

Abstract
The immune response against hepatitis C virus (HCV) is rarely effective at clearing the virus, resulting in ∼170 million chronic HCV infections worldwide. Here we report that ligation of an HCV receptor (CD81) inhibits natural killer (NK) cells. Cross-linking of CD81 by the major envelope protein of HCV (HCV-E2) or anti-CD81 antibodies blocks NK cell activation, cytokine production, cytotoxic granule release, and proliferation. This inhibitory effect was observed using both activated and resting NK cells. Conversely, on NK-like T cell clones, including those expressing NK cell inhibitory receptors, CD81 ligation delivered a costimulatory signal. Engagement of CD81 on NK cells blocks tyrosine phosphorylation through a mechanism which is distinct from the negative signaling pathways associated with NK cell inhibitory receptors for major histocompatibility complex class I. These results implicate HCV-E2–mediated inhibition of NK cells as an efficient HCV evasion strategy targeting the early antiviral activities of NK cells and allowing the virus to establish itself as a chronic infection.
Keywords: natural killer cells, inhibitory signaling, immune evasion, chronic viral infection, tetraspanin
Introduction
All pathogens have evolved specific mechanisms to undermine host defenses. The evasion strategies of those pathogens which give rise to chronic infections can be remarkably sophisticated given the need to circumvent multiple arms of the immune system over years-decades of infection (1). Hepatitis C virus (HCV)* gives rise to chronic liver infections and is the leading cause of chronic liver disease (2). A hallmark of HCV-associated liver pathology is a massive lymphocyte infiltrate in the infected organ (3, 4). Despite this apparently aggressive immune response, spontaneous resolution of chronic HCV is exceedingly rare, suggesting that HCV actively evades and/or directly limits the effectiveness of this immune response. Recent studies have implicated the HCV-E2 protein as a central component of two potential HCV evasion mechanisms (5, 6). Thus, the HCV-E2 protein appears to play at least two distinct roles in thwarting the immune response mounted against the virus.
NK cells are lymphocytes active in innate immune responses against viruses, as well as, bacteria and tumors, due to their potent cytotoxic activities and rapid production of cytokines (7). During the early immune response, NK cell functions are regulated by cytokine feedback loops and direct interaction with infected or transformed cells which are targets of their cytotoxicity. The specificity of NK–target interactions is determined by a balance between stimulatory and inhibitory receptors (8). The NK stimulatory receptors and their ligands on target cells are a heterogeneous class of surface molecules whose usage can vary depending on the target cell type and activation state of the NK cell (8). The less specific interactions between stimulatory NK receptors and their target cell ligands are kept under control by diverse families of well characterized NK inhibitory receptors for MHC class I (8).
The HCV-E2 protein binds the human cell surface molecule CD81 with high affinity, indicating that CD81 may function as a cellular receptor for the virus (9). CD81 belongs to a family of molecules called tetraspanins, characterized by four transmembrane domains forming two extracellular loops (10). The major loop of CD81 is necessary and sufficient for binding to HCV-E2 (9). CD81 is found on the surface of almost all nucleated cells where it is a component of multiple and distinct receptor complexes depending on the cell type (10). Recent studies have shown that ligation of CD81 with antibodies or HCV-E2 costimulates T cells (11). Here we demonstrate that unlike T cells, CD81 ligation by HCV-E2 potently inhibits NK cell functions through a novel negative signaling mechanism. These results provide the first demonstration of an HCV immune escape mechanism specifically targeting the early antiviral responses of NK cells. In addition, they shed new light on fundamental differences between T and NK cell signaling pathways and a potentially dual role of CD81 on the two cell types.
Materials and Methods
Antibodies and Reagents.
Antibodies used were: anti-CD3 (clones OKT-3 and TR-66; American Type Culture Collection), anti-CD16 (3G8, American Type Culture Collection), anti-CD56 (B159.5, provided by G. Trinchieri, Schering Plough Research Institute, Dardilly, France), anti-CD81 (JS-81; BD PharMingen), anti-HLA class I (W6/32; Serotec), anti–HCV-E2 (provided by D. Rosa, Chiron S.p.A., Siena, Italy), anti–IFN-γ (B133.1 and B133.5), anti–TNF-α (B145.9 and B154.7) were both provided by G. Trinchieri. The antiphosphotyrosine (4G10; Upstate Biotechnology, Inc.), anti-p42/p44 erk-2, anti-phospho p42/p44 erk-2 (Cell Signaling Technology), and anti-CD3ζ chain (6B10.2; Santa Cruz Biotechnology, Inc.) were used for immunoprecipitation and Western blot analysis.
Cell Preparation and Cultures.
PBMCs were prepared from peripheral blood by Ficoll-Paque density gradient centrifugation, followed by 1-h incubation in plastic flask to remove adherent monocytes. Cultured NK cells were prepared as described previously (12). All cultures were collected at days 8–10. NK cells (>98% CD56+/CD3−/CD19−/CD14−) were purified by depletion of the magnetically labeled CD3+/CD14+/CD19+ cells using MACS® Separation Columns (Miltenyi Biotec). NK and T cell clones were generated and maintained as described previously (13). In brief, PBMCs were stained with antibodies for CD3, CD56, killer Ig-like receptor (KIR)2DL/S1 (EB6, CD158a; Immunotech), KIR2DL/S2 (GL183, CD158b; Immunotech), KIR3DL1 (NKB1, DX9; Becton Dickinson), KIR3DL2 (NKAT4, DX31, provided by L. Lanier, University of California at San Francisco, San Francisco, CA) TCR Vα24 (C15; Immunotech), and/or TCR Vβ11 (C21; Immunotech) and the desired subpopulations were single-cell sorted using a FACSVantage™ SE® cell sorter (Becton Dickinson) in 60-well Terazaki plates (Robbins Scientific). To expand NK T cells, cells were activated with α-GalCer (100 μg/ml in 100% DMSO) at the final concentration of 30–100 ng/ml (14).
Antibody Coating and Cell Stimulation.
The following purified mAb were used in stimulation experiments: anti-CD16 (3G8), anti-CD3 (TR-66), anti CD81 (JS-81), anti E2 (291A2), anti-CD56 (B159.5), anti-HLA class I (W6/32). 96-well plates (Greiner) were coated as described previously (11). The recombinant purified HCV E2 protein (10 μg/ml in PBS) was incubated in the coated anti-E2 mAb (10 μg/ml) plates for 1 h at 37°C. Prior to the addition of cells the excess HCV-E2 was removed by washing in PBS.
Immunofluorescence Assays.
Antibodies used for flow cytometry were the following: anti-CD3, anti-CD56, anti CD16, NKB1 (KIR3DL1), anti–IFN-γ, and anti–IL-4 (all Becton Dickinson). Anti-CD158a (KIR2DL/S1), anti-CD158b (KIR2DL/S2), anti-TCR Vα24 (C15), and anti-TCR Vβ11 (C21) were from Immunotech. Cells were analyzed on a FACSCalibur™ flow cytometer (Becton Dickinson) using CELLQuest™ software.
Cytokine Production Assays.
Purified NK cells were cultured in 96-well coated plates for 24 and 48 h and the supernatants were assessed by ELISA for IFN-γ and TNF-α. The capture antibodies were anti–IFN-γ mAb B133.1 and the anti–TNF-α mAb B154.9, respectively, which were immobilized on ELISA plates. Detection of each cytokine was achieved using biotinylated anti–IFN-γ mAb B133.5 and anti–TNF-α mAb B154.7, streptravidin-conjugated peroxidase (Sigma-Aldrich), and the chromogenic substrate o-phenylenediamine dihydrochloride (Sigma-Aldrich). Spectrophotometric analysis was performed at 450 nm on a Spectramax 340 spectrophotometer (Molecular Devices) using Softmax Pro software (Molecular Devices).
N-carbobenzoxy-L-thiobenzyl Ester Esterase Assay.
The amount of secreted N-carbobenzoxy-L-thiobenzyl ester (BLT)-esterase was measured using 105 purified cultured NK cells in 100 μl of complete medium in a 96-well ELISA plate coated with the indicated mAb, as described previously (15). After 4-h incubation at 37°C, 5% CO2, 50 μl of cell-free supernatant fluids were added to 100 μl of BLT solution (0.2 mM BLT, 0.22 mM 5,5′-dithio-bis-[2-nitrobenzoic acid]; DTNB) in PBS. BLT-esterase activity was measured by absorbance at 405 nm. Total BLT-esterase activity was determined from lysed (freeze/thaw three times) untreated NK cells.
Proliferation Assays.
NK cell proliferation was assessed by 3[H] thymidine incorporation using 2 × 104 cells per well in mAb-coated 96-well plates. Cultures were pulsed after 48 h with Ci/well 3[H] thymidine, incubated for an additional 12 h, harvested onto filter plates (Packard Instrument Co.), and counts were analyzed using a Top Count NXT β counter (Packard Instrument Co.).
Immunoprecipitation and Immunoblot Analysis.
Purified NK cells (107 per sample for immunoprecipitation or 106 per sample for whole-cell extracts) were incubated on ice for 5 min with anti-CD16 mAb (3G8), anti-CD81mAb (JS-81, 10 μg/ml), or anti-CD56 mAb (B159.5, 10 μg/ml). Cells were then washed and incubated with goat anti–mouse IgG F(ab)′2 fragments (Sigma-Aldrich) at 30 μg/ml at 37°C for the indicated time. Cells were lysed in buffer containing 20 mM Tris-HCl, pH 7.4, 40 mM NaCl, 5 mM EDTA, 1 mM NaF, 20 mM Na4P207, 1 mM Na3VO4, 0.1% BSA, 1 mM PMSF, 5 μg/ml aprotinin, 10 μg/ml leupeptin, and 1% Triton X-100. In some experiments, whole cell extracts were resolved by SDS-PAGE. For immunoprecipitations, cell lysates were incubated for 18 h with the appropriate Ab (4G10 for phosphotyrosines, 6B10.2 antibody for the ζ-chain) and then for 1–2 h with protein A or G –Sepharose (Upstate Biotechnology, Inc.). The immunoprecipitates were resolved by SDS-PAGE and transfered to nitrocellulose membranes (Sigma-Aldrich). Tyrosine-phosphorylated proteins were detected with the 4G10 mAb, followed by sheep anti–mouse IgG coupled to horseradish peroxidase (Amersham Pharmacia Biotech) and the Supersignal detection system (Pierce Chemical Co.). The p42/p44 and phospho p42/p44 mitogen-activated protein kinases (MAPKs) were detected using Cell Signaling Technology antibodies, followed by donkey anti–rabbit IgG coupled to horseradish peroxidase (Amersham Pharmacia Biotech).
Results
The Effect of CD81 Ligation on NK Cell Function.
Previous results from our group indicated a substantial enrichment of NK cells in HCV-infected livers and their altered expression of KIRs (4, 16). Given the role of CD81 as both an HCV attachment receptor with high affinity for HCV-E2 and a functionally relevant molecule on lymphocytes (9–11), we developed an in vitro model to study the effects of HCV-E2 binding to CD81 on NK cells. We first addressed whether cross-linking NK cell CD81 altered NK cell functions induced by the NK cell triggering receptor CD16 (Fig. 1) . CD16 is the low affinity receptor for IgG (FcγRIII) and is considered the best characterized and most potent stimulatory receptor found on NK cells (7). Using highly purified NK cells obtained from bulk culture, we observed that CD16-induced TNF-α and IFN-γ production (Fig. 1 A and B) was inhibited strongly by coligation of NK cell CD81 with HCV-E2. Similarly, HCV-E2 cross-linking of CD81 blocked CD16-mediated induction of NK cell activation marker (CD25) expression (Fig. 1 C) and cytotoxic granule release (Fig. 1 D). In addition, ligation of CD81 on NK cells substantially inhibited their capacity to proliferate in response to IL-2, demonstrating that the inhibitory effect is not only active on CD16-induced signals (Fig. 1 E). A similar inhibitory effect of CD81 engagement was seen on NK cell IFN-γ production in response to treatment with IL-2, IL-12, or a combination of both (data not shown).
Figure 1.

Cross-linking of CD81 by HCV-E2 or anti-CD81 antibody blocks NK cell activation, cytokine production, and cytotoxic granule release induced by CD16 and IL-2 induced proliferation. Purified, cultured NK cells were stimulated for 24 or 48 h and the supernatants were analyzed for cytokine (TNF-α or IFN-γ) production (A and B). NK cells were stimulated for 24 h and then analyzed by flow cytometry to evaluate the expression level of the activation marker CD25 (C). 105 purified NK cells were stimulated for 4 h and supernatants were assayed for BLT-esterase activity which is defined as the percentage of the total BLT-esterase activity obtained from the same number of lysed NK cells (D). For these experiments (A–D) NK cells were cultured in the presence of the indicated concentrations of the anti-CD16 antibody alone (♦) or in combination with 10 μg/ml of: anti-CD56 (▴); anti–HCV-E2 (▪); anti-CD81 (○) or anti–HCV-E2 + rHCV-E2 (□). In E, NK cell proliferation in the presence or absence of rIL-2 was determined by 3[H]thymidine incorporation. NK cells were cultured at the indicated doses of rIL-2 alone (♦) or in combination with 10 μg/ml of: anti-CD56 (▴); anti-HCV-E2 (▪); anti-CD81 (○) or anti–HCV-E2 + rHCV-E2 (□). Experiments to determine the optimal concentrations of anti-CD81 or anti–HCV-E2 + rHCV-E2 required for NK cell inhibition, demonstrated that the negative effect was detectable over a broad range of concentrations (2.5–20 μg/ml), with 10 μg/ml giving the most potent and consistent inhibition compared with controls (data not shown).
In all experiments cross-linking with an anti-CD81 mAb mimicked the inhibition observed with HCV-E2 and no inhibition of NK cell functions induced through CD16 ligation was seen after cotreatment with the control reagents. In addition, anti-CD81 or HCV-E2 cross-linking in the absence of CD16 stimulation had no effect on any of the NK cell functions tested. The inhibitory effect of CD81 cross-linking was not due to toxicity of the HCV-E2 or anti-CD81 reagents because neither treatment effected NK cell survival under all of the conditions tested in Fig. 1. Specifically, flow cytometric analyses of NK cell viability, measured by propidium iodide uptake, or apoptosis determined by annexin V staining, revealed no increased NK cell death in response to CD81 ligation (data not shown).
Resting NK cells were also inhibited by CD81 ligation. Freshly isolated PBMCs were incubated with anti-CD16 mAb alone or in combination with the different anti-CD81 or control reagents used in the experiments of Fig. 2 . The NK cells from the mixed PBMC populations were analyzed by flow cytometry via specific gating on the CD56+/CD3− NK cell subset (Fig. 2). Again, the effect of CD81 cross-linking was potent inhibition of CD16-induced IFN-γ production (Fig. 2 A, C, and E) and activation marker (CD25) expression. (Fig. 2 B, D, and F). Thus, resting NK cells are inhibited by CD81 engagement similar to the purified/activated NK cells obtained from bulk culture. This suggests that early in the immune response against HCV and before NK cells are fully activated, binding of HCV-E2 to NK cell CD81 has the potential to dampen significantly the rapid antiviral responses of NK cells.
Figure 2.

CD81 engagement blocks the functions of resting NK cells. PBMCs freshly purified from healthy donors were cultured in complete medium on plastic plates coated with no antibody (A and B), 1 μg/ml of CD16 mAb alone (C and D), or 1 μg/ml of CD16 mAb plus 10 μg/ml of CD81 mAb (E and F). After 4 h of Brefeldin-A treatment, cells were stained for intracellular IFN-γ production (A, C, and E) and for the surface expression of the activation marker CD25. The plots in (A–F) represent the CD3-CD56+ subpopulation as defined by the staining in H. In G, CD16 stimulation is specific for NK cells as the CD3+ (T cell) PBMC subpopulation did not produce any IFN-γ, as assayed by intracellular staining.
Opposing Effects of CD81 Ligation on NK Cells and T Cells.
NK cells and T cells share many phenotypic and functional features (17). Small subsets of T cells also express characteristic NK cell markers such as CD16 and CD56. In addition, KIR+ T cells can be isolated from the blood of healthy individuals and these T cells are inhibited by KIR recognition of HLA class I ligands, similar to what is routinely observed for NK cells (18). A previous report from Wack et al. demonstrated that bulk populations of T cells were costimulated by CD81 cross-linking, however this study did not investigate T cells bearing NK cell receptors (11). Therefore, we generated T and NK cell clones from healthy individuals and compared their responses after CD81 engagement (Fig. 3) . Similar to our previous results with cultured or resting NK cells, the NK cell clones were all inhibited by CD81 cross-linking either with an anti-CD81 mAb or HCV-E2 (Fig. 3 A). By contrast, none of the T cell clones tested, representing six distinct phenotypic/functional subsets, were inhibited after simultaneous ligation of CD81 and CD3 (Fig. 3 B and C). In fact, 5 out of the 6 subsets tested (TCR αβ+ “classical”; KIR+; NKT; Th1; and Th2 T cell clones) were all costimulated by simultaneous CD81 and CD3 engagement similar to previous results using bulk populations of resting T cells and PHA-activated T cell lines (11). The one exception was found using a rare and understudied population of T cells bearing the characteristic NK marker, CD16 (19). Unlike NK cells and other T cells, the response of these cells to cocross-linking of CD3 and CD81 on their surface was neutral. This failure of CD81 ligation to elicit any response in CD16+ T cells was observed when we measured CD3-induced IFN-γ (Fig. 3 C) and TNF-α production, as well as, CD3-mediated proliferation and activation marker expression (data not shown). Ligation of CD16 on these T cell clones had no effect, consistent with a previous report (20).
Figure 3.

CD81 cross-linking has opposite effects on NK and T cells. NK (A) and T (B) cell clones from the same healthy donor were stimulated for 24 h and the supernatants were analyzed for the presence of IFN-γ. The NK cell clones (A) were stimulated with the indicated concentrations of anti-CD16 alone (•) or in combination with 10 μg/ml of: anti-CD81 (○) or anti-HCV-E2 + rHCV-E2 (□). The “classical” TCR αβ+ T cell clones (B) were stimulated with decreasing concentrations of anti-CD3 alone (•) or in the presence of 10 μg/ml: anti-CD81 (○) or anti–HCV-E2 + rHCV-E2 (□). Control antibodies for anti-CD56 (NK cells) or anti-class I (T cells) had no effect and neither did treatment with the anti-HCV-E2 reagent alone (data not shown). In (C) the effects of CD81 ligation on different T and NK cell subsets is summarized. NKT (gray bar), KIR+ T (stippled bar), CD16+ T (hatched bar), Th1 (striped bar), Th2 (white bar, and NK cell (black bar) clones were obtained from the same healthy donor by single cell sorting. The scheme represents the effect of CD81 cross-linking on these different cell types when activated by the appropriate stimulus (anti-CD16 mAb for NK cells, anti-CD3 mAb for the other T cell types). Cytokine production (IFN-γ: KIR+ T; Th1; CD16+ T, NK or IL-4: Th2 cell clones), or proliferation (NKT) were used as readouts for CD81-mediated costimulation or inhibition. Results are presented as percentage change compared with treatment with 0.3 μg/ml of anti-CD16 (NK cells) or anti-CD3 (T cells). CD16+ T cells were also analyzed for proliferation, their ability to produce TNF-α and their expression of activation markers after CD81 ligation. In all cases this treatment had no effect (data not shown).
Inhibition of CD16-mediated Tyrosine Phosphorylation by CD81 Engagement.
The activation of protein tyrosine kinases (PTKs) and their phosphorylation of diverse substrates is a necessary event in NK cell activation through CD16 and other NK triggering receptors (21, 22). Therefore, we examined the capacity of CD81 cross-linking to block this critical signaling pathway. Analysis of total PTK activity was performed by determining the extent of phosphotyrosine (p-Tyr) substrates induced in NK cells by CD16 cross-linking alone or in combination with anti-CD81 or anti-CD56 treatment. The overall p-Tyr levels induced through CD16 were substantially diminished after simultaneous ligation of CD81 (Fig. 4 A). The examination of specific PTK substrates known to be phosphorylated in NK cells as a result of CD16 stimulation and important for CD16-induced functions yielded similar results (Fig. 4 B and C). Specifically, we chose for this analysis: (i) CD3ζ which is directly associated with CD16 in NK cells and whose phosphorylation is induced immediately proximal to CD16 ligation (23); and (ii) erk-2 that is part of the more downstream MAPK cascade important for NK cell cytokine gene expression and proliferation (24). The tyrosine phosphorylation of both signaling moieties was specifically inhibited after CD81 ligation. In addition, cross-linking with the anti-CD81 or anti-CD56 reagents alone had no effect on the extent of phosphorylated proteins detected in the NK cells in all of the experiments.
Figure 4.
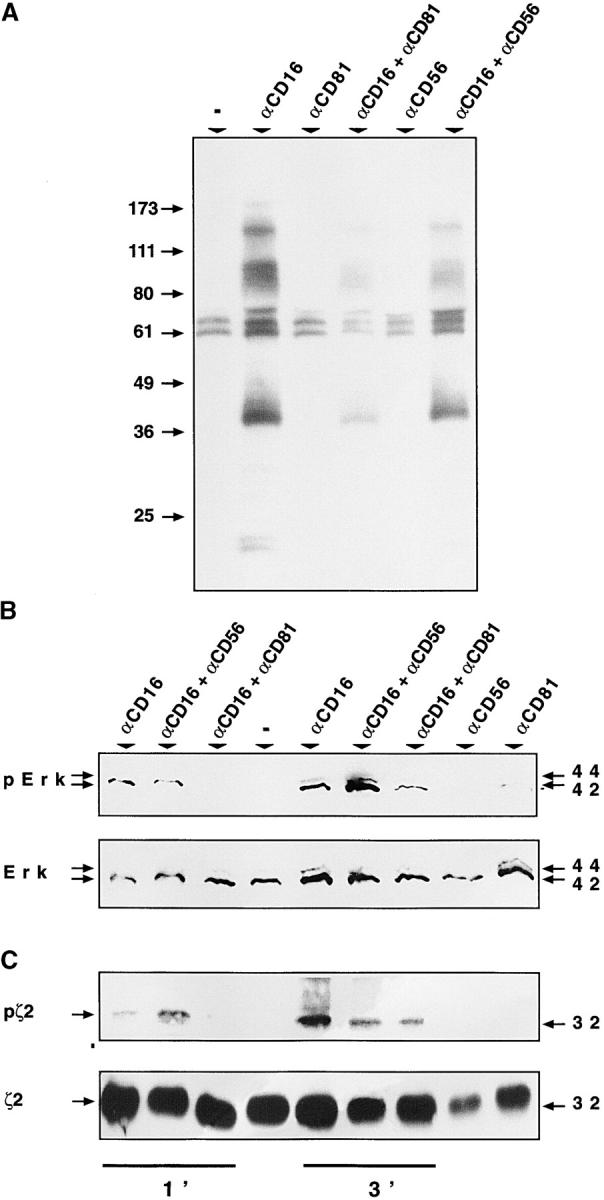
CD81 engagement inhibits specific CD16-triggered tyrosine phosphorylation events. Purified, cultured NK cells (107 per sample) were incubated with the indicated antibodies for 1 min and tyrosine phosphorylated proteins were immunoprecipitated from cell lysates, resolved by SDS-PAGE, transferred to a nitrocellulose membrane and immunoblotted with antiphosphotyrosine mAb (A). NK cells (107 per sample) were stimulated with the indicated cross-linked mAb's for 1 or 3 min, as indicated in the figure (B and C). Total cell lysates were resolved by SDS PAGE and immunoblotted (B) first with a rabbit polyclonal antiphospho-p44/42 MAPK (erk-2) antibody (top) and then reprobed with a rabbit polyclonal p44/42 MAPK (erk-2) antibody (bottom). Under the same conditions cell lysates were subjected to immunoprecipitation with anti-ζ polyclonal antibody (C). Immunoprecipitated proteins were immunoblotted first with antiphosphotyrosine mAb (top) and then with the immunoprecipitating polyclonal antibody (bottom). In these experiments, SDS-PAGE was performed in nonreducing conditions to detect the 32 kD ζ homodimers (ζ2).
We initiated a series of studies investigating the potential association of CD81 with KIR and the inhibitory signaling phosphatases, src homology 2 domain–bearing protein tyrosine phosphatase (SHP)1 and SHP2 (25), by performing coimmunoprecipitation/Western blotting experiments using anti-KIR, anti-SHP, and anti-CD81 anitbodies to detect specific complexes formed on the surface of NK cells. Studies were performed on unstimulated NK cells and those treated with anti-CD16 antibody alone or in combination with anti-CD81. Under all conditions the results of these analyses were negative indicating that a specific association between the HLA class I inhibitory receptor system and CD81 is unlikely (data not shown). This conclusion is also supported by the fact that KIR+ T cells which possess the capacity to be inhibited by HLA class I recognition through the KIR-SHP pathway (18) are not inhibited by CD81 ligation but rather costimulated by it (Fig. 3). In addition, ligation of CD81 blocks NK cell responses to soluble factors which has not been observed for other NK inhibitory receptor systems.
Discussion
NK cells provide a critical first line of defense against viral infections through their rapid and potent cytotoxic activity and production of inflammatory cytokines such as IFN-γ (26). However, no role for NK cells in the anti-HCV immune response has ever been established. We describe that the binding of the HCV envelope protein, HCV-E2, to CD81 directly blocks NK cell functional activation. Thus, HCV possesses a mechanism which specifically downregulates the NK cell response, providing the virus with an efficient immune-escape strategy capable of limiting the antiviral activities of NK cells early in the infectious process. This novel mechanism could be critical in determining the viruses ability to gain a foothold in the host and establish itself as a chronic infection.
The direct inhibition of NK cells via binding of HCV-E2 to CD81 provides a global “off” signal for NK cell activation and efficiently blocks cytokine production, activation marker expression, cytotoxic granule release, and proliferation. CD81-mediated inhibition of NK cells was seen for activated and resting NK cells and in NK cells from healthy donors and patients suffering from chronic HCV infections (data not shown), suggesting that the inhibitory effect is a general function of NK cell CD81 and could occur at all stages of infection. The functional inhibition can be explained by the fact that CD81 engagement blocks tyrosine phosphorylation of important upstream and downstream signaling moieties induced by cross-linking CD16. However, the inhibitory signals generated after CD81 ligation appear independent of the well characterized SHP-mediated negative signaling pathway associated with NK cell and other inhibitory receptor systems (25). Thus, CD81 participates in a previously unrecognized negative signaling pathway of NK cells.
T cell clones representing distinct functional and phenotypic subsets, including those with NK-like properties, were not inhibited by CD81 ligation. In fact, almost all of the different T cell subpopulations studied were costimulated by the interaction, similar to a previous report where bulk populations of T cells were used (11). Furthermore, a different study has shown that CD81 ligation on TCR γδ+ T cells is also costimulatory rather than inhibitory (27). This finding whereby the same surface molecule mediates completely opposing effects on NK cells and T cells is unprecedented and raises fundamental questions about the immunobiology of CD81; NK versus T cell signaling; and how HCV exploits these differences to confound both NK and T cell responses and establish itself as a chronic infection. Intriguingly, a similar inhibitory effect of CD81 ligation on basophils has been reported (28). The mechanism of this inhibition was not well characterized but appeared to be distinct from other known inhibitory pathways and similar to the inhibition of NK cells reported here (28). It is likely that CD81 inhibits NK cells and basophils through a common mechanism. The larger question of why NK cells and T cells which share so many phenotypic and functional characteristics (17) differ so dramatically in their responses to CD81 engagement still remains open for investigation. It is possible that distinct inhibitory or stimulatory receptors which differentially associate with CD81 on the two cell types govern these unique responses. Alternatively, a more fundamental difference in the intracellular signaling requirements of T and NK cells, and an inhibitory bias of the NK cell signaling machinery could explain the dual functional outcomes of CD81 ligation.
Infection with HCV is a major health problem worldwide. There is no therapy which is effective at eliminating established HCV infections in the majority of patients and prophylactic vaccine candidates are just beginning to enter clinical trials (16, 29). The study of HCV and, therefore, the design of effective treatments and vaccines is hampered by many technical obstacles. Two of the most important of which are the lack of in vitro culture systems and small animal models (30). These barriers render it difficult to study the virus and its associated pathologies directly. To overcome these limitations it has been necessary to develop novel experimental systems which shed light on HCV biology and pathogenesis. One such system employed the recombinant HCV-E2 protein to identify the high affinity interactions of this envelop protein and the virus itself with human CD81 (9). Using this model system we have uncovered a role for the HCV-E2:CD81 interaction which could function as an immune escape mechanism for the virus and explain how HCV is so adept at establishing itself as a chronic infection. Although it is currently impossible to measure directly how NK cell responses are affected upon binding to HCV-infected cells or the virus itself, our results presented suggest that such an encounter in vivo would suppress significantly NK cell functions. Given that the HCV-E2 protein is expected to be present at high copy number on HCV viral particles, the cross-linking of CD81 required for inhibition should be comparable to the antibody-mediated cross-linking reported here. However, further study of the potential inhibitory effects of HCV on NK cells is clearly warranted and it will be particularly important to establish the physiologic relevance of NK cell inhibition via HCV-E2 binding to CD81. In conclusion, these findings not only provide important new insights into HCV immune evasion and the role of NK cells in the anti-HCV response, but also into the unique functions of CD81 and the divergent signaling mechanisms of NK and T cells. It will be important in the future to investigate each of these new research avenues both in the context of HCV immune escape and in the more general framework of T and NK cell biology.
Acknowledgments
The authors would like to thank D. Rosa, G. Saletti, P. Dileri, and C. Giovine for providing critical reagents and blood samples.
A. Wack is supported by a Marie Curie Fellowship from the European Union.
A. Stilla's present address is Consorzio Mario Negri Sud, via Nazionale- Santa Maria Imbaro, 66100 Chieti, Italy.
A. D'Andrea's present address is Pharmaceutical Discovery Division, 100-08, SRI International, 333 Ravenswood Ave., Menlo Park, CA 94025.
Footnotes
Abbreviations used in this paper: HCV, hepatitis C virus; KIR, killer Ig-like receptors; MAPK, mitogen-activated protein kinase; PTK, protein tyrosine kinase; SHP, src homology 2 domain–bearing protein tyrosine phosphatase.
References
- 1.Brodsky, F. 1999. Stealth, sabotage and exploitation. Immunol. Rev. 168:5–11. [DOI] [PubMed] [Google Scholar]
- 2.Choo, Q., G. Kuo, A. Weiner, L. Overby, D. Bradley, and M. Houghton. 1989. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science. 244:359–362. [DOI] [PubMed] [Google Scholar]
- 3.Bianchi, L. 1983. Liver biopsy interpretation in hepatitis. Pathol. Res. Pract. 178:180–213. [DOI] [PubMed] [Google Scholar]
- 4.Nuti, S., D. Rosa, N. Valiante, G. Saletti, M. Caratozzolo, P. Dellabona, V. Barnaba, and S. Abrignani. 1998. Dynamics of intra-hepatic lymphocytes in chronic hepatitis C: enrichment for Vα24+ T cells and rapid elimination of effector cells by apoptosis. Eur. J. Immunol. 28:3448–3455. [DOI] [PubMed] [Google Scholar]
- 5.Farci, P., A. Shimoda, A. Coiana, G. Diaz, G. Peddis, J.C. Melpolder, A. Strazzera, D.Y. Chien, S.J. Munoz, A. Balestrieri, et al. 2000. The outcome of acute hepatitis C predicted by the evolution of the viral quasispecies. Science. 288:339–344. [DOI] [PubMed] [Google Scholar]
- 6.Taylor, D.R., S.T. Shi, P.R. Romano, G.N. Barber, and M.M. Lai. 1999. Inhibition of the interferon-inducible protein kinase PKR by HCV E2 protein. Science. 285:107–110. [DOI] [PubMed] [Google Scholar]
- 7.Trinchieri, G. 1989. Biology of natural killer cells. Adv. Immunol. 47:187–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lanier, L. 1998. NK cell receptors. Annu. Rev. Immunol. 16:359–393. [DOI] [PubMed] [Google Scholar]
- 9.Pileri, P., Y. Uematsu, S. Campagnoli, G. Galli, F. Falugi, R. Petracca, W. AJ, M. Houghton, D. Rosa, G. Grandi, and S. Abrignani. 1998. Binding of hepatitis C virus to CD81. Science. 282:938–941. [DOI] [PubMed] [Google Scholar]
- 10.Levy, S., S. Todd, and H. Maecker. 1998. CD81 (TAPA-1): a molecule involved in signal transduction and cell adhesion in the immune system. Annu. Rev. Immunol. 16:89–109. [DOI] [PubMed] [Google Scholar]
- 11.Wack, A., E. Soldaini, C.K. Tseng, S. Nuti, G.R. Klimpel, and S. Abrignani. 2001. Binding of hepatitis C virus envelope protein E2 to CD81 provides a co-stimulatory signal for human T cells. Eur. J. Immunol. 31:166–175. [DOI] [PubMed] [Google Scholar]
- 12.Perussia, B., C. Ramoni, I. Anegon, M.C. Cuturi, J. Faust, and G. Trinchieri. 1987. Preferential proliferation of natural killer cells among perpheral blood mononuclear cells co-cultured with B lymphoblastoid cell lines. Nat. Immun. Cell Growth Reg. 6:171–182. [PubMed] [Google Scholar]
- 13.Litwin, V., J. Gumperz, P. Parham, J.H. Phillips, and L.L. Lanier. 1993. Specificity of HLA class I antigen recognition by human NK clones: evidence for clonal heterogeneity, protection by self and non-self alleles, and influence of the target cell type. J. Exp. Med. 178:1321–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kawano, T., J. Cui, Y. Koezuka, I. Toura, Y. Kaneko, K. Motoki, H. Ueno, R. Nakagawa, H. Sato, E. Kondo, et al. 1997. CD1d-restricted and TCR-mediated activation of vα14 NKT cells by glycosylceramides. Science. 278:1626–1629. [DOI] [PubMed] [Google Scholar]
- 15.Takayama, H., G. Trenn, and M.V. Sitkovsky. 1987. A novel cytotoxic T lymphocyte activation assay. Optimized condition for antigen receptor triggered granule enzyme secretion. J. Immunol. Meth. 104:183–190. [DOI] [PubMed] [Google Scholar]
- 16.Valiante, N.M., A. D'Andrea, S. Crotta, F. Lechner, P. Klenerman, S. Nuti, A. Wack, and S. Abrignani. 2000. Life, activation and death of intrahepatic lymphocytes in chronic hepatitis C. Immunol. Rev. 174:77–89. [DOI] [PubMed] [Google Scholar]
- 17.Valiante, N.M., and P. Parham. 1996. NK cells and CTL: opposite sides of the same coin. Chem. Immunol. 64:146–163. [PubMed] [Google Scholar]
- 18.Phillips, J.H., J.E. Gumperz, P. Parham, and L.L. Lanier. 1995. Superantigen-dependent, cell-mediated cytotoxicity inhibited by MHC class I receptors on T lymphocytes. Science. 268:403–405. [DOI] [PubMed] [Google Scholar]
- 19.Lanier, L.L., A.M. Le, C.I. Civin, M.R. Loken, and J.H. Phillips. 1986. The relationship of CD16 (Leu-11) and Leu-19 (NKH-1) antigen expression on human peripheral blood NK cells and cytotoxic T lymphocytes. J. Immunol. 136:4480–4486. [PubMed] [Google Scholar]
- 20.Uciechowski, P., J. Gessner, R. Schindler, and R. Schmidt. 1992. Fc-γRIII activation is different in CD16+ cytotoxic T lymphocytes and natural killer cells. Eur. J. Immnol. 22:1635–1638. [DOI] [PubMed] [Google Scholar]
- 21.Perussia, B. 1998. Fc receptors on natural killer cells. Curr. Top. Microbiol. Immunol. 230:63–88. [DOI] [PubMed] [Google Scholar]
- 22.Einspahr, K.J., R.T. Abraham, B.A. Binstadt, Y. Uehara, and P.J. Leibson. 1991. Tyrosine phosphorylation provides an early and requisite signal for the activation of natural killer cell cytotoxic function. Proc. Natl. Acad. Sci. USA. 88:6279–6283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lanier, L., G. Yu, and J. Phillips. 1989. Co-association of CD3 ζ with a receptor (CD16) for IgG Fc on human natural killer cells. Nature. 342:803–805. [DOI] [PubMed] [Google Scholar]
- 24.Milella, M., A. Gismondi, P. Roncaioli, L. Bisogno, G. Palmieri, L. Frati, M. Cifone, and A. Santoni. 1997. CD16 cross-linking induces both secretory and extracellular signal-regulated kinase (ERK)-dependent cytosolic phospholipase A2 (PLA2) activity in human natural killer cells: involvement of ERK, but not PLA2, in CD16-triggered granule exocytosis. J. Immunol. 158:3148–3154. [PubMed] [Google Scholar]
- 25.Burshtyn, D.N., A.M. Scharenberg, N. Wagtmanm, S. Rajagopalan, K. Berrada, T. Yi, J.-P. Kinet, and E.O. Long. 1996. Recruitment of tyrosine phosphatase HCP by the killer cell inhibitory receptor. Immunity. 4:77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Biron, C., K. Nguyen, G. Pien, L. Cousens, and T. Salazar-Mather. 1999. Natural killer cells in antiviral defense: function and regulation by innate cytokines. Annu. Rev. Immunol. 17:189–220. [DOI] [PubMed] [Google Scholar]
- 27.Tseng, C., E. Miskovsky, and G. Klimpel. 2001. Crosslinking CD81 results in activation of TCRγδ T cells. Cell. Immunol. 207:19–27. [DOI] [PubMed] [Google Scholar]
- 28.Fleming, T.J., E. Donnadieu, C.H. Song, F.V. Laethem, S.J. Galli, and J.P. Kinet. 1997. Negative regulation of Fcε RI-mediated degranulation by CD81. J. Exp. Med. 186:1307–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Everhart, J., M. Stolar, and J. Hoofnagle. 1997. Management of hepatitis C: a national survey of gastroenterologists and hepatologists. Hepatology. 26:78S–82S. [DOI] [PubMed] [Google Scholar]
- 30.Cerny, A., and F. Chisari. 1999. Pathogenesis of chronic hepatitis C: immunological features of hepatic injury and viral persistence. Hepatology. 30:595–601. [DOI] [PubMed] [Google Scholar]