

Abstract
Dysregulated T cell responses to enteric bacteria have been implicated as a common mechanism underlying pathogenesis in rodent models of colitis. However, the bacterial species and T cell specificities that induce disease have been poorly defined. We have developed a model system in which target antigen, bacterial host, and corresponding T cell specificity are defined. OVA-specific T cells from DO11.RAG-2−/− TCR transgenic mice were transferred into RAG-2−/− recipients whose intestinal tracts were colonized with OVA-expressing or control Escherichia coli. Transfer of antigen-naive DO11.RAG-2−/− T cells into recipients colonized with OVA-E. coli resulted in enhanced intestinal recruitment and cell cycling of OVA-specific T cells; however, there was no development of disease. In contrast, transfer of polarized T helper (Th) 1 and Th2 populations resulted in severe wasting and colitis in recipients colonized with OVA-expressing but not control E. coli. The histopathologic features of disease induced by Th1 and Th2 transfers were distinct, but disease severity was comparable. Induction of disease by both Th1 and Th2 transfers was dependent on bacterially associated OVA. These results establish that a single bacterially associated antigen can drive the progression of colitis mediated by both Th1 and Th2 cells and provide a new model for understanding the immunoregulatory interactions between T cells responsive to gut floral antigens.
Keywords: inflammatory bowel disease, E. coli, CD4, cytokines, TCR transgenic
Introduction
Evidence from a growing number of rodent models of inflammatory bowel disease (IBD)* supports a central role for dysregulated CD4+ T cell responses to the enteric bacterial flora as a common disease mechanism (1). Targeted deletion or overexpression of a variety of genes important in immune regulation or mucosal barrier function, and disruption of normal thymic development have produced chronic inflammatory responses confined to the intestines (2–8). In addition, transfers of normal T cell subsets defined by differential expression of CD45RB into immunodeficient mice demonstrate that different T cell populations can promote or prevent colitis (9, 10). In the majority of these models, the pattern of cytokine expression in inflammatory lesions has implicated a Th1-dominated inflammatory disease, and neutralization of IL-12, IFN-γ, or TNF-α has been therapeutic (11–14). In a few models, a Th2 pattern of cytokine expression has been observed (15, 16), and neutralization of IL-4 or targeted deletion of IL-4–producing cells was protective (16, 17). In human IBD, there is evidence that Crohn's disease is associated with a Th1 pattern of inflammation (18, 19). Elevation of some Th2 cytokines has been detected in ulcerative colitis (18), but whether the disease is associated with Th2 dominant responses is unclear.
Although many of the animal models of IBD have systemic disruptions of T cell regulatory mechanisms, inflammatory disease localizes primarily or exclusively to the large intestine. Because the cecum and colon of the large intestine contain the bulk of the enteric flora, it has been proposed that dysregulated T cell populations in these models are reactive to components of the normal intestinal flora (20, 21). This is supported by the abrogation of colitis development in germ-free HLA-B27 transgenic rats and IL-2−/− and IL-10−/− mice, and the rapid development of colitis upon reconstitution of the enteric flora (4, 22, 23). It has also been shown that T cells developing in the abnormal thymic microenvironment of Tgε26 mice do not induce colitis in the absence of normal bacterial antigen (24). Similarly, depletion of the enteric flora by broad-spectrum antibiotic treatment has ameliorated disease in several murine models of colitis. Antibiotic manipulations in the HLA-B27 transgenic rats and DSS-treated mice have also suggested that complex interactions of aerobic and anaerobic commensal bacteria provide the stimulus for chronic inflammation in these models (25). More direct evidence implicating enteric bacterial antigens is found in data from studies in the spontaneously colitic C3H/HejBir mouse strain (26). CD4 T cells isolated from these mice are reactive to a mixture of cecal bacterial antigens and induce colitis upon transfer to immunodeficient hosts (27). These studies strongly suggest that the enteric bacterial flora provides the stimulus for development of colitis in the setting of altered immunoregulatory mechanisms.
The mechanism by which the bacterial flora stimulates colitis in susceptible models is unknown and is likely to be multifactorial. Bacterial components may act as immune adjuvants or may provide bacterial superantigens that nonspecifically activate an adaptive immune response. Alternatively, proinflammatory bacterial wall components may initiate or direct a pathologic inflammatory response independent of specific immunity. However, the fact that T cell regulatory dysfunction is involved in experimental colitis indicates that specific recognition of bacterial antigens is central to disease pathogenesis. An intriguing finding in this regard has been the relative selectivity of immune responses to a limited number of bacterial species in mouse and rat colitis models (22, 28, 29). Nevertheless, details regarding the types of enteric bacteria that are responsible for initiating and perpetuating a pathogenic T cell response and the specific bacterial antigens recognized remain poorly defined.
A significant impediment to advances in our understanding of homeostatic and pathogenic intestinal T cell responses is the lack of identified target antigens and their corresponding T cell receptor specificities. In none of the existing models are specific bacterial species and their antigens or the responsive T cell specificities defined. The goal of the current studies was the development of a model system for characterization of T cell responses to bacterially associated enteric antigens in which the target antigen, bacterial host, TCR specificity, and T cell developmental stage are each defined. To this end, we have adapted the well-characterized DO11.10 αβ TCR transgenic model to examine the inductive and regulatory effects stimulated by free or bacterially associated cognate antigen, OVA. Adoptive transfers of antigen-naive or polarized Th1 or Th2 cells derived from recombination activating gene (RAG)-2–deficient DO11.10 mice were made into congenic RAG-2–deficient recipients colonized with E. coli engineered to express OVA or a control antigen. Using this system, we have observed for the first time that precommitted Th1 and Th2 cells reactive to a single bacterially associated antigen can be colitogenic, whereas naive cells are not.
Materials and Methods
Mice.
Specific pathogen-free BALB/cBJ, DO11.10 (30), BALB.RAG-2−/−, and DO11.10 TCR transgenic mice crossed with RAG-2−/− mice (DO11.RAG-2−/−) were bred and/or maintained in the Genetically Engineered Mouse facility at University of Alabama at Birmingham. DO11.10 and RAG-2−/− mice had been backcrossed onto the BALB/c background for at least 10 generations before intercrosses to generate mice homozygous for the RAG-2 targeted mutation and hemizygous for the DO11.10 transgene (DO11.RAG-2−/−). Male and female DO11.RAG-2−/− mice were used as donors for adoptive transfer studies; female BALB.RAG-2−/− mice were used as recipients. All experimental mice were 6–10 wk of age.
Generation of OVA-producing Escherichia coli.
Escherichia coli was chosen as the bacterial host for these studies because of the relative ease with which it is genetically modified and its demonstrated role as an “immunodominant” organism in spontaneously colitic mice (28). The full coding sequence of OVA or TET-C (C fragment of tetanus toxoid, as a control) was expressed in E. coli strain DH5α under the control of an inducible bacterial promoter, nir15 (31). The nir15 promoter is an improved expression variant of the nirB promoter of the nitrite reductase gene of E. coli that is strongly induced by environmental nitrites or low oxygen tension (32). This promoter has been used to direct stable in vivo expression of heterologous antigens in vaccine strains of Salmonella (31, 33). The plasmid pTETnir15 (reference 31; referred to henceforth as pnir15.TET) expresses the C fragment of tetanus toxoid and was provided by Jerry McGhee and James Lillard, University of Alabama at Birmingham. To produce an OVA-expressing variant, pnir15.OVA, the TET coding sequence was excised by BglII/BamH1 digestion and replaced with a pcr-generated fragment that included an idealized Shine-Dalgarno sequence upstream of the entire OVA coding sequence (forward primer: 5′ CTTATAGATCTTAATCATCCACAGGAGATATTATTATGAAAAACATCGGCGCAGCAAGCA-TGGAA; reverse primer: 5′ GCTAGCGGATCCTTAAGGGGAAACACATCTGCC). The plasmid VR1020/OVA was used as a template for amplifying the OVA coding sequence (provided by William O. Rogers, Naval Medical Research Center, Bethesda, MD). DH5α E. coli were transformed by each plasmid (pnir15.OVA or pnir15.TET) by electroporation and selected for ampicillin resistance. Clones containing pnir15.OVA were isolated and sequenced to prove authenticity of the OVA coding sequence. Expression of OVA in E. coli was confirmed by Western blot analysis after overnight culture in media containing ampicillin, without shaking in tightly capped tubes at 37°C for anaerobic induction of nir15 promoter. Bacteria were lysed in 2× sample buffer (6% TRIS-Cl, pH 6.8, 20% glycerol, 4% SDS, 2% 2-ME), and heat denatured at 100°C for 5 min. Cell lysates (105 cell equivalents) and purified OVA (positive control) were resolved on a 10% SDS polyacrylamide gel and transferred to nylon membranes (Nytran Plus; Schleicher & Schuell). Membranes were incubated at room temperature with 1% blocking reagent (Boehringer Mannheim) for 1 h, a 1:500 dilution of murine anti-OVA mAb, OVA-3 (IgG2β; Brookwood Biomedical) for 2 h, and were detected with POD-conjugated anti–mouse IgG/anti–rabbit IgG (40 mU/ml; Boehringer Mannheim) using the chemiluminescent substrate luminol (Boehringer Mannheim).
Cell Purification and Flow Cytometry.
CD4+ T cells were isolated from pooled spleens and peripheral lymph nodes of DO11.10 or DO11.RAG-2−/− mice using mouse CD4 (L3T4) Dynabeads® followed by CD4 DETACHaBEAD® per the manufacturer's protocol (Dynal Biotech). The resulting CD4+ enriched cells were stained with FITC-conjugated KJ1–26 (anti-DO11.10 TCR, reference 34) and PE-conjugated anti-CD4 and analyzed by flow cytometry to determine purity of the population. 10,000 total events were acquired on a Becton Dickinson FACScan™, gating on cells with forward and side scatter properties of lymphocytes, and were analyzed using CELLQuest™ software. Isolations routinely resulted in >92% purity of CD4+ cells (DO11.10 donors) or CD4+KJ1–26+ cells (DO11.RAG-2−/−).
In Vitro and In Vivo Analyses of DO11.10 T Cell Response to OVA-expressing E. coli.
For in vitro studies, splenic adherent cells isolated from BALB/c mice were pulsed for 18 h with nothing, 100 μg/ml whole OVA, or the indicated number of gentamycin-killed, anaerobically induced pnir15.TET or pnir15.OVA E. coli. Gentamycin killing was done by incubating bacteria with 100 μg/ml gentamycin for 30 min. Pulsed splenic adherent cells were then irradiated (2,500 rads) before culture with CD4+ T cells isolated from DO11.10 mice. Day 3 proliferative responses were determined by 3[H]-TdR incorporation as described previously (35). For in vivo studies, 5 × 106 CD4+KJ1–26+ T cells isolated from DO11.10 mice were transferred into BALB/c mice by tail vein injection. 1 d after the transfer, 106 bacteria (E. coli-OVA or -TET) were injected IV and mice were killed at days 1, 3, 5, 7, and 9. Cells from spleen and lymph nodes were pooled and analyzed by FACS® for expression of CD4 and DO11.10 TCR and the total number of CD4+KJ1–26+ cells were calculated as described previously (36).
Generation and Characterization of OVA-specific Th1 and Th2 Cells.
Th1 and Th2 cells were generated in vitro from antigen-naive CD4+ T cells as described previously (37). Briefly, purified CD4+ T cells isolated from pooled spleens and peripheral lymph nodes of DO11.RAG-2−/− were cultured at a ratio of 1:10 with irradiated BALB/c splenocytes and 5 μg/ml OVA peptide 323–339. The addition of 50 U/ml IL-12 (R&D Systems) and 10 μg/ml anti–IL-4 (11B11; reference 38) was used to generate Th1 cells, while 1,000 U/ml IL-4 (R&D Systems) and 10 μg/ml anti–IL-12 (C17.8, reference 39) was used to generate Th2 cells (40). Recovered cells were washed and transferred on day 7 at 106 cells/RAG-2−/− recipient mouse. A separate aliquot of 5 × 106 cells was restimulated for 6 h with PMA (50 ng/ml) and ionomycin (750 ng/ml; Sigma-Aldrich) for analysis of cytokine mRNA expression by ribonuclease protection assay. Ribonuclease protection assays were performed with 1 μg of RNA using the template set mCK-1 (RiboQuant kit; BD PharMingen).
Reconstitution of RAG2−/− Mice with CD4 Subsets, Colonization with Bacteria, and Monitoring for Development of Colitis.
Bacterial colonization was performed by gastric gavage of pnir15.OVA-E. coli or pnir15.TET-E. coli (control) to RAG-2−/− mice one day before CD4 T cell transfer. Bacteria were grown in LB medium overnight at 37°C under anaerobic conditions and 1010 CFU in 100 μl PBS were administered orally using a straight gavage needle. Colonization was assessed by plating stool suspensions on LB plates containing ampicillin and was maintained at ∼108 CFU/g of stool by biweekly gavage. 106 naive, Th1, or Th2 DO11.10 CD4+ T cells were transferred by tail vein injections into RAG-2−/− mice a day after the first gavage. Mice were weighed weekly and monitored for appearance, and signs of loose stool and diarrhea. 0.8 mg/ml bromodeoxyuridine (BrdU) was added to the drinking water of all groups for 9 d before killing and intestinal tissues were evaluated histologically.
Histopathologic Assessment and Disease Scoring.
At necropsy, the small intestine, cecum, and colon were separated and Swiss rolls of each prepared. Tissues were fixed in 10% buffered formalin and paraffin 5-μm embedded sections were stained with H&E. Additional sections were stained for identification of polymorphonuclear cells (Leder stain), eosinophils (cyanide-resistant eosinophil peroxidase stain; reference 41), T cells (anti-CD3), and/or BrdU as indicated. An immunohistochemical method for coidentification of CD3 cells and BrdU-labeled cells was developed for these studies and will be described in detail elsewhere (unpublished data). Briefly, 5-μm sections were digested with pepsin, blocked with normal goat serum, and sequentially incubated with polyclonal rabbit anti–human CD3 antibody (0.5 μg/ml, 1:100 dilution, 30 min; Dako), biotinylated anti–rabbit IgG (H+L) antibody (1.5 μg/ml: 1:100 dilution, 30 min), and 0.3% hydrogen peroxide (10 min). Bound antibody was then detected with a preformed avidin and biotinylated horseradish peroxidase macromolecular complex (ABC; Vectastain Elite ABC kit; Vector Laboratories) and diaminobenzadine substrate was used for development (Vector Laboratories). After development of anti-CD3 staining, sections were rinsed with PBS followed by incubation in denaturing solution for 20 min (BrdU Staining kit; Zymed Laboratories). Slides were then sequentially incubated with biotinylated mouse anti-BrdU antibody for 30 min, alkaline phosphatase labeled streptavidin for 20 min, and detected with Fast Red TR chromogen for 30 min (BrdU Staining kit; Zymed Laboratories). Slides were counterstained with hematoxylin and coverslips mounted with crystal mount (Biomeda). All incubations were performed at room temperature and each step was followed by a rinse with PBS. Sections processed without primary antibody served as negative controls.
Histological scoring was performed using a modification of the scoring system reported previously by Sundberg et al. (42), and was performed independently by two pathologists (A.S. Lazenby and C.T. Weaver). Briefly, each section was scored for lesions based on four criteria: severity of inflammation, hyperplasia of mucosal epithelium, mucosal ulceration, and extent of involvement. Severity of inflammation and hyperplasia were graded on a score of 0–3, 0 being normal and 3 being severe. The extent of disease involvement was determined for each and defined as: 0, normal; 1, 1–33%; 2, 34–66%; and 3, 67–100%. The percent area of involvement was determined separately for cecum, proximal colon, middle colon, and distal colon/rectum. The average area of involvement for the colon was calculated as the sum of the three colonic segments divided by three and was used for calculation of the total colon score. The total score was assessed separately for the cecum and colon by adding the scores of severity, hyperplasia, area of involvement, and ulceration. The thickness of mucosal hyperplasia was directly measured in the areas of most severe disease using an ocular grid, but was not included as a component of the disease score. All cell counts were performed at original magnifications: 200× or 400× by point counting using an ocular grid (36); a minimum of 1,000 total cells was counted for each determination.
Isolation and Cytokine Phenotyping of Intestinal Lamina Propria T Cells.
Colon and small intestine were washed thoroughly with PBS to remove debris, opened longitudinally, and then cut into 0.5 cm pieces. The epithelium was removed by incubation with 2 mM DTT (Sigma-Aldrich) and 1 mM EDTA (Sigma-Aldrich) in RPMI 1640 medium supplemented with 2% FCS at 37°C for 30 min with gentle shaking. Tissue was collected, further cut into smaller pieces, and digested with 0.3 mg/ml collagenase type IV (Sigma-Aldrich) at 37°C for 90 min. Lamina propria (LP) cells were harvested by discontinuous 40/70 percoll gradient (BD PharMingen). CD4+ T cells were then purified from LP mononuclear cells by positive selection using mouse CD4 Dynabeads® and mouse CD4 DETACHaBEAD® as described previously (Dynal). Recovered cells were cultured in 96-well plates (Nunc) that were precoated with anti-CD3 mAb (145–2C11; reference 43; 10 μg/ml) in 100 μl PBS at 37°C for 4 h and washed with PBS three times to remove unbound Ab. LP CD4+ T cells (5 × 105 cells per milliliter) were incubated in the presence of coated anti-CD3 and soluble anti-CD28 (37.51, reference 44; final concentration, 1 μg/ml) at 37°C. After 72 h of culture, supernatants were harvested and assayed for IFN-γ, IL-4, and IL-10 by capture ELISA using commercially available ELISA kits (R&D Systems).
Statistical Analyses. Parametric data were analyzed by Student's t test. Nonparametric data were analyzed by the Mann-Whitney test. P values < 0.05 were considered significant.
Results
Generation and Characterization of OVA-producing E. coli.
To generate bacteria that express the antigen recognized by the DO11.10 TCR, a plasmid encoding the full-length OVA cDNA (pnir15.OVA) was introduced into DH5α E. coli (Fig. 1 A). E. coli was chosen as the bacterial host because it is a normal commensal and because it is an immune stimulatory organism in spontaneously colitic mice (28). Western blot analyses of lysates of DH5α E. coli transformed with the pnir15.OVA plasmid indicate that this construct directs excellent cytosolic expression of OVA protein under anaerobic conditions (Fig. 1 B). E. coli transformed with a plasmid in which the coding sequence of OVA was replaced with that of the C fragment of tetanus toxoid (pnir15.TET-E. coli) served as a negative control. Based on densitometric comparisons between immunoblots performed with bacterial lysates of pnir15.OVA-E. coli and purified OVA, the anaerobically induced plasmid expressed ∼1 μg OVA per 106 bacteria under in vitro conditions. The immunogenicity of pnir15.OVA-E. coli was determined in vitro by T cell proliferation assays (Fig. 1 C). Splenic adherent cells pulsed with anaerobically induced, gentamycin-killed pnir15.OVA-E. coli–stimulated proliferation of naive DO11.10 T cells, whereas splenic adherent cells pulsed with pnir15.TET-E. coli did not. The dose of bacteria that stimulated a maximal response (107 organisms) expressed ∼10 μg OVA; the dose of free OVA that was required for a maximal response was 100 μg/ml. Thus, induced pnir15.OVA-E. coli are a potent immunogen for stimulation of DO11.10 cells in vitro.
Figure 1.



Inducible expression of immunogenic OVA by pnir15.OVA-E. coli. (A) Schematic of the pnir15.OVA plasmid. Restriction enzyme sites are: Bgl, Bgl II; RV, Eco RV; B, Bam H1. The ampicillin resistance gene (Ampr) and origin of replication (pBR322ORI) are indicated. (B) Western blot analysis of DH5α E. coli transformed with pnir15.OVA or a control plasmid that codes for expression of the C fragment of tetanus toxoid (pnir15.TET). Cytosolic fractions of lysates of bacteria cultured for 12 h under aerobic (−; noninduced) or anaerobic (+; induced) conditions were prepared and analyzed by Western blotting (see Materials and Methods). The indicated lanes were loaded with 6 × 105 bacterial equivalents or 500 ng purified native OVA. The antibody used for detection was a murine IgG2b monoclonal (OVA-3) that is specific for OVA. Note that the bacterially expressed OVA runs at a lower MW than native OVA due to absent glycosylation. (C) Splenic adherent cells isolated from BALB/c mice were pulsed for 18 h with the indicated number of gentamycin-killed, anaerobically induced pnir15.TET-E. coli (white circles) or pnir15.OVA-E. coli (black circles) then irradiated (2,500 rads) before culture with CD4+ T cells isolated for DO11.10 mice (left). The response of the same cells to APCs pulsed with nothing or an optimal dose of native OVA (100 μg/ml) is shown for comparison (right). Data are the mean ± SD of day 3 proliferative responses determined by 3[H]-TdR incorporation.
The immunogenicity of live pnir15.OVA-E. coli was assessed in vivo using a DO11.10 adoptive transfer system (45). Normal BALB/c mice that received transfers of 5 × 106 DO11.10 CD4+ T cells (DO11.10/BALB mice) were innoculated intravenously with 106 pnir15.OVA-E. coli or pnir15.TET-E. coli, and expansion of the clonotypic T cell population in the spleens of recipient mice was monitored by flow cytometric analysis (Fig. 2) . In agreement with a previous study by Chen et al. (46), intravenous administration of the OVA-producing E. coli stimulated a robust clonal expansion in the spleens and lymph nodes of DO11.10 adoptive transfer recipients. Significant clonal expansion (approximately fivefold) was induced by challenge with pnir15.OVA-E. coli, but not the pnir15.TET-E. coli control. The peak response measured was on day five, with a rapid decline to baseline by day nine. Induction of a comparable peak response with free OVA required intraperitoneal administration of a ∼100-fold higher dose of OVA (100 μg) in complete Freund's or a block copolymer adjuvant (data not shown), indicating that the bacterially associated OVA is more efficient at stimulating an in vivo response. Collectively, these data indicate that the pnir15.OVA plasmid produces high-level expression of OVA that is potently immunogenic for DO11.10 T cells in vitro and in vivo.
Figure 2.


OVA-expressing E. coli induce clonal expansion of DO11.10 T cells in vivo. DO11.10/BALB/c adoptive transfer recipients were challenged with a single intravenous injection of 106 viable pnir15.OVA-E. coli or pnir15.TET-E. coli. Two-color flow cytometric analysis was performed on splenic or pooled peripheral lymph node lymphocytes isolated at the indicated times after immunization as described in Materials and Methods. The percentages of KJ1–26+CD4+ cells in the lymphocyte gate were determined and used to calculate the total number of splenic KJ1–26+ CD4+ as described previously (reference 36). (A) Representative flow cytometric profiles for lymph node lymphocytes isolated 5 d after challenge with pnir15.TET-E. coli (left) or pnir15.OVA-E. coli (right). The percentages of total gated cells in each quadrant are indicated. (B) Kinetics of clonal expansion of total splenic KJ1–26+CD4+ T cells induced by pnir15.OVA-E. coli versus control pnir15.TET-E. coli. Data points are the mean ± SD of 3–4 animals per group.
Rag-2−/− Mice Reconstituted with Th1- or Th2-polarized DO11.RAG-2−/− T Cells Develop Severe Wasting Disease that is Ova-E. coli–dependent; Mice Reconstituted with Antigen-Naive DO11.10 T Cells Do Not Develop Wasting Disease.
To evaluate the in vivo responses of antigen-specific T cells to a floral, bacterially associated antigen, OVA-specific T cells were transferred into congenic, immunodeficient recipients whose intestinal tracts were colonized with E. coli that expressed either OVA (pnir15.OVA-E. coli) or a control antigen, TET (pnir15.TET-E. coli). Three T cell populations were examined: naive CD4+ T cells or Th1 and Th2 cells derived by short-term in vitro culture under selective cytokine conditions. A 1 wk protocol was used for Th1/Th2 derivation because it induced excellent polarization of cytokine phenotype (Fig. 3 A), but did not significantly affect T cell migration to the secondary lymphoid organs compared with Th1 and Th2 cells maintained in vitro for longer periods of time (unpublished data). In previous studies, we found that a fraction of CD4+ T cells that expressed the transgenic TCR in DO11.10 mice coexpressed a second, endogenous TCR that recognized bacterial antigens in the normal flora (35). Hence, the DO11.10 donors used for these experiments were crossed onto a BALB.RAG-2−/− background (DO11.RAG-2−/−) to obviate possible complications due to dual TCR specificities. In preliminary colonization studies, it was found that delivery of 1010 anaerobically induced organisms by gastric gavage resulted in a transient colonization of the colon (half-life of 3–4 d; data not shown). Biweekly gavage maintained the floral colony count at a minimum of 108 CFU per gram of colonic contents and was used in experimental protocols.
Figure 3.


Th1 and Th2 cells derived from DO11.RAG-2−/− mice induce chronic wasting in RAG-2−/− recipients colonized with pnir15.OVA-E. coli. (A) Cytokine analysis of naive and Th1- or Th2-polarized OVA-specific T cells. Th1 and Th2 cells were generated in vitro under polarizing cytokine conditions as described in Materials and Methods. Briefly, CD4+ T cells isolated from DO11.RAG-2−/− were cultured for 7 d with OVA323–339 peptide, irradiated BALB/c splenocytes, and either IL-12 plus anti–IL-4 (Th1) or IL-4 plus anti-IL-12 (Th2). To confirm Th1/Th2 polarization, aliquots were restimulated for 6 h with PMA and ionomycin, and analyzed for cytokine mRNA expression by ribonuclease protection assay (RPA; mCK-1 RiboQuant kit; BD PharMingen) in comparison with naive T cell isolates. (B) Weight changes of BALB.RAG-2−/− mice that were colonized with 1010 CFU pnir15.OVA-E. coli (black symbols) or pnir15.TET-E. coli (white symbols), reconstituted with 106 naive (○), Th1 (▿), or Th2 (□) cells from DO11.RAG-2−/− donors and monitored for weight loss (B). Data are the mean weekly weights and SD (n = 5–6 mice per group).
1 d after gavage with anaerobically induced pnir15. OVA-E. coli or pnir15.TET-E. coli, RAG-2−/− mice received a transfer of 106 naive, Th1, or Th2 DO11.RAG-2−/− CD4+ T cells by tail vein injection. Bacterial colonization was maintained with biweekly gavage (1010 induced organisms per dose), and mice were weighed and monitored for clinical signs of disease (Fig. 3 B). Significant weight loss was evident at week 6 in animals from groups that were colonized with pnir15.OVA-E. coli and reconstituted with either Th1 or Th2 cell transfers. Mice that received naive T cells and were colonized with pnir15. OVA-E. coli did not show weight loss or clinical signs of disease. Similarly, mice that received Th1 or Th2 cells and were colonized with control pnir15.TET-E. coli also showed no clinical evidence of disease. All mice were administered 0.8 mg/ml BrdU in their drinking water for 9 d before sacrifice at week 8 and tissue was processed for histological analyses.
At necropsy, macroscopic evaluation of animals from Th1 and Th2 groups colonized with pnir15.OVA-E. coli was remarkable for short, thickened large intestines. Focal superficial erythema and hemorrhages were present on cecal and colonic mucosal surfaces. The pnir15.TET-E. coli–colonized Th1 and Th2 groups were grossly normal, as were naive T cell recipients colonized with either pnir15.OVA-E. coli or pnir15.TET-E. coli. On microscopic evaluation, the large intestines from Th1 and Th2 groups colonized with pnir15.OVA-E. coli had moderate to severe disease characterized by extensive inflammatory cell infiltrates, marked epithelial cell hyperplasia and depletion of epithelial cell mucin (Fig. 4) . As shown in results from a representative experiment (Table I), the incidence and severity of disease was comparable in Th1 and Th2 groups. The distribution of involvement was also similar in Th1- and Th2-mediated disease (Fig. 5) , and was patchy along the length of the cecum and colon. Disease severity was modestly increased in colon compared with cecum; the rectum was spared. Although there was good population of the small intestinal LP with clonotypic T cells in all groups, no pathologic abnormalities were identified (data not shown). Similarly, there was no inflammation in other organs
Figure 4.
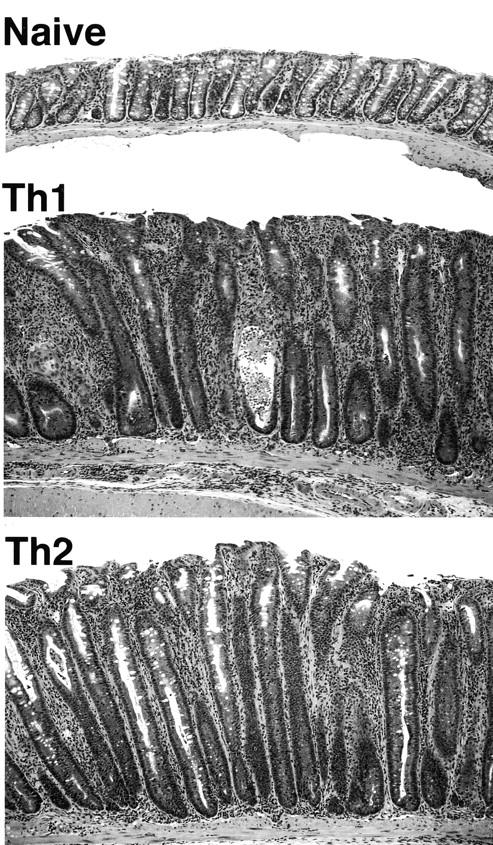
Th1- and Th2-reconstituted mice colonized with pnir15.OVA-E. coli develop antigen-specific colitis. H&E stained sections of proximal colon from pnir15.OVA-E. coli–colonized recipients of naive T cells (top), Th1 cells (middle), or Th2 cells (bottom) (original magnification: 100×).
Table I.
Histological Scoring of Colonic Pathology in DO11.RAG-2−/− /RAG-2−/− Recipients Colonized with E. coli-OVA versus E. coli-TET
| T cells | Intestinal colonization | Colitisa | Severity | Hyperplasia | Extent of involvement | Total score |
|---|---|---|---|---|---|---|
| Naive | pnir15.TET-E. coli | 0/10 | 0.0 ± 0.0b | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 |
| pnir15.OVA-E. coli | 1/10 | 0.1 ± 0.3 | 0.1 ± 0.3 | 0.1 ± 0.3 | 0.3 ± 1.0 | |
| Th1 | pnir15.TET-E. coli | 0/8 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 |
| pnir15.OVA-E. coli | 7/7 | 2.5 ± 0.6 | 2.3 ± 0.5 | 2.5 ± 0.6 | 7.8 ± 1.0 | |
| Th2 | pnir15.TET-E. coli | 0/7 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 |
| pnir15.OVA-E. coli | 6/6 | 2.3 ± 0.6 | 2.7 ± 0.5 | 2.3 ± 0.3 | 7.3 ± 0.6 |
RAG-2−/− recipients were reconstituted with 106 CD4+ T cells of the indicated phenotypes that were isolated or generated from DO11.RAG-2−/− donors. 1 d before T cell reconstitution, recipients were colonized with 1010 CFU of the indicated bacteria and were maintained with biweekly administration of an additional dose of 1010 CFU bacteria. 8 wk after reconstitution, mice were killed and necropsied.
See Materials and Methods for details of scoring system.
All data are the means ± SEM of two independent pathological evaluations of the indicated numbers of mice pooled from three separate experiments. Scores that differed significantly from control (P > 0.05) are indicated in bold.
Figure 5.

Distribution of large intestinal disease mediated by Th1 and Th2 cells. The large intestines of pnir15.OVA-E. coli-colonized RAG-2−/− mice reconstituted with naive, Th1, or Th2 DO11.RAG-2−/− cells were subdivided into the indicated anatomical regions and evaluated histologically for disease involvement as in Table I.
The histologic features of disease in Th1 and Th2 recipient mice colonized with pnir15.OVA-E. coli were distinct. The lesions in mice that received Th1 cells were characterized by predominantly mononuclear cell infiltrates composed of lymphocytes and monocytes/macrophages that expanded the LP and distorted the crypt architecture (Fig. 6 and Table II). Focally, there was extension of the inflammation into the underlying muscularis and serosa. Scattered, small granulomas were evident, but were not a dominant feature. Frequent crypt abscesses were evident, and T cells invaded the epithelium. Neutrophils were present in the areas of most severe epithelial injury, particularly bordering and infiltrating damaged crypts. Eosinophils were rarely seen.
Figure 6.
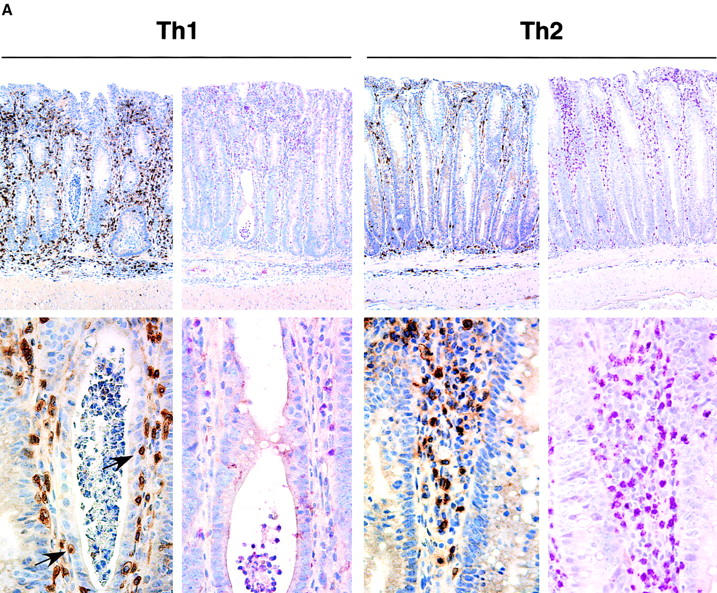


Histological analyses of colonic tissues from mice with Th1- or Th2-mediated colitis. Paraffin-embedded sections of colon from the indicated experimental groups were processed and stained as described in Materials and Methods. (A) Th1- and Th2-reconstituted/pnir15.OVA-E. coli-colonized, serial sections of proximal colon stained with anti-CD3 to identify T cells (left, top, and bottom) or Leder stain to identify neutrophils and eosinophils (right, top, and bottom). Arrows identify intraepithelial Th1 cells in diseased proximal colon. (B) Proximal colon from naive-reconstituted RAG-2−/− mice colonized with pnir15.TET-E. coli (top) or pnir15.OVA-E. coli mice (bottom) stained with anti-CD3. (C) Low and high power images of Th1-reconstituted/pnir15.OVA-E. coli–colonized middle colon double-labeled with anti-CD3 (brown) and anti-BrdU (red). Arrowheads identify CD3+ DO11.RAG-2−/− T cells that have incorporated BrdU after 9 d in vivo administration. Note also the incorporation of BrdU by colonic crypt epithelial cells.
Table II.
Histological Characterization of Inflammatory Lesions in DO11.RAG-2−/−/RAG-2−/− Recipients Colonized with E. coli-OVA versus E. coli-TET
| T cells | Intestinal colonization | CD3+/mm2totala | CD3+/mm2IEL | PMN/mm2total | Percentage of eosinophils (PMNs) |
Percentage of BrdU+(CD3) |
|---|---|---|---|---|---|---|
| Naive | pnir15.TET-E. coli | 77.4 ± 44.2b | 5.5 ± 5.0 | 4.0 ± 3.7 | ND | 1.6 ± 1.7 |
| pnir15.OVA-E. coli | 192.3 ± 73.5 | 7.3 ± 3.6 | 8.5 ± 3.8 | 2.4 ± 2.9 | 24.0 ± 5.2 | |
| Th1 | pnir15.TET-E. coli | 203.7 ± 55.8 | 17.9 ± 8.4 | 17.9 ± 8.4 | ND | 2.5 ± 1.8 |
| pnir15.OVA-E. coli | 796.1 ± 190.3 | 66.5 ± 29.3 | 246.0 ± 97.3 | 5.7 ± 4.8 | 34.8 ± 7.6 | |
| Th1 | pnir15.TET-E. coli | 140.1 ± 50.0 | 13.0 ± 8.3 | 0.5 ± 1.0 | ND | 7.5 ± 1.9 |
| pnir15.OVA-E. coli | 385 ± 96.2 | 33.0 ± 5.2 | 496.7 ± 44.7 | 39.6 ± 8.9 | 38.8 ± 9.6 |
RAG-2−/− recipients were reconstituted with 106 CD4+ T cells of the indicated phenotypes that were isolated/generated from DO11.RAG-2−/− donors. 1 d before T cell reconstitution, recipients were colonized with 1010 CFU of the indicated bacteria and maintained with biweekly administration of an additional dose of 1010 CFU bacteria. 8 wk after reconstitution, mice were killed and necropsied.
The total number of CD3+ T cells per mm2 of colonic mucosa, including LP and IEL compartments (see Material and Methods for details).
Data are the means ± SEM from 3–6 animals per group. Values that differed significantly from control (P > 0.05) are indicated in bold font.
ND, not determined.
While the disease severity was comparable in pnir15. OVA-E. coli-colonized Th2 animals, the histological features were different (Fig. 6 and Table I). In contrast to Th1-mediated disease, the inflammatory infiltrates in Th2 mice included a predominance of polymorphonuclear cells, including a significant fraction of eosinophils (∼40% of total PMNs), with lesser numbers of lymphocytes and even fewer monocytes/macrophages. The crypt architecture was less tortuous, but crypts were markedly elongated and hyperplastic. Indeed, the mean crypt length in Th2 lesions was generally greater than in Th1 lesions (Th2, 0.73 mm, or 3.65 times nondiseased; Th1, 0.58 mm, or 2.64 times nondiseased), consistent with a more hyperplastic epithelial response in Th2 lesions. Compared with Th1-mediated disease, there was less epithelial cell injury and necrosis, and few crypt abscesses were found. There were significantly fewer intraepithelial T cells in the Th2-diseased mucosa. No granulomas were identified.
The distinct patterns of inflammation in diseased colons of Th1- and Th2-reconstituted mice strongly suggested that either effector T cell population could induce a pathogenic response. However, given the short-term in vitro polarizations before transfer, and evidence for some heterogeneity of cytokine profiles of the starting T cell populations (see RPA analysis, Fig. 3), it was possible that a shift in cytokine phenotype might have occurred in vivo. To determine if the T cells present in colitic lesions retained a similar cytokine phenotype to those of the original T cell populations transferred, cytokine analyses were performed on recovered T cells (Fig. 7) . CD4+ T cells isolated from the intestinal LP of pnir15.OVA-E. coli-colonized recipients reconstituted with Th1- or Th2-polarized DO11. RAG-2−/− T cells were restimulated in vitro with plate-bound anti-CD3 plus anti-CD28 and assayed for production of IFN-γ, IL-4, and IL-10. Stimulated T cells from the LP of Th1-reconstituted mice produced abundant IFN-γ and no detectable IL-4 or IL-10; LP T cells recovered from Th2-reconstituted mice produced IL-4 and IL-10, but no detectable IFN-γ. Thus, the distinct histopathologies were associated with strongly polarized Th1 or Th2 cells in the diseased intestinal tissues.
Figure 7.

T cells recovered from colitic mice retain a polarized Th1 or Th2 phenotype. Intestinal tissues from Th1 and Th2 adoptive transfer recipients colonized with pnir15.OVA-E. coli were recovered and processed for isolation of LP lymphocytes 8 wk after transfer (Materials and Methods). CD4+ T cells were purified by magnetic sorting and stimulated with plate-bound anti-CD3 and anti-CD28 for 72 h. Culture supernatants were assayed for IFN-γ, IL-4, and IL-10 by ELISA. Data are the means and SD of triplicate determinations for T cells pooled from 6–10 mice per group. The detection limits of the respective ELISA assays are indicated for those values that were not greater than background (P < 0.01).
Interestingly, naive T cell recipients that were colonized with pnir15.OVA-E. coli failed to develop clinical or histologic evidence of disease (Table I and Figs. 2, 5, and 6). BrdU incorporation studies indicated that this was not due to absence of antigen recognition by naive DO11.RAG-2−/− T cells (Fig. 6 and Table II). Thus, the frequency of antigen-specific mucosal T cells that incorporated BrdU in pnir15.OVA-E. coli-colonized mice reconstituted with naive T cells was only modestly lower than that of Th1 or Th2 mice, and was significantly increased compared with controls that were colonized with pnir15.TET-E. coli. Similarly, the density of T cells populating the colonic LP in naive cell recipients colonized with pnir15.OVA-E. coli was approximately fivefold greater than pnir15.TET-E. coli controls (Table II). These results establish that naive DO11.RAG-2−/− T cells populating pnir15.OVA-E. coli-colonized mice were responsive to bacterially associated OVA and underwent clonal expansion and/or recruitment to the large intestinal tissues, but did not mediate disease development. Thus, the absence of colitis in naive T cell recipients was not due to a lack of antigenic responsiveness (antigenic neglect) nor deficient trafficking of naive cells to the colonic mucosa.
Bacterially Associated Antigen Is Required for Colitis Development in Th1- and Th2-reconstituted RAG-2−/− Mice.
Previous studies have shown that continuous oral delivery of free OVA at a dose of 0.1 μg/ml could activate DO11.10-derived Tr1 regulatory cells to control colitis development in adoptively transferred mice (47). We sought to determine if free OVA delivered to RAG-2−/− mice reconstituted with DO11.RAG-2−/− Th1 and Th2 cells would be sufficient to cause intestinal disease, or whether bacterially associated OVA was required. RAG-2−/− mice were reconstituted with DO11.RAG-2−/− Th1 and Th2 cells and were challenged with free versus bacterially associated OVA. Free OVA was delivered continuously in the drinking water over an 8-wk course, with or without pnir15.TET-E. coli colonization to control for potential nonspecific adjuvanticity of the E. coli organisms used for colonization (Fig. 8) . Control animals received no OVA or pnir15.OVA-E. coli.
Figure 8.

DO11.RAG-2−/−/RAG-2−/− recipients fed OVA do not develop colitis. RAG-2−/− recipients were reconstituted with 106 CD4+ Th1 or Th2 cells generated from DO11.RAG-2−/− donors. 1 d before T cell reconstitution, recipients were colonized with 1010 CFU of the indicated bacteria, and maintained with biweekly administration of an additional dose of 1010 CFU bacteria. 8 wk after reconstitution, mice were killed and necropsied. Colonic and cecal disease was scored as described in Materials and Methods; mean total scores and SEM are indicated (bottom). BrdU (0.8 mg/ml) was added to the drinking water of indicated groups for 9 d before sacrifice; intestinal tissues were processed and evaluated as in Table II for CD3+ cell densities (middle) and BrdU incorporation (top). Data are from two experiments and are the means ± SEM from 4–8 animals per group. Values that differed significantly from control (P < 0.05) are indicated with an asterisk. ND, not done.
In contrast to Th1 and Th2 recipients colonized with pnir15.OVA-E. coli, animals challenged with free OVA did not develop significant disease, irrespective of whether they were also colonized with pnir15.TET-E. coli control organisms (Fig. 8). To determine if this was due to an absence of antigenic exposure of colonic T cells in animals fed OVA, the recruitment and/or proliferation of T cells in the colonic LP were assessed. Th1 recipients challenged with low dose OVA (0.1 μg/ml) did not have appreciably increased densities of T cells in the LP compared with no antigen controls (92.5 vs. 78.0 CD3+ cells/mm2), but a significantly greater fraction of T cells present had incorporated BrdU (4.2 vs. 0.8%). At the higher dose of free OVA (1,000 μg/ml), both the T cell density and frequency of BrdU-labeled cells were increased compared with no antigen controls, albeit less so than in animals colonized with pnir15.OVA-E. coli. Interestingly, Th2-reconstituted mice treated with both low and high doses of free OVA had increases in T cell densities in the colonic LP that were comparable to pnir15.OVA-E. coli–colonized controls. However, although frequencies of BrdU-positive cells were greater than the no antigen controls in both groups treated with free OVA, the frequencies were significantly lower than in the pnir15.OVA-E. coli-colonized group. This suggests that there might be more rapid turnover of colonic T cells stimulated with bacterially associated OVA than free OVA. Nevertheless, the absence of colitis in Th1 and Th2 recipient mice challenged with free OVA was not due to the absence of antigen recognition by the DO11.RAG-2−/− T cells, and was not due to lack of an independent adjuvant effect of the E. coli bacterial host. These data indicate that stimulation by bacterially associated antigen is both necessary and sufficient for the development of colitis in this model.
Discussion
This paper establishes that a single bacterially associated antigen is both necessary and sufficient to cause colitis mediated by a defined, monoclonal T cell population. In this model, transfer of both Th1 and Th2 cells derived from OVA-specific DO11.RAG-2−/− donors was associated with colitis that was dependent on the colonization of RAG-2−/− recipient mice with E. coli that expressed OVA. Importantly, transfer of antigen-naive T cells of identical specificity did not induce appreciable disease, despite in vivo responsiveness to bacterially associated antigen. Further, in vivo recognition of free antigen by either Th1 or Th2 effectors was insufficient to induce colitis. Taken together, these results suggest that both committed effector T cells and bacterially associated antigen are required for disease pathogenesis in this model, and emphasize the fact that unopposed Th1 and Th2 responses to enteric bacterial antigens can be equally deleterious to the host.
In each of the rodent models of colitis studied to date, disease development has been associated with the presence of the enteric flora. However, in none of these models has the relevant bacterial antigens been identified, and it has been difficult to define contributions from distinct bacterial species or specific antigens. The lack of identification of either the antigenic targets or T cell specificities responsible for disease has compromised efforts to understand the pathogenic contribution of the enteric flora. Studies of spontaneously colitic C3H/HeJBir mice have demonstrated a surprisingly selective immune reactivity to the enteric flora, and have suggested that facultative anaerobes such as E. coli and Salmonella species were immune stimulatory organisms, despite comprising <1% of the total bacterial flora (28). Similarly, Bacteroides spp. was a common component of bacterial cocktails that induced colitis in germ-free HLA-B27 transgenic rats colonized with defined flora (22). Helicobacter hepaticus has been proposed as a requisite component of the enteric flora in the CD45RB transfer and IL-10−/− colitis models (48, 49), although this has been controversial (50). However, the diversity and specificities of the T cell populations responsive to these bacteria are yet to be defined in these or other IBD models. Advantages of the DO11.10 OVA colitis model described here include the ability to track a specific T cell population that is responsive to a defined bacterially associated antigen in vivo (45, 46), and the ability to control the developmental stage and effector function of the T cell population. This model also offers the potential to vary the bacterial host and the bacterial compartment in which the target antigen, OVA, is expressed.
A technical impediment confronted at the outset of these studies was difficulty in the development of bacterial strains that could stably express OVA in vivo and could establish colonization of a murine host. Poor retention of plasmids and/or silencing of plasmid promoters have hampered efforts to express heterologous antigens in bacterial vectors in vivo (31). It has also been difficult to establish long-term colonization of laboratory strains of bacteria introduced into the normal flora (33). Adoption of the pnir15 promoter has extended expression of OVA in vivo, and we have been able to document stable plasmid retention and expression of OVA up to 5 wk after a single inoculation of pnir15.OVA-E. coli (data not shown). We have had less success maintaining long-term colonization by the DH5α E. coli strain used in these studies without repeated bacterial inoculation. More recent efforts using E. coli strains derived from the normal flora of mice in our colony have resulted in prolonged colonization (unpublished data) and are being characterized for future studies. Nevertheless, it is clear from our results that the protocol developed for in vivo delivery of bacterially associated OVA is effective.
This study establishes that a single antigenic specificity can drive the development of colitis by both Th1 and Th2 effectors, but does not prove that specific bacterial antigens are responsible for pathogenic T cell responses in other colitis models or in human disease. Indeed, pioneering studies by Braun et al. has identified by representational difference analysis a novel bacterial superantigen, I2, which is associated with inflammatory lesions in patients with Crohn's disease (51). While a causal relationship between this molecule and Crohn's disease is not yet established, this study raises the possibility that bacterial superantigens rather than specific bacterial antigens are important contributors to a pathogenic T cell response. However, TCR repertoire analyses in monozygotic twins with Crohn's disease do not support a major role for superantigens (52). Further, recent studies of the TCR-β repertoire of T cells from colitic animals in the CD45RB and C3H/HeJBir colitis models favor an oligoclonal T cell response to specific bacterial antigens rather than a superantigen (27, 53, 54). In the present model, colonization of adoptive transfer recipients with E. coli that express a control antigen (TET) did not develop disease, and there was no evidence of reactivity of either naive or effector DO11.10 T cells to control E. coli or the endogenous enteric flora of RAG-2−/− hosts. Hence, this model does not involve a contribution by bacterial superantigens, and demonstrates that superantigens are not required for colitis development.
The induction of colitis by both Th1 and Th2 effector cells in this model was somewhat unanticipated. Although elevated Th2 cytokines have been demonstrated in the TCR-α−/− and oxazalone colitis models (15, 16), the majority of the rodent models that have been described demonstrate characteristics of a Th1-mediated disease. In the well-characterized CD45RB colitis model, CD4 T cells recovered from animals with active disease produced Th1 cytokines and disease was attenuated by in vivo neutralization of IL-12, IFN-γ, and TNF-α (11, 13). The colitis that develops in IL-10−/− mice also has a Th1 cytokine profile (14). Not surprisingly, the Th1-mediated disease in our model had histopathologic features that closely resembled those of both CD45RB and IL-10−/− models with respect to the pattern of inflammation, extent, and distribution of intestinal involvement and disease severity. In each of these models, the inflammatory lesions are distributed throughout the cecum and colon and are characterized by focally dense infiltrates of CD4 T cells and monocytes, with lesser numbers of neutrophils.
Unexpectedly, the colitic lesions induced by Th2 transfers in the current model were of comparable severity to those mediated by Th1 cell transfers, albeit with distinct histopathologic features. The composition of inflammatory lesions in Th2 colitis was characterized by lesser numbers of infiltrating T cells accompanied by numerous neutrophils and eosinophils. Interestingly, the acute inflammatory infiltrates (neutrophils and eosinophils) were more prominent in Th2-mediated disease despite a paucity of epithelial necrosis compared with that seen in Th1-mediated lesions. This finding is consistent with the enhanced recruitment of these cells by cytokines and chemokines produced by activated Th2 cells. The Th2-mediated lesions also demonstrated a more striking hyperplasia of the crypt epithelium than in Th1 lesions, although epithelial hyperplasia was a prominent feature of both diseases. Although we have not directly compared the histology in our Th2 model with that of the TCR-α−/− or oxazalone models, the pathologic features most closely resemble the published characterizations of the former (55). Given the association of disease in both of these models with IL-4, it will be important to determine if IL-4 plays a pathogenic role in the current model. In any case, the pathogenic potential of an unopposed Th2 response in this model strikes a cautionary note with respect to therapeutic strategies aimed at reversing Th1-mediated autoinflammatory disease by augmenting Th2 activity. In this regard, it has been shown that IL-4 treatment can exacerbate Th1 type colonic inflammation in the CD45RBhi adoptive transfer model (56). This is in agreement with studies that have demonstrated a pathogenic rather than protective role for Th2 cells in murine models of multiple sclerosis (experimental allergic encephalitis) and diabetes (NOD mice) (57, 58). Similarly, in recent murine models of asthma also based on adoptive transfers of DO11.10 T cells, both Th1 and Th2 cells were pathogenic, and cotransfers of both cell types exacerbated rather than controlled disease (59, 60). Thus, although the nature of the chronic inflammatory lesions induced by chronic Th1 and Th2 activation is different, unopposed responses of either can be pathogenic to the intestinal and extraintestinal tissues.
A curious feature of the current model concerns the tempo of disease development mediated by Th1 and Th2 transfers into OVA-E. coli–colonized mice. Similar to each of the spontaneous colitis models that have been characterized, development of clinically apparent disease after Th1 or Th2 transfers evolved over a period of several weeks to months. In previous models, where the precursor frequency of pathogenic or protective T cells was not defined, it was plausible that this relatively slow development of disease was due to a requirement for clonal expansion of a pathogenic T cell population that was initially limiting in the repertoire. Given the monoclonality of the pathogenic T cells in the current model, it is surprising that disease development is not accelerated relative to other models. It is also provocative that Th1 and Th2 disease development progressed at similar rates. This suggests that the rate-limiting step for disease induction and progression in this model may not be the availability of pathogenic T cells. Rather, the immunogenicity of the relevant bacteria or the rate at which translocations of the relevant enteric bacteria occur might be more critical. Because the OVA-E. coli used in this study are quite potently immunogenic (Figs. 1 and 2), we favor a dominant role for the latter. The rate of bacterial translocation is typically low for commensal organisms, and is related to the density of a given bacterial species in the enteric flora and its representation relative to other bacterial species that comprise the flora (61). In our studies, reduced colonization by OVA-E. coli to levels below ∼107 CFU/g of lumenal contents resulted in a marked decrease in disease severity and incidence despite transfers of the same number of Th1 or Th2 cells (data not shown). This suggests that the rate at which bacteria are transported across the mucosa may be limiting, and might explain the focal nature of lesions in this and other colitis models, as well as the relatively slow progression of disease. Since the specific bacterial species responsible for disease is identified in the DO11.10/OVA-E. coli model, it will be possible to examine this directly. It would be predicted that higher colonization rates or delivery of OVA via more invasive organisms would accelerate disease development, and studies to directly quantitate this are in progress.
The basis for the lack of colitis development in recipients of antigen-naive DO11.RAG-2−/− T cells colonized with OVA-E. coli is not yet known, but it is not due to antigenic ignorance or neglect, nor defective trafficking of reactive T cells to the colonic mucosa. Since in previous models of colitis the differentiation state of the T cell population at the onset of experimental protocols could not be defined, it has been unclear whether a pathogenic population was preexistent in the T cell repertoire or developed in response to stimulation by enteric bacteria. In the CD45RB colitis model, for example, it has not been possible to determine whether the pathogenic Th1 response has been due to the development of a Th1 effector population from uncommitted CD45RBhigh precursors in the absence of CD45RBlow regulatory cells, or whether it was due to the release of committed, memory CD45RBhigh Th1 cells from regulatory control (20). Our data are inconsistent with the former mechanism, since recipients of a uniformly naive (and CD45RBhigh) population of T cells did not develop colitis, despite the absence of a CD45RBlow regulatory population. This suggests that a committed Th1 or Th2 population may be required for disease development and/or may recruit the differentiation of naive precursors activated by bacterial antigens. Alternatively, naive CD4 T cells responsive to bacterially associated antigen in the absence of either pathogenic or regulatory effector cell populations may reestablish a homeostatic balance of both pathogenic and regulatory T cells. Experiments to distinguish these alternatives can be addressed in our model, and are in progress.
Acknowledgments
The authors thank Drs. William H. Benjamin, Jr., R. Pat Bucy, Yingzi Cong, Philip Dean, James Lillard, Jerry McGhee, and Robert Shaw for comments and helpful discussion. We also thank J. Scott Thomas, and M. Lamar Jones for their excellent technical and immunohistochemical expertise and Noelle Nicholls for editorial assistance.
This work was supported by grants DK 44240 and AI35783 from the National Institutes of Health, a career development award from the CCFA (NI), and a grant from Sankyo Co., Ltd.
Footnotes
Abbreviations used in this paper: BrdU, bromodeoxyuridine; IBD, inflammatory bowel disease; LP, lamina propria; RAG, recombination activating gene.
References
- 1.Elson, C. 1998. Experimental models of intestinal inflammation; new insights into mechanisms of mucosal homeostasis. 2nd ed. Mucosal Immunology. P. Ogra, J. Mestecky, M. Lamm, W. Strober, J. McGhee and J. Bienenstock, editors. Academic Press, San Diego. 1007–1024.
- 2.Hammer, R.E., S.D. Maika, J.A. Richardson, J.P. Tang, and J.D. Taurog. 1990. Spontaneous inflammatory disease in transgenic rats expressing HLA-B27 and human β2m: an animal model of HLA-B27-associated human disorders. Cell. 63:1099–1112. [DOI] [PubMed] [Google Scholar]
- 3.Kuhn, R., J. Lohler, D. Rennick, K. Rajewsky, and W. Muller. 1993. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 75:263–274. [DOI] [PubMed] [Google Scholar]
- 4.Sadlack, B., H. Merz, H. Schorle, A.F. Schimpl, A.C., and I. Horak. 1993. Ulcerative colitis-like diseasse in mice with a disrupted interleukin-2 gene. Cell. 75:253–261. [DOI] [PubMed] [Google Scholar]
- 5.Mombaerts, P., E. Mizoguchi, M.J. Grusby, L.H. Glimcher, A.K. Bhan, and S. Tonegawa. 1993. Spontaneous development of inflammatory bowel disease in T cell receptor mutant mice. Cell. 75:275–282. [DOI] [PubMed] [Google Scholar]
- 6.Takeda, K., B.E. Clausen, T. Kaisho, T. Tsujimura, N. Terada, I. Forster, and S. Akira. 1999. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity. 10:39–49. [DOI] [PubMed] [Google Scholar]
- 7.Hermiston, M.L., M.H. Wong, and J.I. Gordon. 1996. Forced expression of E-cadherin in the mouse intestinal epithelium slows cell migration and provides evidence for nonautonomous regulation of cell fate in a self-renewing system. Genes & Development. 10:985–996. [DOI] [PubMed] [Google Scholar]
- 8.Hollander, G.A., S.J. Simpson, E. Mizoguchi, A. Nichogiannopoulou, J. She, J.C. Gutierrez-Ramos, A.K. Bhan, S.J. Burakoff, B. Wang, and C. Terhorst. 1995. Severe colitis in mice with aberrant thymic selection. Immunity. 3:27–38. [DOI] [PubMed] [Google Scholar]
- 9.Powrie, F., M.W. Leach, S. Mauze, L.B. Caddle, and R.L. Coffman. 1993. Phenotypically distinct subsets of CD4+ T cells induce or protect from chronic intestinal inflammation in C. B-17 scid mice. Int. Immunol. 5:1461–1471. [DOI] [PubMed] [Google Scholar]
- 10.Morrissey, P.J., K. Charrier, S. Braddy, D. Liggitt, and J.D. Watson. 1993. CD4+ T cells that express high levels of CD45RB induce wasting disease when transferred into congenic severe combined immunodeficient mice. Disease development is prevented by cotransfer of purified CD4+ T cells. J. Exp. Med. 178:237–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Powrie, F., M.W. Leach, S. Mauze, S. Menon, L.B. Caddle, and R.L. Coffman. 1994. Inhibition of Th1 responses prevents inflammatory bowel disease in scid mice reconstituted with CD45RBhi CD4+ T cells. Immunity. 1:553–562. [DOI] [PubMed] [Google Scholar]
- 12.Neurath, M.F., I. Fuss, B.L. Kelsall, E. Stuber, and W. Strober. 1995. Antibodies to interleukin 12 abrogate established experimental colitis in mice. J. Exp. Med. 182:1281–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simpson, S.J., S. Shah, M. Comiskey, Y.P. de Jong, B. Wang, E. Mizoguchi, A.K. Bhan, and C. Terhorst. 1998. T cell-mediated pathology in two models of experimental colitis depends predominantly on the interleukin 12/Signal transducer and activator of transcription (Stat)-4 pathway, but is not conditional on interferon γ expression by T cells. J. Exp. Med. 187:1225–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davidson, N.J., S.A. Hudak, R.E. Lesley, S. Menon, M.W. Leach, and D.M. Rennick. 1998. IL-12, but not IFN-γ, plays a major role in sustaining the phase of colitis in IL-10-deficient mice. J. Immunol. 161:3143–3149. [PubMed] [Google Scholar]
- 15.Mizoguchi, A., E. Mizoguchi, C. Chiba, G.M. Spiekermann, S. Tonegawa, C. Nagler-Anderson, and A.K. Bhan. 1996. Cytokine imbalance and autoantibody production in T cell receptor-α mutant mice with inflammatory bowel disease. J. Exp. Med. 183:847–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boirivant, M., I.J. Fuss, A. Chu, and W. Strober. 1998. Oxazolone colitis: a murine model of T helper cell type 2 colitis treatable with antibodies to interleukin 4. J. Exp. Med. 188:1929–1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mizoguchi, A., E. Mizoguchi, and A.K. Bhan. 1999. The critical role of interleukin 4 but not interferon γ in pathogenesis of colitis in T-cell receptor α mutant mice. Gastroenterology. 116:320–326. [DOI] [PubMed] [Google Scholar]
- 18.Fuss, I.J., M. Neurath, M. Boirivant, J.S. Klein, C. de la Motte, S.A. Strong, C. Fiocchi, and W. Strober. 1996. Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn's disease LP cells manifest increased secretion of IFN-γ, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J. Immunol. 157:1261–1270. [PubMed] [Google Scholar]
- 19.Parronchi, P., P. Romagnani, F. Annunziato, S. Sampognaro, A. Becchio, L. Giannarini, E. Maggi, C. Pupilli, F. Tonelli, and S. Romagnani. 1997. Type 1 T-helper cell predominance and interleukin-12 expression in the gut of patients with Crohn's disease. Am. J. Pathology. 150:823–832. [PMC free article] [PubMed] [Google Scholar]
- 20.Powrie, F. 1995. T cells in inflammatory bowel disease: protective and pathogenic roles. Immunity. 3:171–174. [DOI] [PubMed] [Google Scholar]
- 21.Sartor, R.B. 1997. The influence of normal microbial flora on the development of chronic mucosal inflammation. Res. Immunol. 148:567–576. [DOI] [PubMed] [Google Scholar]
- 22.Rath, H.C., H.H. Herfarth, J.S. Ikeda, W.B. Grenther, T.E. Hamm, Jr., E. Balish, J.D. Taurog, R.E. Hammer, K.H. Wilson, and R.B. Sartor. 1996. Normal luminal bacteria, especially Bacteroides species, mediate chronic colitis, gastritis, and arthritis in HLA-B27/human β2 microglobulin transgenic rats. J. Clin. Invest. 98:945–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sellon, R.K., S.L. Tonkonogy, H. Grable, J. Kwon, M. Schultz, E. Balish, D. Rennick, and R.B. Sartor. 1997. Development of spontaneous colitis in IL-10 knockout mice requires normal enteric bacterial flora. Gastroenterology. 112:A1088. [Google Scholar]
- 24.Veltkamp, C., S.L. Tonkonogy, Y.P. De Jong, C. Albright, W.B. Grenther, E. Balish, C. Terhorst, and R.B. Sartor. 2001. Continuous stimulation by normal luminal bacteria is essential for the development and perpetuation of colitis in Tg(ε26) mice. Gastroenterology. 120:900–913. [DOI] [PubMed] [Google Scholar]
- 25.Rath, H.C., M. Schultz, R. Freitag, L.A. Dieleman, F. Li, H.J. Linde, J. Scholmerich, and R.B. Sartor. 2001. Different subsets of enteric bacteria induce and perpetuate experimental colitis in rats and mice. Infection & Immunity. 69:2277–2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sundberg, J.P., C.O. Elson, H. Bedigian, and E.H. Birkenmeier. 1994. Spontaneous, heritable colitis in a new substrain of C3H/HeJ mice. Gastroenterology. 107:1726–1735. [DOI] [PubMed] [Google Scholar]
- 27.Cong, Y., S.L. Brandwein, R.P. McCabe, A. Lazenby, E.H. Birkenmeier, J.P. Sundberg, and C.O. Elson. 1998. CD4+ T cells reactive to enteric bacterial antigens in spontaneously colitic C3H/HeJBir mice: increased T helper cell type 1 response and ability to transfer disease. J. Exp. Med. 187:855–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brandwein, S.L., R.P. McCabe, Y. Cong, K.B. Waites, B.U. Ridwan, P.A. Dean, T. Ohkusa, E.H. Birkenmeier, J.P. Sundberg, and C.O. Elson. 1997. Spontaneously colitic C3H/HeJBir mice demonstrate selective antibody reactivity to antigens of the enteric bacterial flora. J. Immunol. 159:44–52. [PubMed] [Google Scholar]
- 29.Rath, H.C., J.S. Ikeda, H.J. Linde, J. Scholmerich, K.H. Wilson, and R.B. Sartor. 1999. Varying cecal bacterial loads influences colitis and gastritis in HLA-B27 transgenic rats. Gastroenterology. 116:310–319. [DOI] [PubMed] [Google Scholar]
- 30.Murphy, K.M., A.B. Heimberger, and D.Y. Loh. 1990. Induction by antigen of intrathymic apoptosis of CD4+CD8+TCRlo thymocytes in vivo. Science. 250:1720–1723. [DOI] [PubMed] [Google Scholar]
- 31.Chatfield, S.N., I.G. Charles, A.J. Makoff, M.D. Oxer, G. Dougan, D. Pickard, D. Slater, and N.F. Fairweather. 1992. Use of the nirB promoter to direct the stable expression of heterologous antigens in Salmonella oral vaccine strains: development of a single-dose oral tetanus vaccine. Bio/Technology. 10:888–892. [DOI] [PubMed] [Google Scholar]
- 32.Oxer, M.D., C.M. Bentley, J.G. Doyle, T.C. Peakman, I.G. Charles, and A.J. Makoff. 1991. High level heterologous expression in E. coli using the anaerobically-activated nirB promoter. Nucl. Acids Research. 19:2889-2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fairweather, N.F., S.N. Chatfield, I.G. Charles, M. Roberts, M. Lipscombe, L.J. Li, D. Strugnell, S. Comerford, J. Tite, and G. Dougan. 1990. Use of live attenuated bacteria to stimulate immunity. Res. Microbiol. 141:769–773. [DOI] [PubMed] [Google Scholar]
- 34.Haskins, K., R. Kubo, J. White, M. Pigeon, J. Kappler, and P. Marrack. 1983. The major histocompatibility complex-restricted antigen receptor on T cells. I. Isolation with a monoclonal antibody. J. Exp. Med. 157:1149–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saparov, A., L.A. Kraus, Y. Cong, J. Marwill, X.Y. Xu, C.O. Elson, and C.T. Weaver. 1999. Memory/effector T cells in TCR transgenic mice develop via recognition of enteric antigens by a second, endogenous TCR. Int. Immunol. 11:1253–1264. [DOI] [PubMed] [Google Scholar]
- 36.Rogers, W.O., C.T. Weaver, L.A. Kraus, J. Li, L. Li, and R.P. Bucy. 1997. Visualization of antigen-specific T cell activation and cytokine expression in vivo. J. Immunol. 158:649–657. [PubMed] [Google Scholar]
- 37.Bucy, R.P., L. Karr, G.-Q. Huang, J. Li, D. Carter, K. Honjo, J.A. Lemons, K.M. Murphy, and C.T. Weaver. 1995. Single cell analysis of cytokine gene co-expression during CD4+ T-cell phenotype development. Proc. Natl. Acad. Sci. USA. 92:7565–7569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ohara, J., and W.E. Paul. 1985. Production of a monoclonal antibody to and molecular characterization of B-cell stimulatory factor-1. Nature 315:333–336. [DOI] [PubMed] [Google Scholar]
- 39.Wysocka, M., M. Kubin, L.Q. Vieira, L. Ozmen, G. Garotta, P. Scott, and G. Trinchieri. 1995. Interleukin-12 is required for interferon-γ production and lethality in lipopolysaccharide-induced shock in mice. Eur. J. Immunol. 25:672–676. [DOI] [PubMed] [Google Scholar]
- 40.Szabo, S.J., N.G. Jacobson, A.S. Dighe, U. Gubler, and K.M. Murphy. 1995. Developmental commitment to the Th2 lineage by extinction of IL-12 signaling. Immunity. 2:665–675. [DOI] [PubMed] [Google Scholar]
- 41.Broide, D.H., S. Sullivan, T. Gifford, and P. Sriramarao. 1998. Inhibition of pulmonary eosinophilia in P-selectin- and ICAM-1-deficient mice. Amer. J. Resp. Cell & Mol. Biology. 18:218–225. [DOI] [PubMed] [Google Scholar]
- 42.Mahler, M., I.J. Bristol, E.H. Leiter, A.E. Workman, E.H. Birkenmeier, C.O. Elson, and J.P. Sundberg. 1998. Differential susceptibility of inbred mouse strains to dextran sulfate sodium-induced colitis. Amer. J. Physiology. 274:G544–G551. [DOI] [PubMed] [Google Scholar]
- 43.Leo, O., M. Foo, D.H. Sachs, L.E. Samelson, and J.A. Bluestone. 1987. Identification of a monoclonal antibody specific for a murine T3 polypeptide. Proc. Natl. Acad. Sci. USA. 84:1374–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gross, J.A., E. Callas, and J.P. Allison. 1992. Identification and distribution of the costimulatory receptor CD28 in the Mouse. J. Immunol. 149:380–388. [PubMed] [Google Scholar]
- 45.Kearney, E.R., K.A. Pape, D.Y. Loh, and M.K. Jenkins. 1994. Visualization of peptide-specific T cell immunity and peripheral tolerance induction in vivo. Immunity. 1:327–339. [DOI] [PubMed] [Google Scholar]
- 46.Chen, Z.M., and M.K. Jenkins. 1998. Revealing the in vivo behavior of CD4+ T cells specific for an antigen expressed in Escherichia coli. J. Immunol. 160:3462–3470. [PubMed] [Google Scholar]
- 47.Groux, H., A. O'Garra, M. Bigler, M. Rouleau, S. Antonenko, J.E. de Vries, and M.G. Roncarolo. 1997. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature. 389:737–742. [DOI] [PubMed] [Google Scholar]
- 48.Cahill, R.J., C.J. Foltz, J.G. Fox, C.A. Dangler, F. Powrie, and D.B. Schauer. 1997. Inflammatory bowel disease: an immunity-mediated condition triggered by bacterial infection with Helicobacter hepaticus. Infection & Immunity. 65:3126–3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kullberg, M.C., J.M. Ward, P.L. Gorelick, P. Caspar, S. Hieny, A. Cheever, D. Jankovic, and A. Sher. 1998. Helicobacter hepaticus triggers colitis in specific-pathogen-free interleukin-10 (IL-10)-deficient mice through an IL-12- and γ interferon-dependent mechanism. Infection & Immunity. 66:5157–5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dieleman, L.A., A. Arends, S.L. Tonkonogy, M.S. Goerres, D.W. Craft, W. Grenther, R.K. Sellon, E. Balish, and R.B. Sartor. 2000. Helicobacter hepaticus does not induce or potentiate colitis in interleukin-10-deficient mice. Infection & Immunity. 68:5107–5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sutton, C.L., J. Kim, A. Yamane, H. Dalwadi, B. Wei, C. Landers, S.R. Targan, and J. Braun. 2000. Identification of a novel bacterial sequence associated with Crohn's disease. Gastroenterology. 119:23–31. [DOI] [PubMed] [Google Scholar]
- 52.Gulwani-Akolkar, B., L. Shalon, P.N. Akolkar, S.E. Fisher, and J. Silver. 1994. Analysis of the peripheral blood T-cell receptor (TCR) repertoire in monozygotic twins discordant for Crohn's disease. Autoimmunity. 17:241–248. [DOI] [PubMed] [Google Scholar]
- 53.Aranda, R., B.C. Sydora, P.L. McAllister, S.W. Binder, H.Y. Yang, S.R. Targan, and M. Kronenberg. 1997. Analysis of intestinal lymphocytes in mouse colitis mediated by transfer of CD4+, CD45RBhigh T cells to SCID recipients. J. Immunology. 158:3464–3473. [PubMed] [Google Scholar]
- 54.Rudolphi, A., K. Bonhagen, and J. Reimann. 1996. Polyclonal expansion of adoptively transferred CD4+ αβ T cells in the colonic lamina propria of scid mice with colitis. Eur. J. Immunol. 26:1156–1163. [DOI] [PubMed] [Google Scholar]
- 55.Bhan, A.K., E. Mizoguchi, R.N. Smith, and A. Mizoguchi. 1999. Colitis in transgenic and knockout animals as models of human inflammatory bowel disease. Immunol. Reviews. 169:195–207. [DOI] [PubMed] [Google Scholar]
- 56.Fort, M., R. Lesley, N. Davidson, S. Menon, F. Brombacher, M. Leach, and D. Rennick. 2001. IL-4 exacerbates disease in a Th1 cell transfer model of colitis. J. Immunol. 166:2793–2800. [DOI] [PubMed] [Google Scholar]
- 57.Lafaille, J.J., F.V. Keere, A.L. Hsu, J.L. Baron, W. Haas, C.S. Raine, and S. Tonegawa. 1997. Myelin basic protein-specific T helper 2 (Th2) cells cause experimental autoimmune encephalomyelitis in immunodeficient hosts rather than protect them from the disease. J. Exp. Med. 186:307–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pakala, S.V., M.O. Kurrer, and J.D. Katz. 1997. T helper 2 (Th2) T cells induce acute pancreatitis and diabetes in immune-compromised nonobese diabetic (NOD) mice. J. Exp. Med. 186:299–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hansen, G., G. Berry, R.H. DeKruyff, and D.T. Umetsu. 1999. Allergen-specific Th1 cells fail to counterbalance Th2 cell-induced airway hyperreactivity but cause severe airway inflammation. J. Clin. Investig. 103:175–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Randolph, D.A., R. Stephens, C.J. Carruthers, and D.D. Chaplin. 1999. Cooperation between Th1 and Th2 cells in a murine model of eosinophilic airway inflammation. J. Clin. Investig. 104:1021–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Berg, R.D. 1983. Host immune response to antigens of the indigenous intestinal flora. Human intestinal microflora in health and disease. D.C. Savage, editor. Academic Press, New York. 101–126.