

Abstract
Low molecular weight fragmentation products of the polysaccharide of Hyaluronic acid (sHA) produced during inflammation have been shown to be potent activators of immunocompetent cells such as dendritic cells (DCs) and macrophages. Here we report that sHA induces maturation of DCs via the Toll-like receptor (TLR)-4, a receptor complex associated with innate immunity and host defense against bacterial infection. Bone marrow–derived DCs from C3H/HeJ and C57BL/10ScCr mice carrying mutant TLR-4 alleles were nonresponsive to sHA-induced phenotypic and functional maturation. Conversely, DCs from TLR-2–deficient mice were still susceptible to sHA. In accordance, addition of an anti–TLR-4 mAb to human monocyte–derived DCs blocked sHA-induced tumor necrosis factor α production. Western blot analysis revealed that sHA treatment resulted in distinct phosphorylation of p38/p42/44 MAP-kinases and nuclear translocation of nuclear factor (NF)-κB, all components of the TLR-4 signaling pathway. Blockade of this pathway by specific inhibitors completely abrogated the sHA-induced DC maturation. Finally, intravenous injection of sHA-induced DC emigration from the skin and their phenotypic and functional maturation in the spleen, again depending on the expression of TLR-4. In conclusion, this is the first report that polysaccharide degradation products of the extracellular matrix produced during inflammation might serve as an endogenous ligand for the TLR-4 complex on DCs.
Keywords: extracellular matrix, glycosaminoglycans, dendritic cells, Hyaluronan, toll-like receptors
Introduction
Hyaluronic acid (HA)*, one of the major glycosaminoglycans of the extracellular matrix, has been shown to undergo rapid degradation at sites of inflammation (1, 2). In its native state, HA is a high-molecular mass (up to 106 Da) nonsulfated, linear polysaccharide consisting of repeating units of (β,1–4)-d-glucuronic acid-(β,1–3)-N-acetyl-D-glucosamine (3). Recently, low-molecular weight degradation products of HA have been found to elicit various proinflammatory responses such as the activation of murine alveolar macrophages as well as the stimulation and invasion of macrophages into affected joints in rheumatoid arthritis (4, 5). We have recently shown that low-molecular weight oligosaccharides of HA (sHA) induced complete and irreversible phenotypic and functional maturation of human DCs, in contrast to high-molecular weight HA (HMW-HA) which had no such effect (5). These effects of sHA were independent of the major HMW-HA receptors RHAMM and CD44 and were highly specific for sHA since other glycosaminoglycans of disaccharide structure similar to HA, such as chondroitin sulfate, heparan sulfate, or their fragmentation products were not effective (5, 6). Furthermore, in contrast to HMW-HA, sHA has also been reported to induce specific intracellular signaling pathways in immunocompetent cells and endothelial cells (7). These include activation of mitogen-activated protein kinases (MAPKs) in vascular endothelium and the translocation of NF-κB to the nucleus in murine alveolar macrophages, in murine kidney tubular epithelium and in rat hepatic cells (4, 8–10).
Recently, the so-called Toll-like receptors (TLRs), a new group of receptors belonging to the IL-1 receptor family have been described (11–14). TLRs were initially discovered in Drosophila and are conserved amongst various species. They are involved in the activation of immunocompetent cells such as macrophages and DCs and thus participate in the innate defense against bacterial infection (11, 12, 14, 15). Several TLRs have been identified in blood macrophages and cells of the myelomonocytic lineage, but TLR-2 and TLR-4 appear to be particularly important for DC activation (13, 14). Unlike other receptors involved in DC activation such as CD14 (16), TLRs have a transmembrane domain (12) that associates with the intracellular adaptor protein MyD88 (17, 18) which mediates TLR-induced signal transduction through its interaction with the serine kinase IRAK (18). LPS-mediated activation of the TLR-4 complex was found to induce specific signaling pathways involving the phosphorylation of p38, p42/p44 MAPKs, c-Jun NH2-terminal kinase, and led to liberation of NF-κB/Rel family members into the nucleus (13, 19). However, activation of the TLR-4 receptor complex is not limited to LPS, and other proinflammatory stimuli such as Taxol and Heat-Shock Protein 60 (HSP-60) have been described as alternative ligands (20, 21).
These recent findings prompted us to determine whether TLR-4 receptors are involved in sHA-mediated DC maturation, and whether this might be associated with the activation of distinct intracellular signaling pathways. In this paper we show that TLR-4 plays a critical role during activation of human and murine DCs by sHA in vivo and in vitro, and that sHA stimulation induces the TLR-4 signal transduction pathway involving p38/p42–44 MAPKs and NF-κB translocation.
Materials and Methods
Experimental Animals.
6–8-wk-old female C57BL/10 and C57BL/10ScCr or C3H/HeN or C3H/HeJ mice (12) as well as TLR-2–deficient C57BL/6 and C57BL/6 wild-type mice (14) were maintained in the specific pathogen-free facility of the Max-Planck Institute for Immunobiology in Freiburg, Germany.
Materials.
The MAPK inhibitors SB203580, PD 98059, Wortmannin, Herbimycin A, and CAPE were purchased from Alexis Corporation. LPS from Salmonella minnesota Re-595, Salmonella abortus equi, or crude cell wall extracts were prepared as described previously (22).
Polymyxin B, a cationic antibiotic that displaces Ca2+ from anionic phospholipids, was obtained from Calbiochem. The monoclonal anti–human TLR-4 antibody was used for FACS® analysis and blocking studies in concentrations described previously (23).
Preparation of the HA Fragments.
HMW-HA (HEALON™) for clinical application with an endotoxin content <0.1 ng/mg was provided by Amersham Pharmacia Biotech. Small HA-fragmentation products (sHA) were generated by enzymatic digestion as described previously (5). After digestion and separation, a stock solution of 1 mg/ml sHA was prepared, which contained <0.1 ng/ml LPS as determined by Limulus amebocyte lysate (LAL) assays. For experimental use this was diluted to a final concentration of 20–50 μg/ml, with a maximal estimated Endotoxin content of 0.005 ng/ml for a 50 μg/ml concentration.
Preparation of Human DCs.
Buffy coats from healthy blood donors were obtained from the Department of Transfusion Medicine, Freiburg University Medical Center, Freiburg, Germany. White blood cells were separated on an endotoxin-free Ficoll-paque plus™ gradient (Amersham Pharmacia Biotech) and incubated with a nonactivating, nonblocking anti–human-CD14 mAb (Clone 26IC; American Type Culture Collection) for 45 min on ice. The cells were washed several times in PBS before incubation with the appropriate secondary anti–mouse MACS®-Beads (Miltenyi Biotech). CD14+ cells were purified by magnetic cell sorting using a VS-column MACS® system™ (Miltenyi Biotech). Purity of separated monocytes was >90% as determined by flow cytometry. Monocytes (5 × 106 cells per well) were seeded into 6-well flat-bottomed plates (Costar) and cultured at 37°C, 5% CO2 for 4 d in RPMI 1640 (GIBCO BRL) supplemented with 5% endotoxin-free human serum albumin (Bayer Diagnostics), GCP/GMP quality 1,000 U/ml GM-CSF (Leukomax™; Novartis), and 100 U/ml IL-4 (provided by Schering-Plough). The CD14 mAb disappeared completely from the cell surface of monocytes on day 3 of culture, as determined by flow cytometry.
Generation of Murine Bone Marrow–derived DCs.
DCs were generated following the method of Inaba et al. (24), with minor alterations. BM was harvested from the tibia and femur of C57BL/6 mice (n = 4). The cells were resuspended at 106 cells per milliliter cRPMI 1640 (GIBCO BRL) with 40 ng/ml GM-CSF and 100 ng/ml IL-4 (both PromoCell). Cells were fed on days 3 and 5 of culture, by replacing half the medium in each well with fresh cRPMI containing GM-CSF and IL-4. On day three, nonadherent cells were aspirated off, after gentle swirling of the plate. On day 6, loosely adherent cells including DCs were harvested by gentle pipetting. DCs were washed once and resuspended at ∼5 × 105 cells per milliliter in cRPMI. Aliquots of the cell suspension (8 ml) were underlayed with 2 ml 14.5% metrizamide (Boehringer Ingelheim) in a 14-ml conical bottomed tube (Becton Dickinson) and centrifuged at room temperature (22°C) for 20 min at 600× g. The low buoyant density cells were collected, washed twice, and resuspended for use. Purity of Iab-positive cells was >70% as determined by flow cytometry
TNF-α ELISA.
Supernatants from unstimulated, LPS- or sHA-stimulated DCs were collected at the indicated time points to determine the content of TNF-α. The ELISA assays were developed according to the manufacturer's instructions (Becton Dickinson) and measured at an extinction of 630 nm in a M r 5000 ELISA reader and analyzed using Bio-Linx™ Software (both Dynatech).
Lymphocyte Preparation/T Cell Proliferation Assay.
Spleen lymphocytes were isolated by mincing the organ with the back of a 10-ml syringe in a dish before passing it through a 60-μm nylon mesh cell strainer. Red blood cells were removed by addition of lysing solution (0.15 M NH4Cl, 1 mM KHCO3, 0.1 mM Na2-EDTA adjusted to a pH of 7.2) for 5 min. Remaining APCs were removed by negative selection with a magnetic bead–labeled mAb against CD4+ for 45 min at 4°C followed by magnetic cell sorting (MACS®; Miltenyi Biotech). Flow cytometry showed purity of the CD4+ cells to be >85% and IA-expressing cells to be <1%. The cells did not respond to PHA stimulation. The magnetic beads were removed from the CD4+ cells by incubation with CD4-detecha-beads and magnetic cell sorting (MACS®; Miltenyi Biotech). Purified CD4+ cells in cRPMI 1640 were plated at 105 cells per well in round-bottomed 96-well plates (Costar) together with 5 × 103 allogeneic DCs per well and incubated at 37°C, 5% CO2. On day 4, 1 μCi/well 3[H]thymidine (Amersham Pharmacia Biotech) was added for the final 18 h of culture. Finally, the plates were harvested onto 96-well glass-fiber filter plates and the radioactivity was determined by liquid scintillation spectroscopy using a TopCount β-counter (all Canberra Packard).
Immunostaining and Flow Cytometry.
DCs were incubated with primary mAb for 30 min at 4°C, then washed, and stained with the appropriate FITC-labeled secondary mAb. To determine cell viability and to exclude dead cells, 1 μg/ml propidium iodide (Sigma-Aldrich) was added. Samples of 5 × 104 cells were analyzed using a FACScan™ together with CELLQuest™ research software (both Becton Dickinson).
Determination of Phosphorylated Forms of p42/44 and p38-MAPK by Western Blot Analysis.
Activated forms of p42/44 MAPK (ERK1/2) and p38 MAPK were detected by Western blot analysis using antibodies to the phosphorylated forms of these kinases. Extracts from untreated cells and from cells cultured with sHA or LPS were made by sonication in ice-cold RIPA buffer (150 mM NaCl, 0.1 mM Tris-HCl, pH 7.2, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 5 mM EDTA) containing a mixture of protease inhibitors and the phosphatase inhibitors sodium orthovanadate (1 mM) and glycerophosphate (50 mM). For Western blot analysis, 100 μg cell extract proteins were electrophoresed in a SDS/12% polyacrylamide gel and electroblotted onto a nitrocellulose filter. Blots were blocked for 1 h at room temperature in PBS containing 0.2% Tween 20 and 5% low fat milk. They were then incubated overnight at 4°C with rabbit polyclonal antibodies recognizing either the unphosphorylated forms of p42/44 MAPK or p38 MAPK, or the double phosphorylated (Thr-202/Tyr-204) p42/44 MAPK or (Thr-180/Tyr-182) p38 MAPK (New England Biolabs). Blots were then washed five times and incubated for 1 h at room temperature with horseradish peroxidase-conjugated goat anti–rabbit IgG (Santa Cruz Biotechnology, Inc.). After further washing, the blots were developed with the Enhanced Chemiluminescence detection (ECD) kit (Amersham Pharmacia Biotech). Bands of p42/44 MAPK or p38 MAPK were visualized after exposing the blots to a Kodak RX film.
Preparation of Nuclear Extracts and EMSA.
NF-κB oligonucleotide (Promega) was labeled using γ-32[P]ATP (3,000 Ci/mmol; Amersham) and T4 polynucleotide kinase (Promega). Nuclear extracts from DC were prepared as described previously (25). In brief, total cell extracts were prepared using a high-salt detergent buffer (20 mM Hepes, pH 7.9, Totex; 350 mM NaCl; 20% [wt/vol] glycerol; 1% [wt/vol] NP-40; 1 mM MgCl2; 0.5 mM EDTA; 0.1 mM EGTA; 0.5 mM DTT; 0.1% PMSF; and 1% aprotinin). Cells were harvested by centrifugation, washed once in ice-cold PBS (Sigma-Aldrich) and resuspended in four cell volumes of Totex buffer. After 30 min on ice, the lysates were centrifuged for 5 min at 13,000× g at 4°C. The protein content of the supernatant was determined and equal amounts of protein (10–20 μg) were added to a reaction mixture containing 20 μg BSA (Sigma-Aldrich), 2 μg of poly (dI dC) (Boehringer Mannheim), 2 μl of buffer D+ (20% mM Hepes, pH 7.9, 20% glycerin, 100 mM KCl, 0.5 mM EDTA, 0.25% NP-40, 2 mM DTT, and 0.1% PMSF), 4 μl of buffer F (20% Ficoll 400, 100 mM Hepes, 300 mM KCl, 10 mM DTT, and 0.1% PMSF), and 100,000 cpm (Cerenkov) of a 32[P]-labeled oligonucleotide in a final volume of 20 μl. Samples were incubated at room temperature for 25 min. NF-κB oligonucleotide (Promega) was labeled using γ-32[P]ATP (3,000 Ci/mmol; Amersham Pharmacia Biotech) and T4 polynucleotide kinase (Promega). The samples were separated on a 6% acrylamide TBE gel, which was dried and subjected to autoradiography.
Assessment of TLR-4 and TLR-2 mRNA Expression.
RT-PCR analysis of human TLR-4 was assessed as described previously (26). Total RNA was isolated from monocyte-derived DCs using the Pharmacia Quick prep kit (Amersham Pharmacia Biotech) according to the manufacturer's instructions. cDNA was synthesized from 5 μg of total RNA using Superscript II reverse transcriptase (GIBCO BRL). The product was subjected to 30 cycles of PCR amplification at 94°C/1 min, 72°C/2 min, 55°C/3 min using Taq-DNA-polymerase (GIBCO BRL). The forward and reverse PCR primers used for human TLR-4 were 5′-TGGATACGTTTCCTTATAAG-3′ and 5′-GAAATGGAGGCACCCCTTC-3′, giving an amplification product of 506 bp. Primers for TLR-2 were used as described previously (27): 5′-TATCGTCTTCCTGGTTCAAGCC-3′ and 5′-AACAGAGCACAGATGCCAGAC-3′, resulting in an amplification product of 1,452 bp. Aliquots of the PCR reaction products (25 μl) were analyzed by electrophoresis on 1% agarose gels.
Skin Organ Cultures.
Full thickness skin containing epidermis plus papillary dermis from C57BL/10ScSn wild-type (TLR+/+) and TLR-4−/− C57BL/10Cr mice (three mice per group) was obtained by mechanical disaggregation of the outer epidermal sheet from the cartilage of the ears. Ear sheets were floated on supplemented RPMI 1640 in 6-well plates (Greiner) with or without addition of 30 μg/ml sHA or 100 ng/ml LPS. After 24 h, cells that had migrated into the culture medium were collected from each group and double stained with FITC-conjugated CD11c and IaB PE-conjugated mAbs. FACS® analysis was performed as described above. All cells obtained from one well (5–6 × 105 cells) were analyzed and large, highly CD11c and IaB-positive cells were considered as DCs and the percentage was calculated using the CELLQuest™ software (Becton Dickinson).
Intravenous Injection Experiments.
C57BL/6ScSn wild-type or SnCr TLR-4–deficient mice (three mice per group) were injected into the tail vein with 100 μg sHA, 10 μg LPS, or 100 μl PBS. After 12 h the mice were killed and the spleen DCs were purified by passing the spleens through a 60-μm nylon mesh cell strainer. Red blood cells were removed as described above. DCs were prepared by negative selection with magnetic bead-labeled mAbs against CD3 (pan T cell) and B220 (pan B cell) for 45 min at 4°C followed by magnetic cell sorting (MACS®; Miltenyi Biotech). 5 × 103 DCs were incubated for 5 d in an allogenic T cell proliferation assay with 105 BALB/c T cells as described above.
Results
Terminal Maturation of both Human and Murine DCs Is Induced by sHA.
We and others have proposed that during inflammation high molecular weight hyaluronate is degraded by enzymes released from activated fibroblasts. These degradation products can deliver activation signals to DC and macrophages (2, 5). sHA fractions containing sugar units of 4–16 oligosaccharide size stimulated phenotypic and functional maturation of human monocyte-derived DCs, in contrast to HMW-HA (5). Maturation included upregulation of MHC class II and costimulatory B7-molecules, an enhanced allostimulatory capacity and an increased production of proinflammatory cytokines. This was independent of LPS-contamination, since preincubation with polymyxin B had no effect (5). Furthermore, LAL assays revealed that the maximal endotoxin content in our sHA preparations was <0.1 ng/ml, a concentration which is unable to induce functional or phenotypic DC maturation (reference 5, and data not shown). To facilitate further analysis of sHA-mediated DC maturation using distinct mouse strains, we set out to determine whether sHA also effectively activates murine DCs. Immature bone marrow–derived DCs from C57/BL-6 mice were coincubated for 48 h with either sHA, HMW-HA or LPS, or were left untreated. Only sHA, but not HMW-HA, induced maturation of murine DCs as shown by marked upregulation of MHC-class II (Iab) and costimulatory B7–2 molecules, resembling our observations in human DCs (Fig. 1 A and C). The maturation of sHA-stimulated DCs was accompanied by a dramatically enhanced capacity to stimulate the proliferation of naive alloreactive T cells in a standard MLR (Fig. 1 B and D). This phenotypic and functional maturation exactly matched the DC maturation induced by 100 ng/ml LPS (Fig. 1 D).
Figure 1.

sHA induces phenotypic and functional maturation of human and mouse DCs. Human monocyte–derived day 4 DCs (A and B) or murine day 6 bone marrow–derived DCs (C and D) were used after 48-h incubation with 20 μg/ml sHA (bold line), 50 μg/ml HMW-HA (solid line), 100 ng/ml LPS (dotted line), or left untreated (broken line). (A and C) DCs were stained with mAbs directed against MHC class II (top) or B7–2 (CD86; lower) and analyzed by flow cytometry. A representative of five independent experiments is shown. (B and D) The cells were coincubated for 4 d with 105 alloreactive T cells at a DC:TC ratio of 1:20. T cell proliferation was determined on day 5 by addition of 1 μCi of 3[H]thymidine for the final 18 h. Results are shown in counts per minute (CPM) ± SD of triplicate wells. * P > 0.001 compared with untreated DCs (−). A representative of four independent experiments is shown.
The Induction of TNF-α by sHA Treatment of DCs Is Time and Dose Dependent.
We have previously shown that DC stimulated with sHA secrete large quantities of proinflammatory cytokines, such as IL-1β, TNF-α, and IL-12, and that sHA-induced DC maturation is TNF-α dependent (5). To further characterize the time and dose dependence of sHA-induced TNF-α expression, we used RT-PCR to analyze TNF-α mRNA expression after sHA treatment of human DCs. Rapid induction of TNF-α mRNA was observed, which was detectable as early as 4 h after stimulation of DCs with either 25 μg/ml sHA or 100 ng/ml LPS (Fig. 2 A). At this dose the sHA-induced TNF-α expression was more long-lasting compared with that induced by LPS (Fig. 2 A). When the overall amount of TNF-α protein secreted into the culture medium after 24 h of stimulation with different concentrations of sHA was quantified by ELISA, there was a clear dose response in TNF-α production, with up to 4 ng/ml being produced in response to 50 μg/ml sHA (Fig. 2 B and C). The kinetics of TNF-α induction by sHA showed a late onset starting after 4–5 h of coincubation, reaching near-peak levels after 12 h, with a slight further increase up to 24 h. This effect was concentration dependent, as 10 μg/ml sHA sufficed to induce a significant TNF-α production, while saturation was observed at concentrations >20 μg/ml (Fig. 2 C). In contrast, TNF-α induction by 100 ng/ml LPS occurred much more rapidly, with significant amounts being produced after 30 min (Fig. 2 B). We conclude that the TNF-α response is a very sensitive parameter to measure the sHA-induced maturation of DC, therefore it was adopted as the major readout-system in further experiments.
Figure 2.



Analysis of TNF-α production by DCs in response to sHA stimulation. (A and B) Human monocyte–derived day 4 DCs were treated with 25 μg/ml sHA or 100 ng/ml LPS for the indicated times. After stimulation, (A) cells were harvested and RNA was prepared for TNF-α–specific RT-PCR, and (B) cell-free supernatants were collected and assayed by TNF-α ELISA. The dotted line shows the response to LPS, the solid line the response to sHA. Data represent the mean TNF-α release of triplicate values; pg/mg total protein ± SD. (C) DCs were incubated for 24 h with the indicated concentrations of sHA or 100 ng/ml LPS and cell-free supernatants were collected and assayed by TNF-α ELISA as detailed above.
sHA-mediated DC Activation Requires p38-MAPK and P42/44-MAPK Activity and Nuclear Translocation of NF-κB.
It has been shown that DC maturation by LPS involves phosphorylation of p38 MAPK and p42/44 MAPK (13). To test whether activation of the MAPK pathway might be also involved in the sHA-induced DC maturation we analyzed p38 and p42/44 MAPK in DC lysates by Western blot analysis. After sHA treatment, but not HMW-HA treatment, p38 and p42/44 MAPK were rapidly phosphorylated within 1–2 h when compared with the total content of these MAPKs (Fig. 3 A and B). Then, we investigated whether p42/44 MAPK activation was required for the TNF-α production leading to DC maturation. DCs stimulated either with 30 μg/ml sHA or 100 ng/ml LPS were preincubated for 7 h with different concentrations of defined kinase inhibitors. Specifically, PD98059, which blocks p42/44 MAPKs, SB-203580, a selective inhibitor of p38-MAPKs, Herbimycin A, an inhibitor of src-like protein tyrosine kinases, and Wortmannin, a phosphatidylinositol (PI)3-kinase inhibitor were used as described previously (28–31). ELISA assays showed a dose-dependent decrease in TNF-α production after preincubation with each of the different MAPK inhibitors, whereas Wortmannin had no effect, suggesting that p38 and p42/44 MAPKs, but not PI3-kinase are involved in the signaling pathway induced by sHA (Fig. 3 C). Since activation of PI3-kinase is involved in the control of intracellular calcium stores, the latter finding is consistent with our results that sHA treatment of DCs does not induce intracellular calcium fluxes as analyzed by Fura labeling (data not shown).
Figure 3.
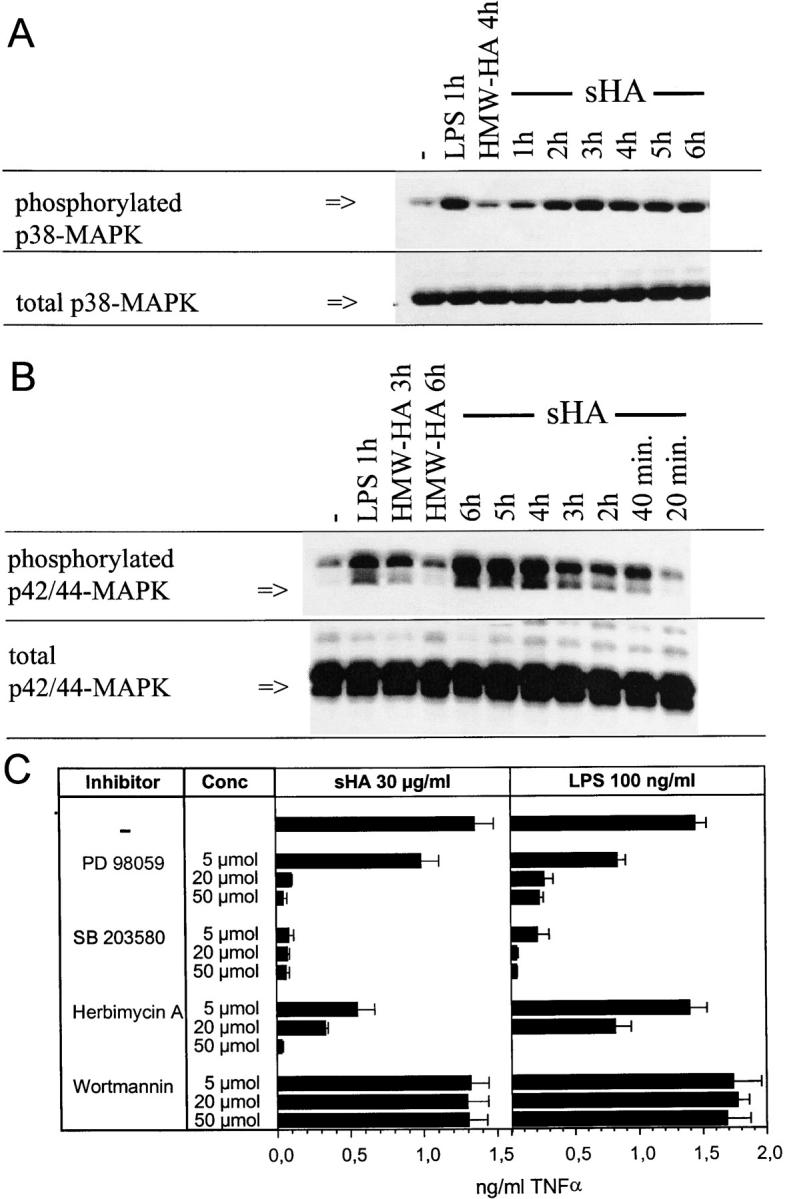
sHA-mediated DC maturation requires p38-MAPK and P42/44-MAPK phosphorylation. (A and B) Human monocyte–derived day 4 DCs were treated with 50 μg/ml sHA, 100 μg/ml HMW-HA, or 100 ng/ml LPS for the times indicated. Western immunoblotting was performed using mAbs specific for the phosphorylated kinase (top) or total kinase content of the cells (bottom). Shown are blots for (A) p38 MAPK and (B) p42/44 MAPK. (C) Supernatants of DCs stimulated with 30 μg/ml sHA or 1 ng/ml LPS in the presence of the indicated concentrations of the MAPK inhibitors PD98059 (p38), SB 203580 (p42/44), Herbimycin A (src-like tyrosine kinases), or Wortmannin (PI3-kinase) were harvested after 7 h and screened for their TNF-α content by ELISA. Data represent the mean TNF-α release of triplicate wells; pg/mg total protein ± SD.
To determine whether the sHA-induced DC maturation involves NF-κB translocation to the nucleus, EMSA of nuclear extracts were performed. As shown in Fig. 4 A, sHA induced NF-κB activation in a time-dependent manner, with a significant signal obtained as early as 30 min after sHA treatment. Furthermore, coincubation with the specific NF-κB inhibitor CAPE dose dependently inhibited the ability of DCs to produce TNF-α and their upregulation of HLA-DR (Fig. 4 B and data not shown), with an almost total inhibitory effect observed at a concentration of 20 μg/ml. As expected, NF-κB liberation from the cytosol was enhanced by LPS, with strong NF-κB signaling being observed after 30 min of incubation with LPS (Fig. 4 A). Taken together, these data suggest that phosphorylation of p38 and p42/44 MAPK as well as nuclear translocation of NF-κB are prerequisites for the sHA-induced TNF-α production by DC.
Figure 4.


The sHA-induced DC maturation is NF-κB dependent. (A) Nuclear extracts were prepared from DCs treated with either HMW-HA (30 μg/ml), sHA (30 μg/ml), LPS (10 ng/ml), or left untreated. A total of 10 μg of protein from each nuclear extract was analyzed for NF-κB activity by EMSA. The arrow indicates specific NF-κB bands. (B) Supernatants of DCs stimulated with 30 μg/ml sHA (white bars) or 10 ng/ml LPS (black bars) in the presence of the indicated concentrations of the NF-κB inhibitor CAPE were harvested after 7 h and screened for their TNF-α content by ELISA. Data represent the mean TNF-α release of triplicate wells; pg/mg total protein ± SD.
TLR-4 Is Involved in the sHA-mediated TNF-α Production by Human DCs and Is Downregulated After Stimulation.
The TLR-4 family, in particular the TLR-4 complex, has recently been described as being involved in the LPS-dependent activation of DCs during bacterial infection (13). We investigated whether TLR-4 might also be involved in sHA-induced activation of DCs. The anti–TLR-4 mAb HTA125 is able to block TLR-4 ligation (23). Therefore, we analyzed what effect this antibody has on TNF-α production by DCs in response to sHA. Preincubation of human DC for 30 min with 10 μg/ml HTA125 mAb, a concentration previously shown to effectively block TLR-4 ligation (23), was able to reduce the sHA-induced TNFα release by these cells for up to 7 h, as found for LPS (Fig. 5 A). A higher concentration of HTA 125 (20 μg/ml) or a longer preincubation time of 60 min could enhance the blocking effect to some extent (Fig. 5 A). These results provide first evidence that TLR-4 is involved in the intracellular signaling and maturation of DCs induced by sHA.
Figure 5.

TLR-4 mediates TNF-α production of sHA-stimulated human DCs and is downregulated after activation. (A) Anti–TLR-4 antibodies inhibit TNF-α production by DCs in response to sHA (left) and LPS (right). DCs were either preincubated for 30 or 60 min with 10 or 20 μg/ml of the anti–TLR-4 mAb HTA 125 as indicated, a matched IgG-isotype control (black circles) or left untreated (white circles). The cells were then stimulated with 30 μg/ml sHA (left) or 100 ng/ml LPS (right) for the times (h) indicated. Supernatants were harvested and screened for their TNF-α content by ELISA as described previously. Data represent the mean TNF-α release of triplicate wells; ng/mg total protein. (B) Total mRNA from human DCs treated either with 100 ng/ml LPS or 30 μg/ml sHA for the indicated times was analyzed by RT-PCR. The top shows TLR-2, the middle TLR-4 regulation, the bottom shows β-actin control. (C) DCs were analyzed by FACS® for their surface-expression of TLR-4 on: untreated control DCs (solid line), HMW-HA–treated DCs (30 μg/ml; dashed line), sHA-treated DCs (30 μg/ml; bold line), and LPS-treated DC (1 ng/ml; dotted line). Dot-dash line: DCs stained with secondary antibody alone. A representative of three independent experiments is shown.
To further investigate the involvement of TLRs with sHA-induced DC maturation, we characterized the expression of TLR-4 and TLR-2 (the latter is a receptor associated with the binding of bacterial lipoproteins; reference 14) on human DCs in response to sHA treatment. RT-PCR analysis revealed that TLR-4 as well as TLR-2 mRNA was clearly expressed in preparations of unstimulated DCs. Interestingly, treatment with 20 μg/ml sHA downregulated both TLR-4 and TLR-2 mRNA expression within 6–12 h, with no signal being detectable after 12 h incubation with sHA (Fig. 5 B). Similar results were obtained when the surface expression of TLR-4 protein on DCs was assessed by FACS® analysis. Again, we observed significant downregulation of the receptor after 24 h of incubation with either sHA or LPS (Fig. 5 C).
From these data we conclude that TLR-4 is involved in sHA-induced activation of DCs, and that TLR-2 and TLR-4 are downregulated in response to sHA treatment. To obtain more direct information about the possible involvement of TLR-2 and TLR-4 in sHA-induced DC maturation, we isolated DCs from the bone marrow of TLR-2– and TLR-4–deficient mice and analyzed the effect of sHA on these cells.
DCs from TLR-4–deficient Mice Do Not Respond to sHA Stimulation.
The mouse strains C3H/HeJ and C57BL10/ScCr are nonresponsive to LPS due to a mutation or lack of the TLR-4 receptor, respectively (19). We used these mice to further characterize the function of TLR-4 during sHA-mediated DC stimulation. Bone marrow–derived DCs from C3H/HeJ, C57BL/10ScCr, and wild-type mice were prepared and immunophenotypically analyzed. We found no differences in expression levels of MHC class II, costimulatory molecules B7–1/B7–2 or CD11c in DCs from TLR-4–deficient or wild-type mice (data not shown). Moreover, the DCs from the mutant mice were functionally active, since they produced significant amounts of TNF-α after coincubation with bacterial crude cell wall extracts which were shown to activate macrophages from TLR-4–deficient mice (17), although this amount was lower than that produced by wild-type DCs (Fig. 6 A). However, while sHA dose dependently stimulated TNF-α production in wild-type DCs as expected, DCs from mutant mice were completely resistant to sHA stimulation as well as to purified LPS from S. abortus equi or S. minnesota R595 (Fig. 6 A). We next examined whether the nonresponsiveness of DCs from TLR-4 mutant mice to sHA-stimulation is functionally relevant to their T cell stimulatory function. In a standard MLR, the allostimulatory capacity of the DCs from mutant mice was significantly lowered on sHA treatment when compared with wild-type DCs (Fig. 6 B). In comparison, HMW-HA had no effect on either the TNF-α release or the T cell stimulatory capacity of both wild-type and mutant DCs, even at concentrations of 100 μg/ml (Fig. 6). Since TLR-4 is the major receptor for LPS and concentrations as low as 50 pg/ml have been described to activate cells of the myelomonocytic lineage (32), we wished to exclude the possibility of endotoxin contamination in the sHA preparations used in this study. LAL assays showed a maximum endotoxin content of 0.1 ng/ml in our 1 mg/ml sHA stock solutions, resulting in a maximum possible contamination throughout the assays of 0.005 ng/ml in the 50 μg/ml concentration of sHA. Titration experiments with purified LPS from S. abortus equi were performed, showing that DCs under the culture conditions used in our experiments did not respond to LPS concentrations <0.2 ng/ml (data not shown). Furthermore, blocking experiments were performed by preincubating the DCs with 10 μg/ml of the LPS-inhibitor polymyxin B before sHA stimulation (Fig. 6 C). Polymyxin B had no effect on the sHA-induced upregulation of IAb, B7–1, or B7–2, but in the same experiment inhibited the LPS effect almost completely (Fig. 6 C).
Figure 6.

DCs derived from TLR-4–deficient mice are not sensitive to sHA stimulation. (A) Bone marrow–derived DCs were obtained from either C3H/HeN (wild-type) and C3H/HeJ (TLR-4 mutant) or C57BL/10ScSn (wild-type) and C57BL/10ScCr (TLR-4 mutant). On day 6 of culture the DCs were either incubated with 100 ng/ml of bacterial cell wall lysates containing LPS and other substances such as lipoprotein, or 100 ng/ml highly purified LPS from Salmonella abortus equi or Salmonella minessota strain R595 as positive and negative controls. Alternatively, 10 or 50 μg/ml sHA or 100 μg/ml HMW-HA were added. After 48 h of treatment the cell free supernatants were screened for their TNF-α content by ELISA. Data represent the mean TNF-α release of triplicate wells; pg/mg total protein ± SD. (B) DCs were treated for 48 h with the same substances described in A, washed, and coincubated for 4 d with 105 alloreactive T cells at a DC/TC ratio of 1:20. T cell proliferation was determined on day 5 by addition of 1 μCi of 3[H]thymidine for the final 18 h. Results are shown in counts per minute (CPM) ± SD of triplicate wells. (C) Day 6 bone marrow–derived DCs of either C57BL/10Sn (black bars) or C57BL/10Cr (white bars) were incubated for 24 h with 30 μg/ml sHA or 100 ng/ml LPS and/or pretreated with 10 mg/ml polymyxin B (polyB) 1 h before stimulation. After 24 h of incubation DCs were stained for Iab, B7–1, and B7–2 expression and analyzed by flow cytometry. Results are shown a mean fluorescence intensities (MFI).
sHA-mediated DC Stimulation Is Not Dependent on TLR-2 Expression.
TLR-2 is a receptor that has been associated with macrophage activation by membrane lipoproteins/lipopeptides from Borrelia burgdorferi, Treponema pallidum, and Mycoplasma fermentans (14, 33). To examine whether TLR-2 plays a role in sHA-induced DC maturation, DCs from TLR-2–deficient (14) and wild-type mice were generated from bone marrow. These cells were treated with either LPS or sHA and their ability to secrete TNF-α in response to these substances was compared. TLR-2–deficient DCs responded well to LPS and sHA (Fig. 7 A). These data were confirmed at the functional level, since in a standard MLR, the allostimulatory capacity of the DCs from mutant mice was significantly enhanced upon sHA and LPS treatment (Fig. 7 B). Taken together, these data suggest that sHA-mediated DC stimulation is dependent on TLR-4 but not on TLR-2, since the presence of a nonfunctional mutated TLR-4 receptor in DCs could not support sHA-induced DC maturation, while TLR-2–deficient DCs responded to sHA in a similar manner to wild-type DCs.
Figure 7.


sHA-mediated DC stimulation is not dependent on TLR-2 expression. Day 6 bone marrow–derived DCs from C57BL/6 or C57BL/6TLR-2−/− were incubated for 48 h with the indicated concentrations of highly purified LPS from Salmonella abortus equi or sHA. (A) The cell free supernatants were screened for their TNF-α content by ELISA. Data represent the mean TNF-α release of triplicate wells; pg/mg total protein ± SD. (B) DCs were treated for 48 h with the same substances described in A, washed, and coincubated for 4 d with 105 alloreactive T cells at a DC/TC ratio of 1:20. T cell proliferation was determined on day 5 by addition of 1 μCi of 3[H]thymidine for the final 18 h. Results are shown in counts per minute (CPM) ± SD of triplicate wells.
In Vivo Relevance of the sHA-induced DC Activation.
The data presented so far suggesting that sHA-induced DC maturation is dependent on TLR-4 were produced using in vitro methods. Therefore, we set out to determine the in vivo relevance of sHA-induced DC maturation. In a first attempt, full thickness skin organ cultures were floated on medium containing 30 μg/ml sHA or 100 ng/ml LPS (Fig. 8 A). After 24 h, cells that had migrated into the culture medium were collected and double stained for CD11c and IaB expression. FACS® analysis of high forward scatter, highly CD11c/IaB-positive cells showed that a significant higher percentage of DCs appeared in the culture medium when sHA was added (Fig. 8 A). In contrast, ear skin from TLR-4–deficient mice did not show an enhanced DC emigration in response to sHA or LPS (Fig. 8 A). In a second set of experiments, 100 μg/animal sHA or 10 μg LPS were injected intravenously into C57BL/10ScCr and wild-type mice (Fig. 8 B). The mice were killed after 12 h and forward scatter high, highly CD11c-expressing cells were quantified in spleen cell suspensions for their IaB/B7–2 expression (Fig. 8 B). sHA induced an significant enrichment of highly Iab and B7–2 positive, activated DCs (Fig. 8 B). In contrast, injection of sHA into TLR-4–deficient mice did not result in an upregulation of Iab and B7–2 on splenic DCs (Fig. 8 B). To quantify their functional activity, DCs were purified from spleens and tested for their allostimulatory function using naive T cells from BALB/c mice as responders (Fig. 8 B). Thymidine incorporation clearly demonstrated an enhanced T cell proliferation induced by DCs isolated from spleens of wild-type mice that were injected with sHA compared with PBS treated controls. In contrast, DCs isolated from TLR-4–deficient C57BL/10ScCr injected with sHA did not induce an enhanced T cell proliferation (Fig. 8 B). From these results we conclude that exogenously applied sHA is able to induce DC activation in vivo and this activation is again mediated by TLR-4–dependent mechanisms.
Figure 8.
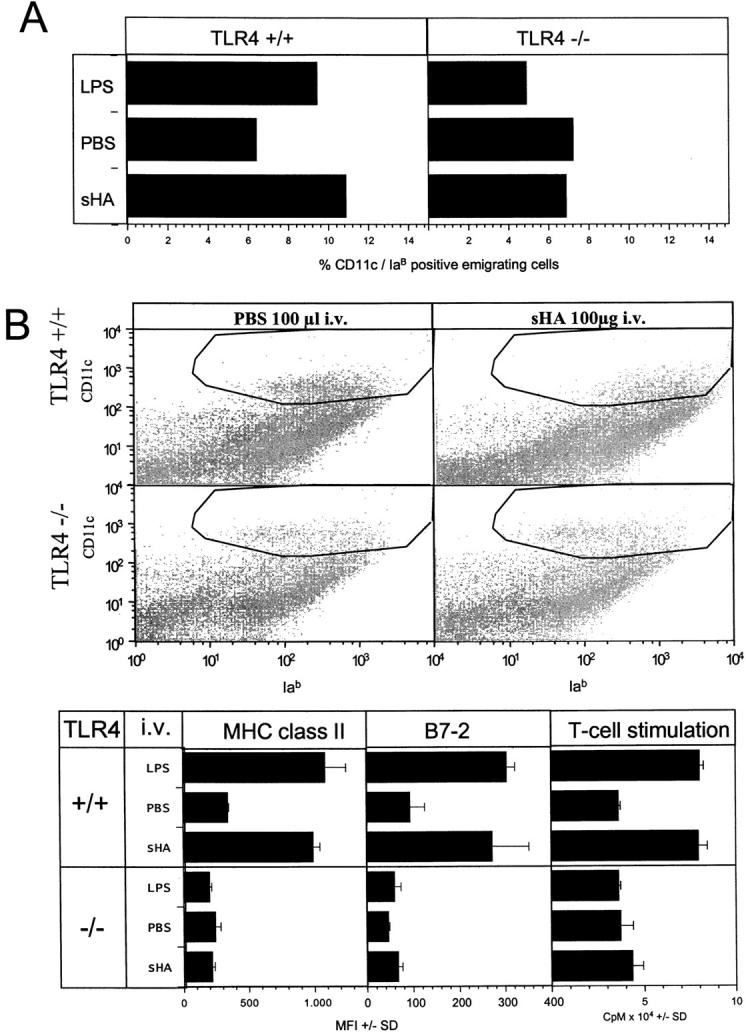
In vivo relevance of the sHA-induced DC stimulation. (A) Full thickness skin was obtained from the outer epidermal sheet of the ears of C57BL/10ScSn wild-type (TLR+/+) and TLR-4−/− C57BL/10Cr mice at three animals per group. Ear sheets were floated on supplemented RPMI 1640 with or without addition of 30 μg/ml sHA or 100 ng/ml LPS. After 24 h, cells that had migrated into the culture medium were collected and double stained for CD11c and IaB expression. Large, highly CD11c and IaB-positive cells were considered as DCs and the percentage was calculated by flow cytometry. A representative of two independent experiments is shown. (B) 100 μg/animal sHA or 100 μl PBS was injected intravenous into the tail vein of C57BL/10ScSn wild-type (TLR+/+) and TLR-4 −/− C57BL/10Cr mice at three animals per group. The mice were killed after 12 h and large, highly CD11c-positive cells in the spleens were analyzed for their IaB and B7–2 expression by flow cytometry. Results are shown as mean fluorescence intensities (MFI) in the marked gates ± SD. Further, DCs were isolated from spleen cells and 5 × 103 DCs were restimulated for 4 d with 105 allogenic T cells from BALB/c mice. T cell proliferation was determined on day 4 by addition of 1 μCi of 3[H]thymidine for the final 18 h. Results are shown as counts per minute (CPM) ± SD of triplicate wells. A representative of two independent experiments is shown.
Discussion
The activation of DCs plays a critical role in the development of T cell–mediated immune responses. Our previous work has made it clear that low-molecular weight degradation products of HA (sHA) produced at sites of inflammation are key players in DC activation (5). The exciting outcome of the study presented here is that the maturation and activation of human and murine DCs in response to sHA is dependent on a functional TLR-4 receptor complex. Thus, sHA was able to mediate activation of bone marrow–derived DCs from wild-type C3H/HeN and C57BL/10Sn mice but not of DCs from the respective closely related C3H/HeJ and C57BL/10ScCr mice, which on account of defects in TLR-4 are LPS nonresponder and exhibit no inflammatory response to endotoxin. In contrast, DCs from TLR-2–deficient mice retained the ability to respond to sHA. Furthermore, blockade of TLR-4 on human monocyte–derived DCs abrogated their ability to produce TNF-α in response to sHA. The stimulatory effects of sHA on DCs were mediated by components of the signaling pathway activated by TLR-4, including the phosphorylation of p38 and p42/p44 MAPK and the translocation of NF-κB to the nucleus.
It is becoming increasingly evident that subcomponents of the extracellular matrix, especially of those built of repeating disaccharide units, can have immunomodulatory functions. NF-κB and intracellular adhesion molecule 1 are induced in endothelial cells by dermatan sulfate, a substance with high structural homology to HA (34), and low molecular weight heparan sulfate can modulate the migration of epidermal Langerhans cells, the immature precursors of skin DCs (35). Nevertheless, it remains unclear whether these low molecular weight fragmentation products use the same or different receptors to exert their effects compared with the intact parental macromolecule. HMW-HA binds to CD44 and RHAMM (36), but we and others could exclude binding of sHA fragments to these receptors (5, 37). Here we show for the first time that small fragmentation products of HA use receptors and signaling cascades which are different from those used by HMW-HA.
The concentrations of sHA needed to induce TLR-4–mediated signaling in DCs are several orders of magnitude higher than for LPS. This makes sense, as HA is a physiological constituent of the organism and minute amounts of degradation products enter into the bloodstream to be further degraded in the liver. Interestingly, higher levels of circulating HA are found during sepsis, correlating with disease severity and prognosis (38). For these reasons, it seems most plausible that only high concentrations of sHA, occurring locally and exclusively at sites of inflammation, induce proinflammatory signals. Our preliminary data using suction blister fluid (24) suggest that small glycosaminoglycan fragments of 4–6 oligosaccharides in size are indeed produced locally in inflamed skin (data not shown), consistent with this hypothesis.
There are many similarities between activation of DCs by LPS and by sHA. For example, DCs from TLR-4–deficient mice are nonresponsive to both LPS and sHA, but TLR-2–deficient DCs are still activated by both substances. Therefore, we performed extensive controls to exclude the possibility that our sHA preparations were contaminated with LPS. In this regard, it is also significant that while there are similarities between the signaling pathways induced by LPS and sHA, a critical difference is that LPS induces PI3-kinase (39), whereas we show here that sHA does not. Moreover, ligation of the TLR-4 complex by LPS in mouse macrophages occurs very rapidly, with the receptor being downregulated within 1 h after stimulation (40). In contrast, we found that both the intracellular signaling and TNF-α protein synthesis by sHA-stimulated DCs are notably delayed for ∼5–6 h in comparison to LPS. Thus, although we cannot completely rule out the possibility that sHA might act synergistically with trace amounts of LPS present in our sHA preparations which are below the threshold of detection and which alone cannot mediate DC maturation, a much more likely interpretation of our results is that sHA is an independent inducer of DC activation which signals via TLR-4.
It remains to be shown how sHA induces TLR-4 signaling at the molecular level. Recent results demonstrate that TLR signaling is dependent on a complex series of protein interactions, which is well exemplified by LPS signaling via TLR-4 (13, 18). In addition to TLR-4, the serum protein LBP and the GPI-linked cell surface protein CD14 are required for the host response to LPS (16). LPS binds to LBP, which in turn binds to CD14. The LPS-LBP-CD14 complex is then thought to interact with TLR-4, perhaps directly (15, 41), which in turn induces TLR-4 signaling via MyD88 and IRAK. TLR-4 also appears to contact LPS directly within the multiprotein complex. A further protein called MD-2 is also required for LPS-induced signaling (23, 41). This secreted extracellular protein binds to the extracellular portion of TLR-4 and possibly stabilizes TLR-4 dimers (23, 42). The complex molecular interactions involved in TLR signaling appears define the range of substances to which a given TLR can recognize, with the specificity determined at least in part by the TLR accessory molecules which bind to the ligand (42).
In sHA-induced TLR-4 signaling, sHA might bind to known TLR-4 accessory molecules, but more likely to as-yet unidentified ones. This latter possibility is underscored by structural considerations: LPS and sHA share almost no homology, except for the highly diverse repeating sugar units at the end of the LPS molecule (43). However, the binding of LPS to the TLR-4 complex and its activating effect on macrophages is mediated by the Lipid A moiety of the molecule (43). Furthermore, other substances known to bind the TLR-4 complex show high diversity. Human heat shock protein 60 can induce a TLR-4 complex–dependent activation of macrophages by upregulation of adhesion molecules and the release of proinflammatory mediators (20). Taxol, a dipertene purified from the bark of Taxus brevifolia, also mediates a TLR-4– and MD-2–dependent activation of murine macrophages. Interestingly this effect could only be observed with murine cells, but not with human LPS responsive cells (21). Therefore, it is significant that we found sHA to effectively activate both murine and human DCs which is suggestive of a highly conserved mechanism, since human TLR-4 has been described as being more effective in the discrimination of possible ligands (21, 44). Future work will focus on dissecting the way in which sHA interacts with components of the TLR-4 complex to induce the signal transduction which leads to DC maturation.
It is clear from the data presented here and from our previous studies (5) that the sHA-induced DC activation is dependent on the induction of TNF-α secretion. Our data suggest that TLR-4 is required for this induction, as TLR-4–deficient DCs did not produce TNF-α in response to sHA, and an anti–TLR-4 antibody was able to block sHA-mediated TNF-α release by wild-type DCs. However, the anti–TLR-4 antibody had no effect on TNF-α production if given 7 h after sHA treatment (Fig. 5 A). This coincides with downregulation of TLR-4 expression in response to sHA, although there is clearly some TLR-4 on the surface of DCs 24 h after sHA treatment (Fig. 5 B and C). These data are consistent with the notion that TNF-α produced by DCs in response to sHA binds to the TNF receptor I on the same cells, which in turn delivers a signal that produces more TNF-α (45).
The current understanding of TLRs and their associated molecules suggests that these multi-protein complexes developed in lower organisms such as Drosophila to provide innate immunity by recognizing conserved motifs present on pathogens. Therefore, it is fascinating that the data presented here suggest that the same molecular machinery involved in the innate immune response has perhaps been evolutionarily coopted to activate the immune response in response to degradation of endogenous extracellular matrix components during inflammation. Further study of the TLRs and the molecules they interact with will undoubtedly reveal many more new and exciting insights into the molecular mechanisms which regulate immunity.
Acknowledgments
This work was supported by a grant from the Deutsche Forschungsgemeinschaft (DFG TE 284/2-1).
Footnotes
Abbreviations used in this paper: DC, dendritic cell; HA, Hyaluronic acid; LAL, Limulus amebocyte lysate; MAPK, mitogen-activated protein kinase; PI, phosphatidylinositol; sHA, low-molecular weight oligosaccharides of HA; TLR, Toll-like receptor.
References
- 1.Agren, U.M., R.H. Tammi, and M.I. Tammi. 1997. Reactive oxygen species contribute to epidermal hyaluronan catabolism in human skin organ culture. Free Radic. Biol. Med. 23:996–1001. [DOI] [PubMed] [Google Scholar]
- 2.Weigel, P.H., G.M. Fuller, and R.D. LeBoeuf. 1986. A model for the role of hyaluronic acid and fibrin in the early events during the inflammatory response and wound healing. J. Theor. Biol. 119:219–234. [DOI] [PubMed] [Google Scholar]
- 3.Laurent, T.C., and J.R. Fraser. 1992. Hyaluronan. FASEB J. 6:2397–2404. [PubMed] [Google Scholar]
- 4.Noble, P.W., C.M. McKee, M. Cowman, and H.S. Shin. 1996. Hyaluronan fragments activate an NF-κB/I-κBα autoregulatory loop in murine macrophages. J. Exp. Med. 183:2373–2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Termeer, C., J. Hennies, U. Voith, T. Ahrens, J.M. Weiss, P. Prehm, and J.C. Simon. 2000. Oligosaccharides of Hyaluronan are potent activators of dendritic cells. J. Immunol. 165:1863–1870. [DOI] [PubMed] [Google Scholar]
- 6.Siegelman, M.H., H.C. DeGrendele, and P. Estess. 1999. Activation and interaction of CD44 and hyaluronan in immunological systems. J. Leukoc. Biol. 66:315–321. [DOI] [PubMed] [Google Scholar]
- 7.Deed, R., P. Rooney, P. Kumar, J.D. Norton, J. Smith, A.J. Freemont, and S. Kumar. 1997. Early-response gene signalling is induced by angiogenic oligosaccharides of hyaluronan in endothelial cells. Inhibition by non-angiogenic, high-molecular-weight hyaluronan. Int. J. Cancer. 71:251–256. [DOI] [PubMed] [Google Scholar]
- 8.Oertli, B., B. Beck-Schimmer, X. Fan, and R.P. Wuthrich. 1998. Mechanisms of hyaluronan-induced up-regulation of ICAM-1 and VCAM-1 expression by murine kidney tubular epithelial cells: hyaluronan triggers cell adhesion molecule expression through a mechanism involving activation of nuclear factor-κB and activating protein-1. J. Immunol. 161:3431–3437. [PubMed] [Google Scholar]
- 9.Rockey, D.C., J.J. Chung, C.M. McKee, and P.W. Noble. 1998. Stimulation of inducible nitric oxide synthase in rat liver by hyaluronan fragments. Hepatology. 27:86–92. [DOI] [PubMed] [Google Scholar]
- 10.Slevin, M., J. Krupinski, S. Kumar, and J. Gaffney. 1998. Angiogenic oligosaccharides of hyaluronan induce protein tyrosine kinase activity in endothelial cells and activate a cytoplasmic signal transduction pathway resulting in proliferation. Lab. Invest. 78:987–1003. [PubMed] [Google Scholar]
- 11.Rock, F.L., G. Hardiman, J.C. Timans, R.A. Kastelein, and J.F. Bazan. 1998. A family of human receptors structurally related to Drosophila Toll. Proc. Natl. Acad. Sci. USA. 95:588–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Poltorak, A., X. He, I. Smirnova, M.Y. Liu, C.V. Huffel, X. Du, D. Birdwell, E. Alejos, M. Silva, C. Galanos, et al. 1998. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in TLR4 gene. Science. 282:2085–2088. [DOI] [PubMed] [Google Scholar]
- 13.Rescigno, M., F. Granucci, S. Citterio, M. Foti, and P. Ricciardi-Castagnoli. 1999. Coordinated events during bacteria-induced DC maturation. Immunol. Today. 20:200–203. [DOI] [PubMed] [Google Scholar]
- 14.Takeuchi, O., K. Hoshino, T. Kawai, H. Sanjo, H. Takada, T. Ogawa, K. Takeda, and S. Akira. 1999. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 11:443–451. [DOI] [PubMed] [Google Scholar]
- 15.Lien, E., T.K. Means, H. Heine, A. Yoshimura, S. Kusumoto, K. Fukase, M.J. Fenton, M. Oikawa, N. Qureshi, B. Monks, et al. 2000. Toll-like receptor 4 imparts ligand-specific recognition of bacterial lipopolysaccharide. J. Clin. Invest. 105:497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kitchens, R.L. 2000. Role of CD14 in cellular recognition of bacterial lipopolysaccharides. Chem. Immunol. 74:61–82. [DOI] [PubMed] [Google Scholar]
- 17.Takeuchi, O., K. Takeda, K. Hoshino, O. Adachi, T. Ogawa, and S. Akira. 2000. Cellular responses to bacterial cell wall components are mediated through MyD88-dependent signaling cascades. Int. Immunol. 12:113–117. [DOI] [PubMed] [Google Scholar]
- 18.Medzhitov, R., P. Preston-Hurlburt, E. Kopp, A. Stadlen, C. Chen, S. Ghosh, and C.A.J. Janeway. 1998. MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol. Cell. 2:253–258. [DOI] [PubMed] [Google Scholar]
- 19.Cario, E., I.M. Rosenberg, S.L. Brandwein, P.L. Beck, H.C. Reinecker, and D.K. Podolsky. 2000. Lipopolysaccharide activates distinct signaling pathways in intestinal epithelial cell lines expressing Toll-like receptors. J. Immunol. 164:966–972. [DOI] [PubMed] [Google Scholar]
- 20.Ohashi, K., V. Burkart, S. Flohe, and H. Kolb. 2000. Cutting edge: heat shock protein 60 is a putative endogenous ligand of the toll-like receptor-4 complex. J. Immunol. 164:558–561. [DOI] [PubMed] [Google Scholar]
- 21.Kawasaki, K., S. Akashi, R. Shimazu, T. Yoshida, K. Miyake, and M. Nishijima. 2000. Mouse toll-like receptor 4.MD-2 complex mediates lipopolysaccharide-mimetic signal transduction by Taxol. J. Biol. Chem. 275:2251–2254. [DOI] [PubMed] [Google Scholar]
- 22.Engelhardt, R., A. Mackensen, and C. Galanos. 1991. Phase I trial of intravenously administered endotoxin (Salmonella abortus equi) in cancer patients. Cancer Res. 51:2524–2530. [PubMed] [Google Scholar]
- 23.Shimazu, R., S. Akashi, H. Ogata, Y. Nagai, K. Fukudome, K. Miyake, and M. Kimoto. 1999. MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J. Exp. Med. 189:1777–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Inaba, K., M. Inaba, M. Deguchi, K. Hagi, R. Yasumizu, S. Ikehara, S. Muramatsu, and R.M. Steinman. 1993. Granulocytes, macrophages, and dendritic cells arise from a common major histocompatibility complex class II-negative progenitor in mouse bone marrow. Proc. Natl. Acad. Sci. USA. 90:3038–3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.von Leoprechting, A., P. van der Bruggen, H.L. Pahl, A. Aruffo, and J.C. Simon. 1999. Stimulation of CD40 on immunogenic human malignant melanomas augments their cytotoxic T lymphocyte-mediated lysis and induces apoptosis. Cancer Res. 59:1287–1294. [PubMed] [Google Scholar]
- 26.Zhang, F.X., C.J. Kirschning, R. Mancinelli, X.P. Xu, Y. Jin, E. Faure, A. Mantovani, M. Rothe, M. Muzio, and M. Arditi. 1999. Bacterial lipopolysaccharide activates nuclear factor-κB through interleukin-1 signaling mediators in cultured human dermal endothelial cells and mononuclear phagocytes. J. Biol. Chem. 274:7611–7614. [DOI] [PubMed] [Google Scholar]
- 27.Visintin, A., A. Mazzoni, J.H. Spitzer, D.H. Wyllie, S.K. Dower, and D.M. Segal. 2001. Regulation of Toll-like receptors in human monocytes and dendritic cells. J. Immunol. 166:249–255. [DOI] [PubMed] [Google Scholar]
- 28.Waterman, W.H., T.F. Molski, C.K. Huang, J.L. Adams, and R.I. Sha'afi. 1996. Tumour necrosis factor-α-induced phosphorylation and activation of cytosolic phospholipase A2 are abrogated by an inhibitor of the p38 mitogen-activated protein kinase cascade in human neutrophils. Biochem. J. 319:17–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alessi, D.R., A. Cuenda, P. Cohen, D.T. Dudley, and A.R. Saltiel. 1995. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J. Biol. Chem. 270:27489–27494. [DOI] [PubMed] [Google Scholar]
- 30.Graber, M., C.H. June, L.E. Samelson, and A. Weiss. 1992. The protein tyrosine kinase inhibitor herbimycin A, but not genistein, specifically inhibits signal transduction by the T cell antigen receptor. Int. Immunol. 4:1201–1210. [DOI] [PubMed] [Google Scholar]
- 31.Powis, G., R. Bonjouklian, M.M. Berggren, A. Gallegos, R. Abraham, C. Ashendel, L. Zalkow, W.F. Matter, J. Dodge, and G. Grindey. 1994. Wortmannin, a potent and selective inhibitor of phosphatidylinositol-3-kinase. Cancer Res. 54:2419–2423. [PubMed] [Google Scholar]
- 32.Weinstein, S.L., C.H. June, and A.L. DeFranco. 1993. Lipopolysaccharide-induced protein tyrosine phosphorylation in human macrophages is mediated by CD14. J. Immunol. 151:3829–3838. [PubMed] [Google Scholar]
- 33.Lien, E., T.J. Sellati, A. Yoshimura, T.H. Flo, G. Rawadi, R.W. Finberg, J.D. Carroll, T. Espevik, R.R. Ingalls, J.D. Radolf, and D.T. Golenbock. 1999. Toll-like receptor 2 functions as a pattern recognition receptor for diverse bacterial products. J. Biol. Chem. 274:33419–33425. [DOI] [PubMed] [Google Scholar]
- 34.Penc, S.F., B. Pomahac, E. Eriksson, M. Detmar, and R.L. Gallo. 1999. Dermatan sulfate activates nuclear factor-κb and induces endothelial and circulating intercellular adhesion molecule-1. J. Clin. Invest. 103:1329–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O'Sullivan, G.M., C.M. Boswell, and G.M. Halliday. 2000. Langerhans cell migration is modulated by N-sulfated glucosamine moieties in heparin. Exp. Dermatol. 9:25–33. [DOI] [PubMed] [Google Scholar]
- 36.Entwistle, J., C.L. Hall, and E.A. Turley. 1996. HA receptors: regulators of signalling to the cytoskeleton. J. Cell. Biochem. 61:569–577. [DOI] [PubMed] [Google Scholar]
- 37.Tammi, R., D. MacCallum, V.C. Hascall, J.P. Pienimaki, M. Hyttinen, and M. Tammi. 1998. Hyaluronan bound to CD44 on keratinocytes is displaced by hyaluronan decasaccharides and not hexasaccharides. J. Biol. Chem. 273:28878–28888. [DOI] [PubMed] [Google Scholar]
- 38.Berg, S. 1997. Hyaluronan turnover in relation to infection and sepsis. J. Intern. Med. 242:73–77. [DOI] [PubMed] [Google Scholar]
- 39.Ardeshna, K.M., A.R. Pizzey, S. Devereux, and A. Khwaja. 2000. The PI3 kinase, p38 SAP kinase, and NF-κB signal transduction pathways are involved in the survival and maturation of lipopolysaccharide-stimulated human monocyte-derived dendritic cells. Blood. 96:1039–1046. [PubMed] [Google Scholar]
- 40.Nomura, F., S. Akashi, Y. Sakao, S. Sato, T. Kawai, M. Matsumoto, K. Nakanishi, M. Kimoto, K. Miyake, K. Takeda, and S. Akira. 2000. Cutting edge: endotoxin tolerance in mouse peritoneal macrophages correlates with down-regulation of surface toll-like receptor 4 expression. J. Immunol. 164:3476–3479. [DOI] [PubMed] [Google Scholar]
- 41.Akashi, S., R. Shimazu, H. Ogata, Y. Nagai, K. Takeda, M. Kimoto, and K. Miyake. 2000. Cutting edge: cell surface expression and lipopolysaccharide signaling via the toll-like receptor 4-MD-2 complex on mouse peritoneal macrophages. J. Immunol. 164:3471–3475. [DOI] [PubMed] [Google Scholar]
- 42.Aderem, A., and R.J. Ulevitch. 2000. Toll-like receptors in the induction of the innate immune response. Nature. 406:782–787. [DOI] [PubMed] [Google Scholar]
- 43.Jann, K., and B. Jann. 1998. Bacterial polysaccharides related to manmmalian structures. Polysaccharides: Structural Diversity and Functional Versatility. S. Dumitriu, editor. Marcel Dekker, Inc., New York. 211–229 pp.
- 44.Poltorak, A., P. Ricciardi-Castagnoli, S. Citterio, and B. Beutler. 2000. Physical contact between lipopolysaccharide and Toll-like receptor 4 revealed by genetic complementation. Proc. Natl. Acad. Sci. USA. 97:2163–2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen, B., Y. Shi, J.D. Smith, D. Choi, J.D. Geiger, and J.J. Mule. 1998. The role of tumor necrosis factor α in modulating the quantity of peripheral blood-derived, cytokine-driven human dendritic cells and its role in enhancing the quality of dendritic cell function in presenting soluble antigens to CD4+ T cells in vitro. Blood. 91:4652–4661. [PubMed] [Google Scholar]