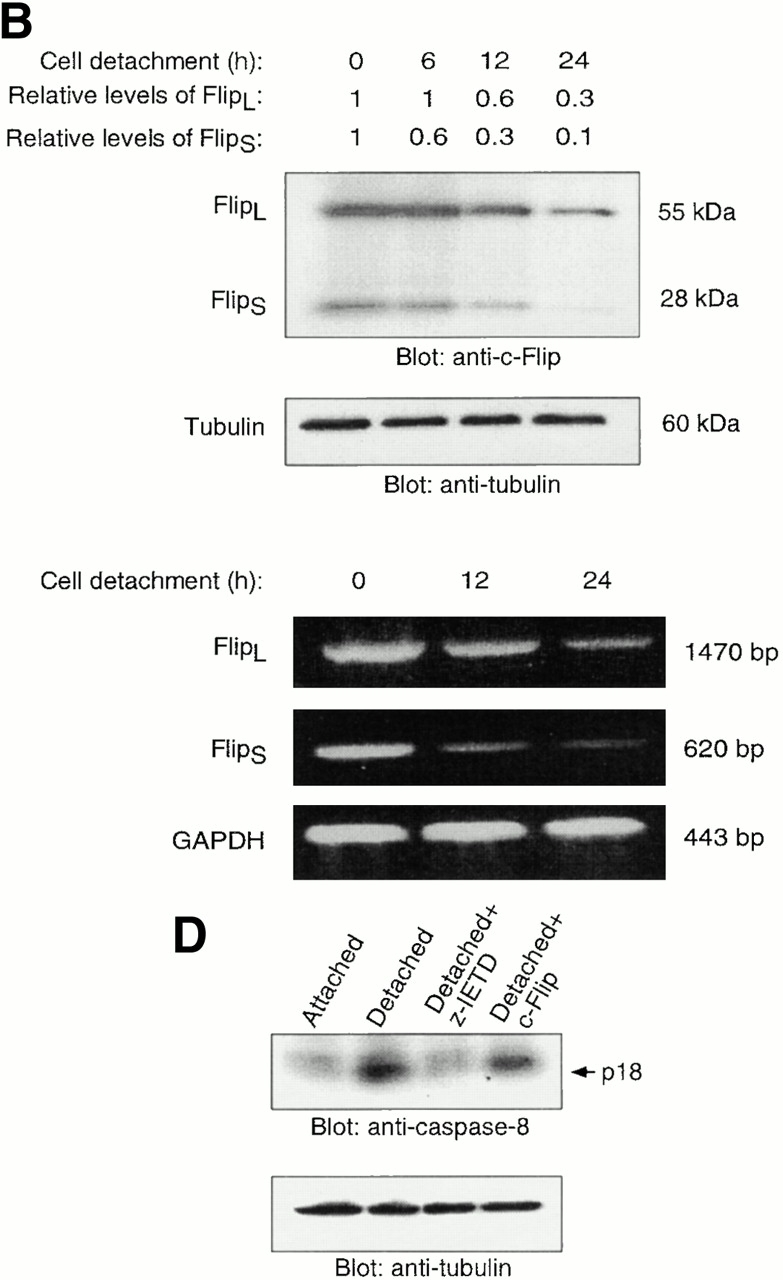
Cell detachment results in downmodulation of c-Flip expression. Exogenous expression of c-Flip protects HUVECs from anoikis and prevents caspase-8 activation in detached cells. (A) Cell lysates were prepared from adherent HUVECs and from HUVECs that had been kept in suspension for 8 h, and the lysates were subjected to immunoblotting analysis by antibodies against c-Flip (top), caspase-8 (5F7; middle), and tubulin (bottom). (B) c-Flip expression was determined in attached cells or in cells that had been kept in suspension for the indicated times by immunoblot analysis (top two panels) and by RT-PCR (bottom three panels). As a control, protein expression levels in the lysates were analyzed by antitubulin immunoblotting (top), and mRNA expression levels by RT-PCR analysis with primers specific for GAPDH (bottom). The relative expression levels of Flip proteins as quantitated with a densitometric analysis are indicated. (C) Exogenous expression of c-Flip protects HUVECs from anoikis. HUVECs were cotransfected with an empty vector (control) or with plasmid encoding c-FlipL, together with plasmid coding for GFP. 24 h after transfection, the cells were either kept adherent or in suspension for 12 h. Apoptosis analysis by FACS® was carried out in the double positive cell population for propidium iodide and fluorescent GFP as described above. Bars indicate SD in a representative experiment done in triplicate. (D) Exogenous expression of c-Flip blocks caspase-8 activation in detached HUVECs. HUVECs were transiently transfected with an empty vector or with plasmid encoding c-FlipL. 24 h after transfection, the cells were kept adherent or in suspension for 12 h. In some of the experiments, the cells were simultaneously treated with 20 μM of the caspase-8 inhibitor z-IETD-fmk, as indicated. The cell lysates were analyzed by immunoblotting with an anti–caspase-8 antibody (Ab-1) that detects the p18 active form of the enzyme. The membrane was stripped and reprobed with an anti–α-tubulin control antibody to confirm equal loading (bottom).
To analyze the putative functional role of c-Flip in adhesion-dependent regulation of anoikis and Fas-mediated apoptosis, two lines of experimentation were undertaken. First, exogenous c-Flip was expressed in detached HUVECs at levels that mimic the c-Flip/caspase-8 ratio observed in adherent cells, and anoikis was monitored in the resulting cells. As shown in Fig. 5 C, exogenous expression of c-Flip blocks anoikis by ∼75%. As shown in Fig. 5 D, expression of c-Flip inhibits caspase-8 activation in detached cells, up to 30% of the inhibition that is observed when detached cells are treated with the specific caspase-8 inhibitor, z-IETD-fmk. Note that caspase-8 activity, which is measured by the appearance of the p18 active form of caspase-8, is monitored in the total pool of c-Flip–transfected cells, and we estimate that the transient transfection efficiency achieved in the cells is ∼30% (not shown). Thus, c-Flip appears to be a highly efficient inhibitor of caspase-8 in HUVECs. Taken together, these studies indicate that downmodulation of c-Flip during cell detachment may be a necessary and causal event in detachment-induced caspase-8 activation and HUVEC anoikis.
In a reciprocal experiment, exogenous caspase-8 was expressed in attached HUVECs to mimic the c-Flip/caspase-8 ratio observed in detached cells, and the susceptibility of the cells for Fas-induced apoptosis was analyzed. As shown in Fig. 6, exogenous expression of caspase-8 alone does not induce apoptosis in HUVEC cells, nor does it make the cells sensitive to CH11-induced apoptosis. However, exogenous expression of both caspase-8 and Fas renders HUVECs sensitive to CH11-mediated killing (Fig. 6). Note, though, that exogenous expression of Fas and caspase-8 does not induce apoptosis in non–CH11-treated cells, despite the fact that the cells express endogenous Fas-L (see Discussion). In conclusion, it appears that cell adhesion regulates anoikis and Fas-mediated apoptosis (at least) at two different levels: first, by regulating the cell surface levels of Fas, thereby controlling the availability of the death receptor for its ligand; and second, by regulating the expression levels of c-Flip, thereby controlling death receptor–induced activation of caspase-8.
Roles of the PI 3′-kinase/Akt and Erk Survival Pathways in HUVECs
In many cell types, matrix attachment has been shown to result in activation of the PI 3′-kinase/Akt and MAPK/Erk pathways, whereas cell detachment is known to downregulate these activities (for review see Giancotti and Ruoslahti 1999; Schlaepfer et al. 1999). Furthermore, activation of the PI 3′-kinase/Akt and the Erk pathways has been shown to protect MDCK and CCL39 cells, respectively, against detachment-induced cell death (Khwaja et al. 1997; Le Gall et al. 2000). We found that matrix attachment activates and matrix detachment inactivates the two pathways in HUVECs (results for the Erk pathway are shown in Fig. 8 B), and we therefore decided to examine the role of these two survival pathways in adhesion-mediated protection against Fas-induced apoptosis and anoikis in endothelial cells.
Figure 8.

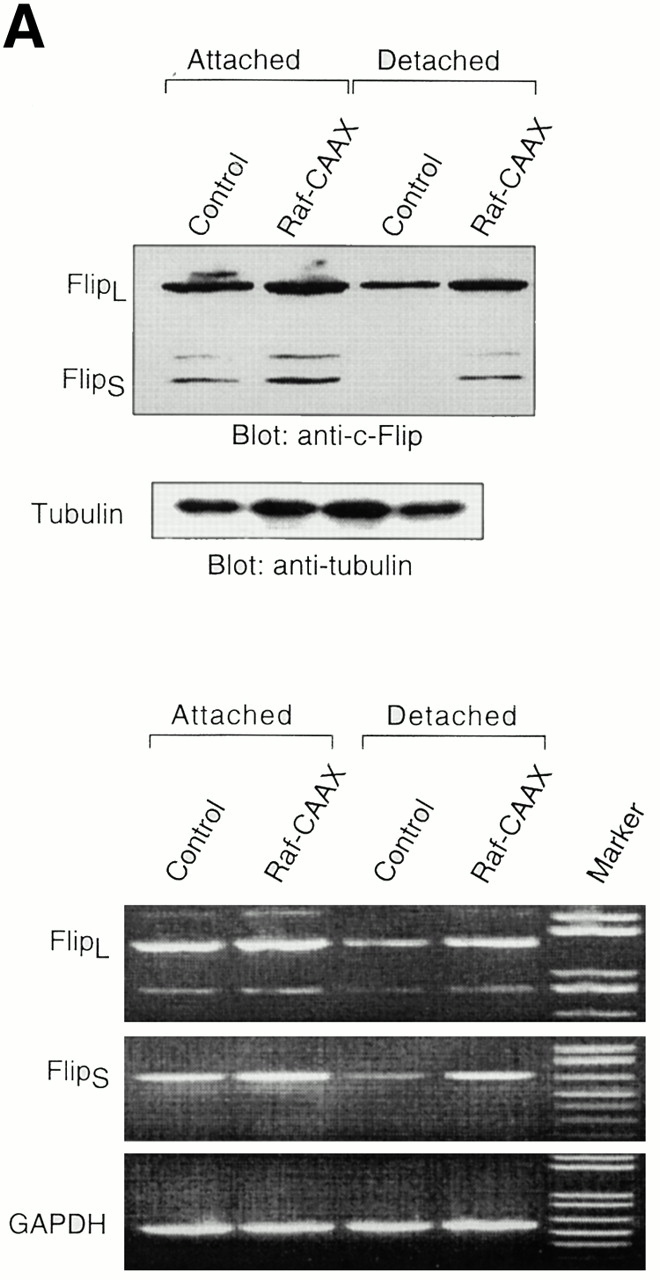
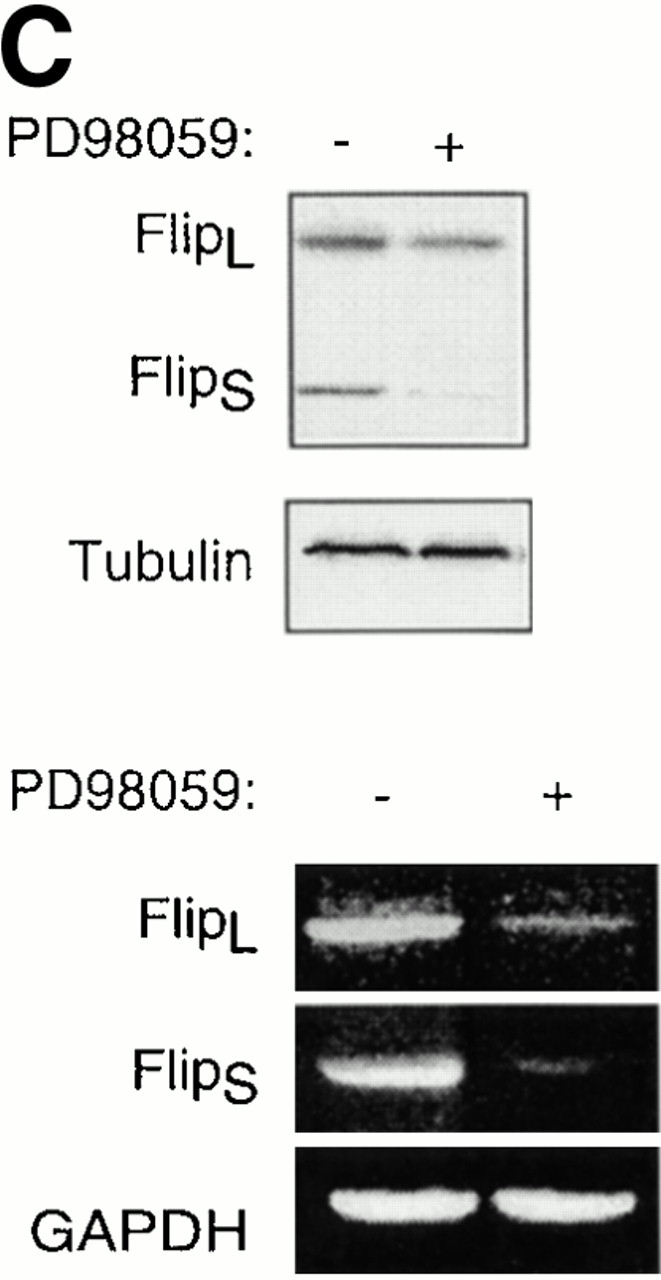


The Erk pathway functions as a survival pathway in attached HUVECs and modulates c-Flip expression. (A) Exogenous expression of activated Raf-1 enhances c-Flip expression in detached HUVECs. HUVECs were transfected with an empty vector (Control) or with a plasmid encoding activated form of c-Raf-1 (Raf-CAAX). After transfection, the cells were kept adherent or in suspension for 24 h. c-Flip expression was determined by immunoblot analysis (top) and by RT-PCR (bottom). (B) Cell detachment results in the inactivation of the MAPK/Erk pathway in a time-dependent manner. HUVECs were kept adherent or in suspension for the indicated times. Cell lysates were prepared and Erk activation was determined by an immunoblot analysis with an anti–phospho-Erk1/2 antibody (top). The membrane was stripped and reprobed with an anti-Erk2 antibody to confirm equal loading (bottom). As indicated, in one of the experiments, the effectiveness of the MEK inhibitor PD98059 was analyzed by treating adherent HUVECs for 16 h before cell lysis. (C) Inhibition of the Erk pathway in adherent HUVECs downregulates c-Flip expression. HUVECs were treated or not with the MEK inhibitor PD98059 for 16 h and c-Flip expression was determined by immunoblotting (top) and by RT-PCR (bottom). (D) Inhibition of the Erk pathway in adherent HUVECs sensitizes the cells to Fas-mediated apoptosis. HUVECs were transiently transfected with an empty control vector or with a plasmid encoding Fas, together with plasmid coding for GFP. After transfection, the cells were treated or not with 25 μM of the MEK inhibitor PD98059 in the presence or absence of 1 μg/ml of the anti-Fas antibody CH11 for 24 h. Apoptosis analysis by FACS® was carried out in the double positive cell population for propidium iodide and fluorescent GFP as described above. Bars indicate SD in a representative experiment done in triplicate. (E) Exogenous expression of activated Raf-1 enhances Erk phosphorylation in HUVECs. HUVECs were transfected with an empty vector (Control) or with a plasmid encoding activated form of c-Raf-1 (Raf-CAAX). After transfection, the cells were kept adherent or in suspension for 12 h. Cell lysates were prepared and Erk activation was determined by an immunoblot analysis with an anti–phospho-Erk1/2 antibody (top). The membrane was stripped and reprobed with an anti-Erk2 antibody to confirm equal loading (bottom).
We first examined whether constitutive activation of the PI 3′-kinase/Akt pathway or the Erk pathway in detached HUVECs would protect these cells against anoikis. As shown in Fig. 7, expression of constitutively activated PI 3′-kinase (p110*), Akt (myr-Akt), or c-Raf-1 (Raf-CAAX; this construct constitutively activates the Erk pathway) (Kazama and Yonehara 2000) inhibits anoikis by 50–80% compared with control transfected cells, demonstrating that exogenous activation of either the PI 3′-kinase/Akt or the Erk pathway mediates survival signaling that is sufficient to block anoikis in HUVECs.
Figure 7.

Ectopic activation of the PI 3′-kinase/Akt and Erk pathways prevents anoikis in HUVECs. HUVECs were cotransfected with an empty vector (control) or with plasmids encoding activated forms of PI 3′-kinase (p110*), Akt (Myr-Akt), and c-Raf-1 (Raf-CAAX), together with a plasmid coding for GFP. After transfection, the cells were kept adherent or in suspension for 24 h. Apoptosis was determined by using FACS® in the double positive cell population for GFP and propidium iodide as described above. Bars indicate SD in an experiment done in triplicate.
The Erk Pathway Regulates c-Flip Expression in HUVECs
Exogenous expression of activated PI 3′-kinase or Akt had no effect on the expression levels of Fas, Fas-L, or c-Flip, suggesting that the PI 3′-kinase/Akt pathway exerts its cell survival effect on anoikis through a different mechanism (data not shown; see Discussion). Also, exogenous expression of activated c-Raf-1 had no effect on the expression levels of Fas and Fas-L in HUVECs (data not shown). As shown in Fig. 8 A, exogenous expression of activated c-Raf-1 results in a slight increase in the mRNA and protein levels of both FlipL and FlipS in attached cells. Importantly, expression of activated c-Raf-1 almost completely prevents downregulation of FlipL and FlipS mRNA and protein levels in detached cells. As a control experiment, we found that expression of activated c-Raf-1 results in an increase in Erk phosphorylation in HUVEC cells; notably, expression of activated c-Raf-1 significantly blocks downregulation of Erk activity in detached cells (Fig. 8 E). Keeping in mind the finding that activated PI 3′-kinase and Akt block anoikis in HUVECs without affecting the expression levels of c-Flip, it is unlikely that the effect of c-Raf-1 on c-Flip expression is secondary to enhanced survival of the cells. As a further support for this, we found that the broad spectrum caspase inhibitor z-VAD blocks anoikis in HUVEC cells, but has no effect on Flip expression in detached cells (not shown). Thus, we conclude that the suppressive effect that the Raf/Erk pathway has on anoikis is likely to be due, at least in part, to Raf/Erk-mediated upregulation of c-Flip.
As a corollary to these findings, inactivation of the Erk pathway upon cell detachment may be necessary for downregulation of c-Flip expression levels, caspase-8 activation, and anoikis to take place in HUVECs. Furthermore, the Raf/Erk pathway may in turn mediate upregulation of c-Flip expression in attached HUVECs, and thereby function as a survival pathway in adherent cells, protecting them against Fas-mediated apoptosis. As shown in Fig. 8 B, Erk activity is downregulated in a time-dependent manner upon HUVEC detachment, and, importantly, this downregulation precedes downregulation of c-Flip expression (see Fig. 5) and induction of anoikis (see Fig. 1) in detached HUVECs. To characterize the putative functional role of the Erk pathway in regulating c-Flip expression and Fas-mediated apoptosis in attached HUVECs, we used the Mek1 inhibitor PD90839 to block attachment-induced Erk activation in adherent endothelial cells. As shown in Fig. 8, treatment of attached HUVECs with PD90839 results in a strong inhibition of Erk kinase activity (Fig. 8 B) and in reduction of both mRNA and protein levels of c-Flip in attached endothelial cells (Fig. 8 C). Thus, Erk activity appears to be necessary for the upregulation of c-Flip expression in adherent endothelial cells. We further found that Erk activity is required for attachment-mediated protection against Fas-induced apoptosis. Attached HUVECs were transiently transfected to express Fas, the cells were treated or not treated with the Mek1 inhibitor, and the susceptibility of the cells to CH11-mediated apoptosis was analyzed. As shown in Fig. 8 D, treatment of Fas-expressing cells with the Mek1 inhibitor sensitizes attached HUVECs to Fas-mediated apoptosis. Thus, the Erk pathway functions as a survival pathway in attached HUVECs to protect against Fas-mediated apoptosis, and this protection, at least in part, is likely to be due to regulation of c-Flip expression by the Erk kinase cascade.
Discussion
Recent experimental evidence has demonstrated that inhibition of endothelial cell apoptosis is an essential prerequisite to maintain vascular remodeling and angiogenesis. Appropriate endothelial cell–extracellular matrix interactions are known to be required for endothelial cell viability both in vitro and in vivo, but at present, molecular mechanisms that couple adhesion-dependent survival signaling to the antiapoptotic machinery in endothelial cells remain largely unknown. Our present findings provide several novel insights into the mechanisms that regulate endothelial cell survival and apoptosis. First, we demonstrate that cell–matrix interaction has a profound effect in regulating Fas-mediated apoptosis in HUVECs. Thus, we find that adherent endothelial cells are resistant to Fas-mediated apoptosis, even when significant levels of Fas receptor are expressed on the cell surface. Second, we identify the molecular mechanisms as to how matrix attachment regulates Fas-mediated apoptosis, namely by regulating the expression levels of Fas, thereby controlling the availability of the death receptor for its ligand, and by regulating the expression levels of c-Flip, thereby controlling death receptor–induced activation of caspase-8. Third, we find that detachment-induced cell death, or anoikis, takes place as a result of activation of the Fas/Fas-L death pathway, and our results therefore provide a molecular mechanism for anoikis in endothelial cells.
Fas, also known as APO-1 or CD95, is one of the best characterized members of the death receptor family. It has been suggested that the increased sensitization to Fas-mediated apoptosis may play an important role in regulation of pathological tissue destruction, but the mechanisms by which stimuli sensitize cells to Fas-mediated apoptosis have remained to be elucidated. Our finding that matrix attachment is a powerful regulator of Fas-mediated apoptosis provides one of the first insights into the regulatory mechanisms that determine the responsiveness of cells to Fas-induced killing. As discussed below in more detail, our results indicate that c-Flip, an antagonist of caspase-8, has a crucial role in adhesion-dependent survival signaling. Taking into consideration the central role of caspase-8 in induction of apoptosis by several tumor necrosis factor family death receptors, it is plausible to assume that matrix attachment regulates apoptosis triggered by other death receptors as well. Further, recent studies have indicated that cell attachment mediates cell survival against serum starvation and growth factor withdrawal–induced apoptosis (see Introduction), and in many cases these forms of cell death are initiated and activated at the level of mitochondria, rather than by cell surface death receptors. Our finding that cell attachment to ECM provides survival signaling against Fas, and possible other death receptor–induced apoptosis, supports an emerging role for ECM as a crucial survival factor against most, if not all, forms of programmed cell death.
While cell attachment to matrix provides survival signaling against various apoptotic stimuli, detachment of cells, without addition of exogenous death stimuli, induces apoptosis in epithelial and endothelial cells. This mechanism, known as anoikis, ensures that these cells do not normally survive in the absence of the correct interaction with ECM and therefore are unable to proliferate in an inappropriate location (Frisch and Ruoslahti 1997). Until recently, the molecular mechanisms that regulate anoikis have remained largely unknown. In the present study, we demonstrate by several biochemical means that activation of the Fas/Fas-L pathway takes place upon endothelial cell detachment and that activation of this pathway precedes anoikis in HUVECs. By using various inhibitory reagents against Fas-L, Fas, and the components of the downstream signaling pathway, we show that Fas-L–mediated activation of the Fas pathway is causal to anoikis in HUVECs. Previously, involvement of a death receptor pathway has been implicated in anoikis in epithelial cells (Frisch 1999; Rytomaa et al. 1999). In these studies, it was shown that anoikis can be blocked by dnFADD and by inhibitors of caspase-8. However, soluble extracellular domains of various death receptors failed to block anoikis, and the authors therefore concluded that ligand-dependent activation of death receptors is unlikely to be involved in anoikis (Rytomaa et al. 1999). In our studies, we have not used soluble extracellular domains as inhibitors, but rather applied inhibitory antibodies against Fas and Fas-L. These antibodies have been widely used as specific inhibitors against Fas-mediated apoptosis by several investigators (Kayagaki et al. 1995; Srivastava et al. 1999). Our studies demonstrate that the bulk of anoikis in endothelial cells is mediated by the Fas/Fas-L interaction, although we cannot rule out the possibility that other death receptors could also have some role in anoikis in endothelial cells. It also remains to be determined whether our findings are applicable to all types of endothelial cells. While the detailed mechanism of anoikis is not known in epithelial cells, it is possible that endothelial and epithelial cells utilize slightly different pathways to activate the death machinery upon cell detachment.
Our results indicate that matrix adhesion regulates the expression levels of Fas-L, Fas, and c-Flip, but not of several other components of the Fas pathway, such as caspase-8 and FADD (not shown). Most significantly, detachment of cells was found to result in an enhancement of Fas expression and in downregulation of Flip expression, and both of these events preceded anoikis in HUVECs. We further found that enhanced Fas expression is necessary, but not sufficient, to sensitize attached HUVECs to Fas-induced apoptosis. This finding is in agreement with a recent report, in which it was shown that interferon-induced Fas expression is not sufficient to sensitize HUVECs to Fas-induced cell death (Sata et al. 2000). In addition to enhanced Fas expression, we found that modulation of the caspase-8/c-Flip ratio to favor caspase-8 activation is necessary for Fas-induced apoptosis to occur in attached HUVECs. In a reciprocal experiment, we found that enhanced expression of c-Flip in detached cells is sufficient to inhibit caspase-8 activation and anoikis in HUVECs. Our results therefore identify regulation of Fas and Flip expression levels as a key event in adhesion-dependent control of Fas-mediated apoptosis.
Recent studies using Flip-null fibroblasts have demonstrated that Flip proteins, which structurally resemble caspase-8 except that they lack proteolytic activity, have a crucial role in death factor–induced apoptosis (Yeh et al. 2000). At present, the relative roles of the two splice variants of c-Flip, FlipL and FlipS, remain to be elucidated. It has been recently demonstrated that in T cell receptor–restimulated T cells, amounts of FlipL are slightly changed, whereas there is a significant change in the amount of FlipS protein expression, and the authors conclude that it is the change in the levels of FlipS that is responsible for the Fas-resistant phenotype in this model system (Kirchhoff et al. 2000). We have similarly found that cell attachment mainly affects the levels of FlipS (see Fig. 5), and it may be that also in our model system FlipS has a more dominant role in the inhibition of Fas-mediated apoptosis. Relatively little is also known about the physiological and pathological stimuli that modulate Flip expression. Our results demonstrate, for the first time, Flip regulation by cell adhesion receptors. Our findings also add to the emerging notion that regulation of Flip may have a crucial role in controlling endothelial cell survival. Sata and Walsh 1998a have recently demonstrated that endothelial cell apoptosis induced by oxidized low density lipoprotein is associated with downregulation of Flip. Modulation of Flip has also been linked to angiogenesis; a recombinant fragment derived from the α2 chain of type IV collagen, termed canstatin, has antiangiogenic activity, and it has been reported that canstatin-induced endothelial cell apoptosis coincides with downregulation of Flip (Kamphaus et al. 2000). By using several lines of investigation, we found that the MAPK/Erk pathway regulates Flip expression and Fas-mediated apoptosis in HUVECs. In addition to our findings here, a recent report indicates that the Erk pathway regulates c-Flip also in T lymphocytes (Yeh et al. 1998). Of note, the αvβ3 integrin, which mediates endothelial cell survival in vivo, has been shown to be necessary for sustained Erk activation during angiogenesis (Eliceiri et al. 1998). It remains to be determined whether the αvβ3 integrin regulates c-Flip expression in vivo, and whether the Erk pathway has a central role in controlling Flip in cellular systems other than endothelial cells and lymphocytes. While we found that adhesion-dependent modulation of the Erk pathway is both necessary and sufficient to regulate c-Flip levels in endothelial cells, our results indicate that this pathway is not involved in regulating the expression levels of Fas and Fas-L (data not shown). Experiments addressing how matrix attachment regulates Fas and Fas-L expression in HUVECs are currently ongoing.
In epithelial cells, disruption of the PI 3′-kinase/Akt pathway in attached cells leads to apoptosis, whereas exogenous expression of activated forms of PI 3′-kinase and Akt protects epithelial cells against anoikis (Khwaja et al. 1997). We found here that exogenous expression of activated PI 3′-kinase and Akt blocks anoikis in endothelial cells. However, treatment of attached HUVECs with inhibitors that disrupt the PI 3′-kinase/Akt pathway did not lead to apoptosis or to sensitization of the cells to anti-Fas–induced apoptosis (data not shown). However, further activation of the PI 3′-kinase/Akt pathway in attached cells provided additional survival signaling in endothelial cells. Thus, we found that expression of activated forms of PI 3′-kinase and Akt suppressed CH11-induced apoptosis in Fas- and caspase-8–transfected, attached HUVECs (not shown). It is also known that treatment of HUVECs with vascular endothelial growth factor or angiopoietin-1 results in enhanced Akt activation and protection against serum withdrawal–induced apoptosis (Fujikawa et al. 1999; Fujio and Walsh 1999; Kim et al. 2000; Papapetropoulos et al. 2000). At present, we do not know how the PI 3′-kinase/Akt signaling pathway inhibits anoikis in HUVECs, but it does not do so by inhibiting the expression of Fas and Fas-L or by upregulating the expression of c-Flip (data not shown). Previous studies have identified several targets for the antiapoptotic function of Akt, and include inactivation of the proapoptotic proteins Bad and Bax and of caspase-9 (Datta et al. 1999), all of which participate in mitochondria-regulated apoptosis. Our hypothesis is that activation of the mitochondrial death pathway downstream of caspase-8 activation is required for anoikis and Fas-mediated apoptosis to take place in endothelial cells; indeed, oxidized LDL-induced Fas-mediated death in HUVECs can be blocked by Bcl-xL expression, which functions prosurvival at the level of mitochondria (Sata and Walsh 1998a). Bcl-xL has also been found to protect against anoikis in epithelial cells (Rytomaa et al. 1999; Rosen et al. 2000). Thus, activated PI 3′-kinase and Akt molecules may block anoikis in detached cells by preventing activation of the mitochondrial death pathway in HUVECs, whereas inactivation of this survival pathway in and of itself in attached endothelial cells does not activate death signaling.
Ilic and coworkers 1998 have recently demonstrated that when serum is absent, correct matrix attachment transduces survival signals via FAK to suppress p53-regulated apoptosis in primary fibroblasts and endothelial cells. The same authors have also found that activation of the c-Jun NH2-terminal kinase (JNK) pathway downstream of FAK is required for anchorage-dependent survival under these conditions (Almeida et al. 2000). In our experimental system, in which apoptosis is induced in endothelial cells via stimulation of Fas or by cell detachment in the presence of serum, we found no indication that the FAK/JNK/p53 pathway would have a crucial role in regulating cell survival. Thus, dominant negative forms of FAK and JNK kinase (JNKK) (Ilic et al. 1998; Almeida et al. 2000) failed to sensitize cells to Fas-mediated apoptosis, even when the Fas receptor was exogenously expressed in the cells. Along the same lines, expression of dominant negative forms of FAK and JNKK had no effect on the expression levels of c-Flip. This finding is in accordance with the observation that FAK does not mediate adhesion-dependent Erk activation in HUVECs (data not shown; Barberis et al. 2000). Also, we used a dominant negative form of p53 (GSE 56) (Ossovskaya et al. 1996) to inhibit p53 function in HUVECs and found that suppression of p53 activity does not rescue HUVECs from Fas-induced or detachment-induced apoptosis. Taken together, our results indicate that the commitment to the Fas-mediated death pathway, which in our model system is activated either by treating cells with the agonistic Fas-antibody CH11 or by cell detachment in the presence of serum, appears to be distinct from commitment to p53-regulated apoptosis that is initiated upon withdrawal of ECM under serum-free conditions. This notion is supported by the earlier finding that CrmA, which blocks Fas-induced apoptosis, does not block p53-regulated apoptosis triggered either by FAK inactivation or by culture of cells on an inappropriate ECM in the absence of serum (Ilic et al. 1998). Along the same lines, we find that detachment-induced cell death in the absence of serum is not inhibitable by caspase-8 inhibitors (not shown).
Our results indicate that attached HUVECs that overexpress Fas and caspase-8, despite the fact that they constitutively express Fas-L on the cell surface, are not apoptotic unless the agonistic Fas antibody CH11 is added to the cultures. Our studies further indicate that Fas-L and Fas do not interact in attached cells (data not shown). It has been demonstrated recently that epithelial cell detachment activates a metalloproteinase that cleaves Fas-L to produce soluble Fas-L, which then binds to Fas and induces apoptosis (Powell et al. 1999). We have tested the possibility that cleavage of Fas-L would take place upon cell detachment and that this processing would be required for anoikis in endothelial cells, and we found that this is unlikely to be the case in HUVECs. First, the only form of Fas-L that can be immunoprecipitated from attached or detached HUVEC lysates or conditioned media has a molecular mass of 37 kD, which corresponds to the membrane form of Fas-L. Second, addition of metalloproteinase inhibitors that have been shown to block processing of soluble Fas-L has no effect on the cell surface expression levels of Fas-L; inhibition of soluble Fas-L formation by these inhibitors should result in an increase in the amount of membrane-bound Fas-L (Kayagaki et al. 1995). Finally, addition of metalloprotease inhibitors had, unlike in epithelial cells (Powell et al. 1999), no effect on anoikis in HUVECs (data not shown). It is possible that when cultured as a monolayer, the cell–cell contacts in HUVECs are not optimal to allow Fas/Fas-L interaction to occur. When endothelial cells are grown in suspension or on polyhema to induce anoikis, the cells become round and form cell aggregates via a yet-to-be-determined mechanism (data not shown), and it may be that this aggregation is crucial for Fas/Fas-L interaction and induction of Fas-mediated apoptosis in detached cells. The possibility that induction of anoikis in endothelial cells would involve a previously unidentified processing and/or activation mechanism at the level of Fas/Fas-L interaction cannot be ruled out and warrants further studies.
Acknowledgments
We thank Steven Frisch, Andrei Gudkov, John Hancock, Dusko Ilic, Donald Nicholson, Jerrold Olefsky, John Reed, Taka-Aki Sato, Guy Salvesen, and Phil Tsichlis for providing reagents used in this study.
This work was supported by National Institutes of Health grants CA71560 and CA76037 to K. Vuori. K. Vuori is a Pew Scholar in Biomedical Sciences.
Footnotes
Abbreviations used in this paper: AICD, activation-induced cell death; DISC, death-inducing signaling complex; dn, dominant negative; ECM, extracellular matrix; Erk, extracellular signal–regulated kinase; FADD, Fas-associated death domain; FAK, focal adhesion kinase; GFP, green fluorescent protein; HUVEC, human umbilical vein endothelial cell; JNK, c-Jun NH2-terminal kinase; JNKK, JNK kinase; MAPK, mitogen-activated protein kinase; MEK, MAP kinase kinase; PI 3′-kinase, phosphatidylinositol 3′-kinase; RT, reverse transcriptase.
References
- Almeida E.A., Ilic D., Han Q., Hauck C.R., Jin F., Kawakatsu H., Schlaepfer D.D., Damsky C.H. Matrix survival signalingfrom fibronectin via focal adhesion kinase to c-Jun NH2-terminal kinase. J. Cell Biol. 2000;149:741–754. doi: 10.1083/jcb.149.3.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoudjit F., Vuori K. Engagement of the alpha2beta1 integrin inhibits Fas ligand expression and activation-induced cell death in T cells in a focal adhesion kinase-dependent manner. Blood. 2000;95:2044–2051. [PubMed] [Google Scholar]
- Barberis L., Wary K.K., Fiucci G., Liu F., Hirsch E., Brancaccio M., Altruda F., Tarone G., Giancotti F.G. Distinct roles of the adaptor protein shc and focal adhesion kinase in integrin signaling to ERK. J. Biol. Chem. 2000;275:36532–36540. doi: 10.1074/jbc.M002487200. [DOI] [PubMed] [Google Scholar]
- Brooks P.C., Montgomery A.M., Rosenfeld M., Reisfeld R.A., Hu T., Klier G., Cheresh D.A. Integrin αvβ3 antagonists promote tumor regression by inducing apoptosis of angiogenic blood vessels. Cell. 1994;79:1157–1164. doi: 10.1016/0092-8674(94)90007-8. [DOI] [PubMed] [Google Scholar]
- Datta S.R., Brunet A., Greenberg M.E. Cellular survivala play in three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- Eliceiri B.P., Klemke R., Stromblad S., Cheresh D.A. Integrin alphavbeta3 requirement for sustained mitogen-activated protein kinase activity during angiogenesis. J. Cell Biol. 1998;140:1255–1263. doi: 10.1083/jcb.140.5.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch S.M. Evidence for a function of death-receptor-related, death-domain-containing proteins in anoikis. Curr. Biol. 1999;9:1047–1049. doi: 10.1016/s0960-9822(99)80455-2. [DOI] [PubMed] [Google Scholar]
- Frisch S.M., Ruoslahti E. Integrins and anoikis. Curr. Opin. Cell Biol. 1997;9:701–706. doi: 10.1016/s0955-0674(97)80124-x. [DOI] [PubMed] [Google Scholar]
- Frisch S.M., Vuori K., Ruoslahti E., Chan-Hui P.Y. Control of adhesion-dependent cell survival by focal adhesion kinase. J. Cell Biol. 1996;134:793–799. doi: 10.1083/jcb.134.3.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujikawa K., de Aos Scherpenseel I., Jain S.K., Presman E., Christensen R.A., Varticovski L. Role of PI 3-kinase in angiopoietin-1-mediated migration and attachment-dependent survival of endothelial cells. Exp. Cell Res. 1999;253:663–672. doi: 10.1006/excr.1999.4693. [DOI] [PubMed] [Google Scholar]
- Fujio Y., Walsh K. Akt mediates cytoprotection of endothelial cells by vascular endothelial growth factor in an anchorage-dependent manner. J. Biol. Chem. 1999;274:16349–16354. doi: 10.1074/jbc.274.23.16349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giancotti F.G., Ruoslahti E. Integrin signaling. Science. 1999;285:1028–1032. doi: 10.1126/science.285.5430.1028. [DOI] [PubMed] [Google Scholar]
- Gilmore A.P., Metcalfe A.D., Romer L.H., Streuli C.H. Integrin-mediated survival signals regulate the apoptotic function of Bax through its conformation and subcellular localization. J. Cell Biol. 2000;149:431–446. doi: 10.1083/jcb.149.2.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hungerford J.E., Compton M.T., Matter M.L., Hoffstrom B.G., Otey C.A. Inhibition of pp125FAK in cultured fibroblasts results in apoptosis. J. Cell Biol. 1996;135:1383–1390. doi: 10.1083/jcb.135.5.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilic D., Almeida E.A., Schlaepfer D.D., Dazin P., Aizawa S., Damsky C.H. Extracellular matrix survival signals transduced by focal adhesion kinase suppress p53-mediated apoptosis. J. Cell Biol. 1998;143:547–560. doi: 10.1083/jcb.143.2.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irmler M., Thome M., Hahne M., Schneider P., Hofmann K., Steiner V., Bodmer J.L., Schroter M., Burns K., Mattmann C. Inhibition of death receptor signals by cellular FLIP. Nature. 1997;388:190–195. doi: 10.1038/40657. [DOI] [PubMed] [Google Scholar]
- Kamitani T., Nguyen H.P., Yeh E.T.H. Activation-induced aggregation and processing of the human Fas antigen. Detection with cytoplasmic domain-specific antibodies. J. Biol. Chem. 1997;272:22307–22314. doi: 10.1074/jbc.272.35.22307. [DOI] [PubMed] [Google Scholar]
- Kamphaus G.D., Colorado P.C., Panka D.J., Hopfer H., Ramchandran R., Torre A., Maeshima Y., Mier J.W., Sukhatme V.P., Kalluri R. Canstatin, a novel matrix-derived inhibitor of angiogenesis and tumor growth. J. Biol. Chem. 2000;275:1209–1215. doi: 10.1074/jbc.275.2.1209. [DOI] [PubMed] [Google Scholar]
- Karsan A., Harlan J.M. Modulation of endothelial cell apoptosismechanisms and pathophysiological roles. J. Atheroscler. Thromb. 1996;3:75–80. doi: 10.5551/jat1994.3.75. [DOI] [PubMed] [Google Scholar]
- Kayagaki N., Kawasaki A., Ebata T., Ohmoto H., Ikeda S., Inoue S., Yoshino K., Okumura K., Yagita H. Metalloproteinase-mediated release of human Fas ligand. J. Exp. Med. 1995;182:1777–1783. doi: 10.1084/jem.182.6.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazama H., Yonehara S. Oncogenic K-Ras and basic fibroblast growth factor prevent Fas-mediated apoptosis in fibroblasts through activation of mitogen-activated protein kinase. J. Cell Biol. 2000;148:557–566. doi: 10.1083/jcb.148.3.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khwaja A., Rodriguez-Viciana P., Wennstrom S., Warne P.H., Downward J. Matrix adhesion and Ras transformation both activate a phosphoinositide 3-OH kinase and protein kinase B/Akt cellular survival pathway. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:2783–2793. doi: 10.1093/emboj/16.10.2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I., Kim H.G., So J.N., Kim J.H., Kwak H.J., Koh G.Y. Angiopoietin-1 regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Circ. Res. 2000;86:24–29. doi: 10.1161/01.res.86.1.24. [DOI] [PubMed] [Google Scholar]
- Kirchhoff S., Muller W.W., Krueger A., Schmitz I., Krammer P.H. TCR-mediated up-regulation of c-FLIP(short) correlates with resistance toward CD95-mediated apoptosis by blocking death-inducing signaling complex activity. J. Immunol. 2000;165:6293–6300. doi: 10.4049/jimmunol.165.11.6293. [DOI] [PubMed] [Google Scholar]
- Krammer P.H. The CD95 (APO/Fas) receptor/ligand systemdeath signals and diseases. Cell Death Differ. 1996;3:159–160. [PubMed] [Google Scholar]
- Le Gall M., Chambard J.C., Breittmayer J.P., Grall D., Pouyssegur J., Van Obberghen-Schilling E. The p42/p44 MAP kinase pathway prevents apoptosis induced by anchorage and serum removal. Mol. Biol. Cell. 2000;11:1103–1112. doi: 10.1091/mbc.11.3.1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meredith J.E., Jr., Fazeli B., Schwartz M.A. The extracellular matrix as a cell survival factor. Mol. Biol. Cell. 1993;4:953–961. doi: 10.1091/mbc.4.9.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulian N., Renvoize C., Desodt C., Serraf A., Berrih-Aknin S. Functional Fas expression in human thymic epithelial cells. Blood. 1999;93:2660–2670. [PubMed] [Google Scholar]
- Nagata S., Golstein P. The Fas death factor. Science. 1995;267:1449–1456. doi: 10.1126/science.7533326. [DOI] [PubMed] [Google Scholar]
- O'Brien V., Frisch S.M., Juliano R.L. Expression of the integrin alpha 5 subunit in HT29 colon carcinoma cells suppresses apoptosis triggered by serum deprivation. Exp. Cell Res. 1996;224:208–213. doi: 10.1006/excr.1996.0130. [DOI] [PubMed] [Google Scholar]
- Ossovskaya V.S., Mazo I.A., Chernov M.V., Chernova O.B., Strezoska Z., Kondratov R., Stark G.R., Chumakov P.M., Gudkov A.V. Use of genetic suppressor elements to dissect distinct biological effects of separate p53 domains. Proc. Natl. Acad. Sci. USA. 1996;93:10309–10314. doi: 10.1073/pnas.93.19.10309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papapetropoulos A., Fulton D., Mahboubi K., Kalb R.G., O'Connor D.S., Li F., Altieri D.C., Sessa W.C. Angiopoietin-1 inhibits endothelial cell apoptosis via the Akt/survivin pathway. J. Biol. Chem. 2000;275:9102–9105. doi: 10.1074/jbc.275.13.9102. [DOI] [PubMed] [Google Scholar]
- Perlman H., Pagliari L.J., Georganas C., Mano T., Walsh K., Pope R.M. FLICE-inhibitory protein expression during macrophage differentiation confers resistance to fas-mediated apoptosis. J. Exp. Med. 1999;190:1679–1688. doi: 10.1084/jem.190.11.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell W.C., Fingleton B., Wilson C.L., Boothby M., Matrisian L.M. The metalloproteinase matrilysin proteolytically generates active soluble Fas ligand and potentiates epithelial cell apoptosis. Curr. Biol. 1999;9:1441–1447. doi: 10.1016/s0960-9822(00)80113-x. [DOI] [PubMed] [Google Scholar]
- Re F., Zanetti A., Sironi M., Polentarutti N., Lanfrancone L., Dejana E., Colotta F. Inhibition of anchorage-dependent cell spreading triggers apoptosis in cultured human endothelial cells. J. Cell Biol. 1994;127:537–546. doi: 10.1083/jcb.127.2.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen K., Rak J., Leung T., Dean N.M., Kerbel R.S., Filmus J. Activated Ras prevents downregulation of Bcl-X(L) triggered by detachment from the extracellular matrix. A mechanism of Ras-induced resistance to anoikis in intestinal epithelial cells. J. Cell Biol. 2000;149:447–456. doi: 10.1083/jcb.149.2.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rytomaa M., Martins L.M., Downward J. Involvement of FADD and caspase-8 signalling in detachment-induced apoptosis. Curr. Biol. 1999;9:1043–1046. doi: 10.1016/s0960-9822(99)80454-0. [DOI] [PubMed] [Google Scholar]
- Salvesen G.S. Programmed cell death and the caspases. Apmis. 1999;107:73–79. doi: 10.1111/j.1699-0463.1999.tb01528.x. [DOI] [PubMed] [Google Scholar]
- Sata M., Walsh K. Endothelial cell apoptosis induced by oxidized LDL is associated with the down-regulation of the cellular caspase inhibitor FLIP J. Biol. Chem 273 1998. 33103 33106a [DOI] [PubMed] [Google Scholar]
- Sata M., Walsh K. Oxidized LDL activates fas-mediated endothelial cell apoptosis J. Clin. Invest 102 1998. 1682 1689b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sata M., Walsh K. TNFalpha regulation of Fas ligand expression on the vascular endothelium modulates leukocyte extravasation Nat. Med 4 1998. 415 420c [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sata M., Suhara T., Walsh K. Vascular endothelial cells and smooth muscle cells differ in expression of Fas and Fas ligand and in sensitivity to Fas ligand-induced cell deathimplications for vascular disease and therapy. Arterioscler. Thromb. Vasc. Biol. 2000;20:309–316. doi: 10.1161/01.atv.20.2.309. [DOI] [PubMed] [Google Scholar]
- Scaffidi C., Schmitz I., Krammer P.H., Peter M.E. The role of c-FLIP in modulation of CD95-induced apoptosis. J. Biol. Chem. 1999;274:1541–1548. doi: 10.1074/jbc.274.3.1541. [DOI] [PubMed] [Google Scholar]
- Scatena M., Almeida M., Chaisson M.L., Fausto N., Nicosia R.F., Giachelli C.M. NF-kappaB mediates alphavbeta3 integrin-induced endothelial cell survival. J. Cell Biol. 1998;141:1083–1093. doi: 10.1083/jcb.141.4.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer D.D., Hauck C.R., Sieg D.J. Signaling through focal adhesion kinase. Prog. Biophys. Mol. Biol. 1999;71:435–478. doi: 10.1016/s0079-6107(98)00052-2. [DOI] [PubMed] [Google Scholar]
- Sethi T., Rintoul R.C., Moore S.M., MacKinnon A.C., Salter D., Choo C., Chilvers E.R., Dransfield I., Donnelly S.C., Strieter R., Haslett C. Extracellular matrix proteins protect small cell lung cancer cells against apoptosisa mechanism for small cell lung cancer growth and drug resistance in vivo. Nat. Med. 1999;5:662–668. doi: 10.1038/9511. [DOI] [PubMed] [Google Scholar]
- Srivastava R.K., Sasaki C.Y., Hardwick J.M., Longo D.L. Bcl-2–mediated drug resistanceinhibition of apoptosis by blocking nuclear factor of activated T lymphocytes (NFAT)-induced Fas ligand transcription. J. Exp. Med. 1999;190:253–265. doi: 10.1084/jem.190.2.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh J.H., Hsu S.C., Han S.H., Lai M.Z. Mitogen-activated protein kinase kinase antagonized fas-associated death domain protein–mediated apoptosis by induced FLICE-inhibitory protein expression. J. Exp. Med. 1998;188:1795–1802. doi: 10.1084/jem.188.10.1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh W.C., Itie A., Elia A.J., Ng M., Shu H.B., Wakeham A., Mirtsos C., Suzuki N., Bonnard M., Goeddel D.V., Mak T.W. Requirement for Casper (c-FLIP) in regulation of death receptor-induced apoptosis and embryonic development. Immunity. 2000;12:633–642. doi: 10.1016/s1074-7613(00)80214-9. [DOI] [PubMed] [Google Scholar]
- Zhang Z., Vuori K., Reed J.C., Ruoslahti E. The alpha 5 beta 1 integrin supports survival of cells on fibronectin and up-regulates Bcl-2 expression. Proc. Natl. Acad. Sci. USA. 1995;92:6161–6165. doi: 10.1073/pnas.92.13.6161. [DOI] [PMC free article] [PubMed] [Google Scholar]