

Abstract
Development of T helper cell (Th)1 or Th2 cytokine responses is essential for effector and regulatory functions of T helper cells. We have compared cytokine profiles of myelin basic protein (MBP) Ac1-16 peptide-specific T helper cells from inbred mouse strains expressing identical k haplotype-derived MHC class II molecules B10.A and B10.BR. B10.BR T cell lines (TCL) produced Th1 cytokines (including high levels of TNF-α) and induced experimental autoimmune encephalomyelitis after adoptive transfer. In contrast, B10.A TCL produced Th2 cytokines (including low levels of TNF-α) and were poorly encephalitogenic. The contributions of the genetic origin of the T cells and the APC were explored. Serial restimulations of the B10.BR TCL with B10.A or (B10.A × B10.BR) F1 splenic antigen presenting cells (APC) during the establishment of TCL markedly reduced both Th1 cytokine production and encephalitogenicity. In addition, a single restimulation with B10.A splenic APC reduced IFN-γ and TNF-α production by established Th1 MBP-specific Ak-restricted B10.BR TCL and by a Th1 KLH-specific, Ek-restricted B10.BR T cell clone. These studies suggest that B10.A and B10.BR APC differ in their ability to stimulate IFN-γ and TNF-α production by mature Th1 cells and also influence their Th1/Th2 commitment in vivo. The nature of the downregulatory activity of B10.A APC on IFN-γ and TNF-α production was explored. 2-hour supernatants from antigen-activated B10.A APC/TCL cultures or from B10.A APC activated by LPS had the same inhibitory effects on IFN-γ and TNF-α production by B10.BR TCL. The downregulatory effects of B10.A APC are independent of TNF-α, IL-4, IL-10, IL-12p40, IFN-γ, IL-13, TGF-β, and PGE2. Thus, genetic difference(s) between B10.A and B10.BR APC appear(s) to control the production or activity of a novel soluble cytokine regulatory factor that influences Th1/Th2 commitment and controls production of IFN-γ and TNF-α by mature Th1 cells.
Most mature CD4+ T helper cells express one of two cytokine profiles: Th1 or Th2. Th1s secrete IL-2, IL-3, IFN-γ, TNF-β, GM-CSF, and high levels of TNF-α. Th2s express IL-3, IL-4, IL-5, IL-6, IL-9, IL-10, IL-13, GM-CSF, and low levels of TNF-α (1–3). These cytokine profiles determine T cell regulatory and effector functions in immune responses (4–8). The Th1 subset promotes delayed-type hypersensitivity, cell-mediated immunity, and immunoglobulin class switching to IgG2a. The Th2 subset induces humoral immunity by activating B cells, promoting antibody production, and inducing class switching to IgG1 and IgE. Skewing the T cell responses toward Th1 is thought to result in susceptibility to autoimmune and inflammatory diseases; skewing toward Th2 cytokines promotes allergic reactions.
Several factors have been shown to influence commitment to Th1 or Th2 profiles. The best characterized regulators are cytokines. IL-12 and IFN-γ are positive Th1 and negative Th2 regulators (9–15). IL-12 promotes IFN-γ production, and IFN-γ provides positive feedback for IL-12. IL-4 and IL-10 appear to be required for the establishment of the Th2 cytokine profile and to downregulate Th1 cytokine production; the effects of IL-4 have been demonstrated to be dominant over those of IL-12 (16–21). IL-13 was shown to inhibit expression of inflammatory cytokines, including IL-12 and TNF-α, by LPS-induced monocytes in a way similar to IL-4 (22–24). The IL-12 p40 homodimer binds to the IL-12 receptor and antagonizes IL-12 biological activity (25, 26); thus, it blocks the pro-Th1 effects of IL-12. TGF-β is also implicated in Th1/Th2 regulation, although its role remains controversial. It was implicated in the suppression of the Th1 response in autoimmune encephalomyelitis (27); however, TGF-β was also shown to enhance the Th1 phenotype (28, 29). Recently, opposing effects of TGF-β on Th1 development were shown to correlate with the amounts of IL-2 produced in the presence of this molecule (30).
Signals through the TCR and co-stimulatory molecules have also been shown to influence Th commitment. Th1 development was found to be associated with high affinity binding of a peptide antigen to MHC class II and strong signaling through the TCR, whereas lower affinity antigen–MHC II interactions and weaker signaling through TCR were reported to result in Th2 cytokine responses (31, 32). Several cell surface molecules expressed by APC and T cells have been suggested to influence commitment to Th1 or Th2 response, including CD40-CD40 ligand interactions and B7.1 versus B7.2 signaling (33–40). At the cellular level, at least in some cases, macrophages and dendritic cells appear to promote the Th1 response, whereas B cells upregulate Th2 cytokines (41–43). However, neither the B7.1/B7.2 effects, nor the effects of different APC types in supporting Th1 or Th2, are absolute (3).
The mechanisms regulating development and expression of Th1 and Th2 cytokine phenotypes have not been fully described. The identification and characterization of genes influencing Th1/Th2 commitment are likely to provide key insights into this process. Studies of Leishmania infections and autoimmunity implicate both MHC and nonMHC genes in the development of specific cytokine responses in humans and animals (8, 44–48). C57BL/6, B10.D2, and CH3/HeN mouse strains, among many others, are predisposed to Th1 responses, and the BALB/c background is known to promote the Th2 cytokine profile in response to a wide variety of parasites and antigens (47, 49–50). Recently, the predisposition of BALB cells to develop the Th2 profile was associated with the failure to respond to IL-12 (51).
This study began with a comparison of encephalitogenicity of myelin basic protein (MBP)1–specific T cell lines (TCL) from two Ak-expressing H-2 congenic mouse strains, B10.A and B10.BR, previously described as being resistant to Leishmania tropica major (44), i.e., predisposed to the Th1 cytokine profile. Experimental autoimmune encephalomyelitis (EAE) is a neurological disease considered to be an animal model of multiple sclerosis. It can be induced in two ways: by direct immunization with MBP, proteolipid protein, or their peptides in adjuvant combined with intravenous injections of Pertussis toxin, or by adoptive transfer using activated T cells specific for these antigens. Effector CD4+ T cells are thought to induce a DTH-like reaction in the central nervous system which results in demyelination and paralysis (8). Th1 cytokine responses, including high TNF expression and high levels of VLA-4, have been shown to be necessary for development of EAE (52–56).
Adoptive transfer experiments show that MBP-specific TCL from B10.BR mice induce severe EAE, whereas TCL from B10.A and (B10.BR × B10.A) F1 mice are weakly encephalitogenic. Our data further suggest that the differences in encephalitogenicity correlate with Th1/Th2 cytokine profiles. Genetic difference(s) between these strains appears to control a novel secreted factor regulating cytokine responses of T cells specific for mouse MBP (mMBP) and probably for other antigens as well.
Materials and Methods
Animals.
B10.A, B10.BR, and B10.PL mice, 6–9 wk old, were obtained from Jackson Laboratory (Bar Harbor, ME); (B10.A × B10.BR) F1, F2, and (B10.BR × F1) backcross mice were bred in our facility.
Antibodies.
Anti–IL-4 mAb 11B11 was provided by J. O'hara and W.E. Paul (National Institutes of Health). Anti–IFN-γ mAb XMG1.2 was provided by T. Mosmann (University of Alberta, Edmonton, Canada). Anti–IFN-γ mAb HB170 was obtained from American Type Culture Collection (Rockville, MD). Anti–IL-4 mAb BVD4 and BVD6, and anti–IL-10 mAb 2A5 and Sxc.1 were provided by M. Howard (DNAX Research Institute, Palo Alto, CA). Anti–IL-12 p40 mAb C17.8 and C15.6 were provided by G. Trinchieri (Wistar Institute of Anatomy and Biology, Philadelphia, PA). Anti–TNF-α mAb was provided by Robert Schreiber (Washington School of Medicine, St. Louis, MO). Anti–IL-13 and anti–TGF-β pan-neutralizing antibodies were purchased from R&D Sys., Inc. (Minneapolis, MN).
Peptides.
mMBP Ac1-16 (Ac-ASQKRPSQRSKYLATA) was purchased from the Protein and Nucleic Acids Facility (Stanford University, Stanford, CA).
TCL.
Mice were immunized in the base of the tail with 100 μM mMBP Ac1-16 in CFA H37 Ra (Difco, Detroit, MI). 8 d later superficial inguinal and sacral lymph nodes were removed, and lymph node cells were stimulated with 30 μM mMBP Ac116 in RPMI 1640 (GIBCO BRL, Gaithersburg, MD) with 2% syngeneic mouse serum. Every 10–14 d thereafter, 0.5–1 × 106 T cells were restimulated with 30 μM mMBP Ac1-16 and 5 × 106 irradiated (3,000 rad) splenocytes per ml. TCL were cultured starting from the third restimulation, in RPMI 1640 with 10% FCS (GIBCO BRL) and 10% supernatant from rat splenocytes activated overnight by 2 μg/ml of Con A added every other restimulation.
BR E7 T Cell Clone.
Every 7–10 d, 2–5 × 105 T cells per ml were restimulated with 2–5 × 106 irradiated (3,000 rad) B10.BR splenic APC per ml, 3 μg/ml KLH (Calbiochem Corp., San Diego, CA), and 10% rat Con A supernatant. Culture supernatants were harvested 24 h later and assayed for cytokines.
Induction and Scoring of EAE.
3 d after the last restimulation with 30 μM mMBP Ac1-16, 5 × 106 irradiated splenic APC, and IL-2, 2–5 × 107 T cells were washed three times in serum free RPMI and injected intravenously in the tail veins of nonirradiated recipient mice 6–9 wk old. Mice were monitored daily, and EAE severity was measured on the 0–5 scale: 0, no symptoms; 0.5, straight tail; 1, limp tail; 2, paraparesis; 3, paraplegia; 4, quadriplegia; 5, moribund. Mice with scores between 4 and 5 were killed.
Induction of Cytokine Responses.
At the time of restimulation, aliquots of 0.25–1 × 106 T cells were washed and plated in 1 ml of RPMI 1640, 10% FCS. 30 μm mMBP Ac1-16, 5 × 106 irradiated (3,000 rad) splenocytes, and 10% rat Con A supernatant (if <1 × 106 T cells were used) were added for 24 h, after which supernatant fluids were removed and assayed for levels of cytokines. For measuring effects of 2 h supernatants on the levels of IFN-γ and TNF-α, supernatant fluids were obtained from culture of 1 × 106 B10.A and B10.BR T cells per ml restimulated with 30 μm mMBP Ac1-16 and 5 × 106 splenocytes per ml for 2 h. Alternatively, 107 freshly isolated spleen cells per ml were incubated for 2 h with 10 μg/ml LPS (Escherichia coli 0127:B8; Difco). The 2 h supernatants from each strain (fresh or stored frozen at −80°C) were then used as culture media in which 0.5–1 × 106 B10.BR T cells per ml were restimulated with 5 × 106 irradiated B10.BR splenocytes per ml and 30 μm mMBP for 24 h. In some experiments, 5 × 105 T cells per ml were restimulated with 30 μm mMBP Ac1-16 and 5 × 105 per ml irradiated (10,000 rad) transfected L cell fibroblasts expressing Ak class II proteins (LAk cells) (see reference 58).
Depletion of Splenic T Cells.
Spleen cells were dissociated in serum-free RPMI 1640, resuspended at 4 × 107 per ml, and incubated for 10 min at 4°C with 100 μl/ml (NH4)2SO4-concentrated anti–Thy-1 mAb (Tib 99; American Type Culture Collection). After incubation for 30 min at 37°C with 1:8 diluted Low-Tox Baby Rabbit Complement (Cedarlane Labs., Westbury, NY), surviving cells were washed three times in RPMI 1640 with 10% FCS and analyzed by FACS® with anti–CD3 mAb or with isotypecontrol hamster IgG (Caltag, Burlingame, CA).
Addition or Depletion of Cytokines.
Cytokines were added as follows: mouse recombinant IL-4 (Boehringer Mannheim Corp., Indianapolis, IN) at 100 U/ml, mouse recombinant IL-12 (PharMingen, San Diego, CA) at 10 U/ml, mouse recombinant IFN-γ (Boehringer Mannheim Corp.) at 100 ng/ml, mouse recombinant TNF-α (Genentech Inc., San Francisco, CA) at 2 ng/ml. To deplete cytokines from the 2 h culture supernatants, anticytokine antibodies were added to 1 ml of 2-h supernatants from B10.BR and B10.A cultures at the following concentrations: anti–IL-4 (11B11) at 10 and 20 μg/ml, anti–IL-10 (Sxc.1) at 20 and 40 μg/ml, anti–IL-12p40 (C17.8) at 20 μg/ml, anti–IL-13 (goat polyclonal) at 50 and 75 μg/ml, anti–IFN-γ (XMG1.2) at 10 and 20 μg/ml, anti–TNF-α (TN3.19.12) at 50 and 100 μg/ml; anti–TGFβ (rabbit polyclonal) at 40 and 80 μg/ml. After 30 min incubation at 4°C, 100 μl of protein G–coated agarose beads (Pierce Chemical Co., Rockford, IL) were added to the supernatants. After incubation for 30 min at room temperature, beads were removed from supernatants by centrifugation at 3,000 rpm for 3 min, and supernatants were then used as a media for the restimulation of T cells.
ELISA.
Double-antibody sandwich ELISA assays were used. Plates were coated overnight with a monoclonal antibody specific for a particular cytokine, washed, and then incubated with culture supernatant or a cytokine standard overnight. A second monoclonal antibody, which was biotin-conjugated and which recognizes an independent epitope of the same cytokine, was used for detection. Antibodies were biotinylated with D-biotinN-hydroxysuccinimide ester (Sigma Chemical Co., St. Louis, MO). Biotinylated antibodies on the plate were detected with Streptavidin-horseradish peroxidase (Pierce, Rockford, IL) plus o-phenylenediamine (Sigma Chemical Co.). Optical densities were determined at wavelength = 490 nm using a plate reader (Molecular Devices Corp., Sunnyvale, CA). Anti–IL-4 mAb BVD4 or 11B11 was used for coating and BVD6 for detection of IL-4. Mouse recombinant IL-4 (DNAX Research Institute) was used as a standard. Anti–IL-10 mAb 2A5 (coating) and Sxc.1 (detection) were used for IL-10. Mouse recombinant IL-10 (DNAX Research Institute) was used as a standard. Anti–IL-12p40 mAb C17.8 (coating) and C15.6 (detection) were used to determine the IL-12 levels. Recombinant murine IL-12, (provided by S. Wolf, Genetics Institute, Cambridge, MA), was used as a standard. Anti-IFN-γ mAb HB170 (coating) and XMG1.2 (detection) were used for the IFN-γ ELISA. Recombinant murine IFN-γ (Genentech, Inc.) was used as a standard.
IL-2 Bioassay.
50 μl of supernatants from TCL restimulated with serial dilutions of mMBP Ac1-16 and irradiated splenic APC for 24 h were added to 1 × 104 HT-2 cells per well in 96-well plates in a final volume of 200 μl. IL-4 was neutralized with 11B11 anti–IL-4 mAb. Murine recombinant IL-2 (Sigma Chemical Co.) was used as a standard. 16–18 h later [3H]thymidine (1 μCi per well in 20 μl) was added to plates. After 6–7 h, incubation plates were harvested (PHD Cell Harvester; Cambridge Technology, Inc., Cambridge, MA), and incorporation of [3H]thymidine was measured by scintillation counting.
TNF Bioassay.
The TNF bioassay was carried out as described (57). Briefly, 2 × 104 L929 cells per well were plated in 96-well plates and incubated overnight. Serial dilutions of supernatants from TCL and 1 μg/ml actinomycin C1 (Boehringer Mannheim Corp.) were added to wells in a final volume of 200 μl. A standard curve was prepared using recombinant murine TNF-α (Genentech, Inc.). After 24 h incubation, 10 μl of 5 mg/ml 3-[4, 5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide (MTT; Sigma Chemical Co.) was added to each well. 4 h later, 100 μl of solubilization solution containing 50% dimethylformamide and 20% SDS (Sigma Chemical Co.) were added to each well for 1 h, and the optical densities were determined using a plate reader (wavelength = 570 nm; Molecular Devices Corp., Sunnyvale, CA).
Results
T Cells from B10.BR Mice Are Significantly More Encephalitogenic in Adoptive Transfer of EAE than B10.A-derived T Cells.
Initial comparison of susceptibility of strains B10.A and B10.BR to EAE induced in vivo by immunization with MBP NH2-terminal peptides were uninformative, as both strains demonstrated comparably low susceptibility to EAE (average score = 0.5, data not shown). Freshly isolated B10.A and B10.BR lymph node cells gave very low proliferative responses to MBP compared to those from Au strain B10.PL, which is susceptible to EAE (46). The differences in susceptibility to EAE and in proliferation of Au- versus Ak-restricted MBP-primed lymph node cells most likely reflects higher affinity binding of MBP NH2-terminal peptides by Au compared to Ak class II proteins (58).
For adoptive transfer of EAE, TCL specific for mMBP Ac1-16 were generated from B10.PL, B10.A, B10.BR, and (B10.A × B10.BR) F1 animals using splenic APC in the serial restimulations. After several in vitro restimulations T cells were transferred into nonirradiated recipients using the EAE adoptive transfer model (59). In two adoptive transfer experiments, two independently derived sets of TCL specific for mMBP Ac1-16 (TCL 1 and TCL 2) were generated from B10.PL, B10.A, B10.BR, and (B10.A × B10.BR) F1 animals. Strain B10.PL was used as a positive control for the encephalitogenicity of MBP-specific TCL. The first set of TCL (TCL 1) was restimulated nine times in vitro and the second set of TCL (TCL 2) was restimulated five times in vitro before transfer into syngeneic recipients. TCL from B10.A and B10.BR strains were also transferred into (B10.A × B10.BR) F1 animals. The severity of EAE was scored on a standard scale from 0 to 5.
The results of these adoptive transfer experiments are presented in Table 1. The positive control B10.PL TCL induced severe EAE. Both TCL from B10.BR mice were also highly encephalitogenic in syngeneic recipients and caused disease in F1 recipients. In contrast, both B10.Aderived TCL failed to induce significant disease in either B10.A or F1 recipients. F1 T cells transferred into F1 mice induced weak EAE, though marginally more severe disease than that induced by B10.A T cells. These results show that B10.A and B10.BR strains display a significant phenotypic difference in that TCL from B10.BR induce severe EAE, whereas TCL from B10.A fail to do so. Low encephalitogenicity appears to be dominant in the (B10.A × B10.BR) F1 TCL specific for MBP.
Table 1.
MBP-specific TCL from B10.A or (B10.A × B10.BR) F1 Mice Are Less Encephalitogenic than MBP-specific TCL from B10.BR Strain
| Clinical signs of EAE | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Strain | Maximum score | Day of onset | ||||||||
| Donor TCL* | No. | Recipient mice | Incidence | Average (range) | Average (range) | |||||
| B10.PL | TCL 1 | B10.PL | 5/5 | 1.9 (1–3) | 7 (5–10) | |||||
| B10.BR | TCL 1 | B10.BR | 5/5 | 2.7 (2–4) | 10.6 (7–14) | |||||
| TCL 2 | B10.BR | 5/5 | 3.4 (3–4) | 7.2 (6–10) | ||||||
| B10.BR | TCL 1 | (A × BR)F1§ | 3/5 | 1.3 (0.5–2) | 13.3 (8–17) | |||||
| TCL 2 | (A × BR)F1 | 5/5 | 3.8 (3–4) | 8.6 (7–10) | ||||||
| B10.A | TCL 1 | B10.A | 4/5‡ | 0.75 (0.5–1) | 7 (4–12) | |||||
| TCL 2 | B10.A | 4/5 | 0.5 (0.5–1) | 8 (7–15) | ||||||
| B10.A | TCL 1 | (A × BR)F1 | 3/4 | 0.66 (0.5–1) | 9 (6–11) | |||||
| TCL 2 | (A × BR)F1 | 3/5 | 0.5 | 16 (12–20) | ||||||
| (A × BR)F1 | TCL 2 | (A × BR)F1 | 4/5 | 1 (0.5–1.5) | 14.2 (9–20) | |||||
TCL 1 cells were re-stimulated nine times in vitro before transfer, using syngeneic APC for B10.PL TCL and a random mixture of B10.A and B10.BR APC for B10.A and B10.BR TCL. TCL 2 cells were restimulated five times before transfer, using syngeneic splenic APC.
One out of five B10.A mice injected with B10.A T cells died shortly after the experiment, likely due to injury or stress.
(A × BR)F1 is an abbreviation of (B10.A × B10.BR) F1.
We noted that the two B10.BR TCL differed in the severity of EAE induced in F1 recipients. B10.BR TCL 1, which had been re-stimulated with a random mixture of B10.BR and B10.A splenocytes, induced less severe EAE in (B10.A × B10.BR) F1 mice than did B10.BR TCL 2, which had been restimulated with B10.BR APC only. This suggested that the genetic origin of the APC influenced the encephalitogenicity of TCL. To test this hypothesis, additional TCL (TCL 3) were generated, in which lymph node T cells from B10.A and B10.BR mice were restimulated as separate lines with B10.A, B10.BR, or (B10.A × B10.BR) F1 splenic APC. After nine restimulations with mMBP Ac1-16 presented by splenic APC of syngeneic or semisyngeneic F1 genetic origin, the TCL were adoptively transferred into recipients. (TCL restimulated with the reciprocal parental APC did not expand sufficiently in number to transfer.) As shown in Table 2, neither B10.A TCL caused significant EAE, consistent with the results with TCL1 and TCL2. Strikingly, while B10.BR TCL restimulated with B10.BR splenic APC induced severe disease, the B10.BR TCL restimulated with F1 APC did not cause EAE. These results indicate that B10.BR splenic APC are required for the development of encephalitogenicity of B10.BR TCL and that the non-EAE-promoting phenotype of B10.A is dominant in F1 APC.
Table 2.
Encephalitogenicity of B10.BR MBP-specific T Cells Is Reduced by Re-stimulation with (B10.A × B10.BR) F1 Splenic APC
Cytokine Profiles, but Not Other Parameters Required for Encephalitogenicity, Differ among B10.A, B10.BR, and (B10.A × B10.BR) F1 TCL.
Encephalitogenic B10.BR TCL and nonencephalitogenic B10.A and F1 TCL were characterized for parameters previously shown to be required for the induction of EAE to determine which parameter is affected by the genetic differences between these strains. The activation state of TCL following stimulation with mMBP Ac1-16 was assayed by examining proliferation, cell surface levels of Mel-14, LFA-1, VEA (CD69), CD3, and CD4 antigens, and by cell cycle analysis. None of these indices of activation differed between the B10.A and B10.BR TCL; all TCL were virtually 100% CD4+ and proliferated vigorously in response to MBP Ac1-16 (not shown). The proliferative responses of all TCL were strictly mMBP Ac1-16–specific and dose-dependent. The TCL restimulated with APC of syngeneic, F1, or reciprocal parent strain origin displayed no allogeneic reactivity. The potential for homing to the central nervous system was examined by measuring cell surface levels of α4β1 integrin, VLA-4; no consistent differences were found between B10.A and B10.BR TCL. FACS® and reverse transcriptase-PCR analysis demonstrated that the predominant T cell receptor Vβ regions expressed by B10.A and B10.BR TCL were extremely similar (predominantly Vβ 4, 8, and 13), and somewhat more heterogeneous than that of the B10.PL MBP TCL (50% Vβ 8) (not shown). The levels and sequences of the Ak class II presenting element for MBP Acl-16 were also examined for strains B10.A and B10.BR. The levels of Ak expressed by splenic B cells, splenic macrophages, and peritoneal macrophages from B10.A and B10.BR mice were found to be identical by flow cytometry. Splenic cDNA for Aαk and Aβk chains were amplified from both strains and sequenced using direct PCR sequencing and were found to be identical over the entire protein coding sequence (not shown). These results indicate that the class II presenting elements, mMBP Ac1-16–specific TCR repertoire, and indices of activation are identical between B10.A and B10.BR.
In contrast, comparison of the cytokine profiles of B10.A and B10.BR TCL revealed differences which correlated with their encephalitogenicity. Data for the TCL 2 lines are shown in Fig. 1. The B10.A TCL stimulated with peptide and B10.A splenic APC displayed the Th2 phenotype, expressing very low levels of IFN-γ, IL-2, and TNF-α and high levels of IL-4 and IL-10. Levels of Th1-promoting IL-12 (made by APC) were undetectable. In contrast, the B10.BR TCL displayed the Th1 phenotype, secreting very little IL-4 and IL-10, and high levels of IFN-γ, IL-2, and TNF-α. IL-12 levels were also significantly higher. TNF-α ELISA results (not shown) were nearly identical (within 10%) to the TNF bioassay results, and as TNF biological activity was totally blocked with the anti–TNF-α mAb TN3-19.12 (see below), all of the TNF activity detected in these TCL supernatants appeared to be TNF-α. Supernatants from the (B10.A × B10.BR) F1 TCL stimulated with peptide and F1 APC contained intermediate amounts of the tested cytokines, but the levels were closer to those from B10.A than B10.BR TCL. Thus, TCL from B10.A mice produced a Th2, low TNF-α response, T cells from B10.BR produced a Th1, high TNF-α response, and the Th2, low TNF-α phenotype was dominant in the (B10.A × B10.BR) F1 T cell cultures. These results indicated that genetic difference between B10.A and B10.BR controls a regulator of Th cytokines.
Figure 1.


Cytokine levels of MBP-specific TCL stimulated overnight with syngeneic APC. Established lymph node TCL (TCL 2) from mMBP Ac116–immunized B10.A, B10.BR, and (B10.A × B10.BR) F1 mice were restimulated for the fifth time with mMBP Ac1-16 and irradiated splenic APC. After 24 h, culture supernatants were harvested. Additional cells harvested 3 d after the same fifth restimulation were used for adoptive transfer experiment EAE (see Table 1). Data presented for TNF are means and standard deviations of two bioassays performed on triplicate wells for each sample (n = 6). Data presented for IFN-γ, IL-12, IL-4, and IL-10 are means and standard deviations of triplicate wells for each sample (n = 3). Data presented for IL-2 are means and standard deviations of two IL-2 bioassays performed in duplicate wells for each sample (n = 4).
The Cytokine Profile of the B10.BR TCL Is Influenced by the Genetic Origin of the APC Used for In Vitro Restimulation.
The cytokine profiles of the third set (TCL 3) of MBP-specific T cell lines serially restimulated with splenic APC of varying genetic origins were also examined. The cytokine responses of the six TCL were determined after the third, fourth, fifth, ninth, and tenth restimulations. We found that the genetic origin of the splenic APC used for restimulations greatly influenced the cytokine profile of the B10.BR TCL. Thus, the Th1, high TNF-α phenotype of the B10.BR TCL was maintained only by syngeneic B10.BR splenic APC; very little IFN-γ, TNF-α, or IL-2 was produced by the B10.BR TCL serially restimulated with F1 or B10.A APC (Fig. 2). For IL-4, all three of the B10.A TCL had higher levels than the B10.BR TCL (Fig. 2 C), and B10.BR T cells maintained on B10.BR APC produced the lowest IL-4 levels. IL-10, B10.A, and F1 APC, but not B10.BR APC, supported levels similar to those produced by B10.A TCL (Fig. 2 D). A Th2, low TNF-α profile was seen for all B10.A TCL irrespective of the source of APC.
Figure 2.
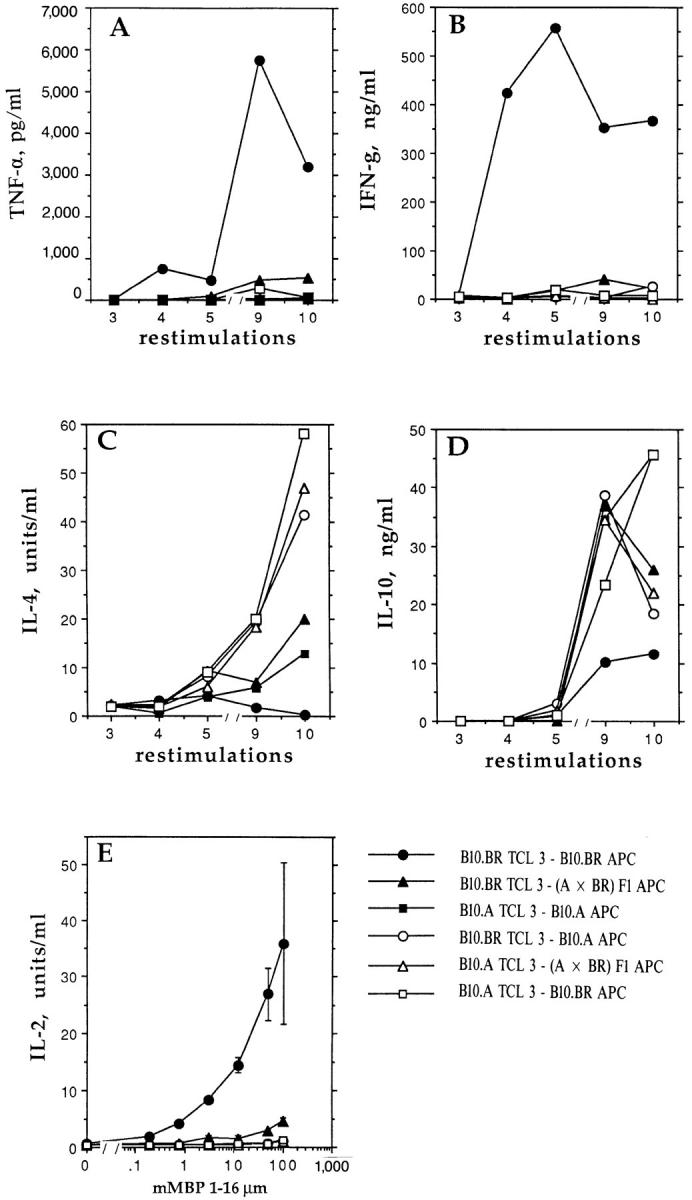
Cytokines produced by TCL (TCL 3) from B10.A and B10.BR mice are influenced by the source of APC (B10.A, B10.BR, or F1) used for in vitro restimulations. (A–D) After the third, fourth, fifth, ninth, and tenth restimulations of TCL with mMBP Ac1-16 and irradiated splenic APC of the indicated origin, T cells were restimulated for 24 h with peptide and APC of the same origin. Adoptive transfer of EAE was performed after the ninth restimulation (see Table 2). Supernatant fluids were analyzed for cytokines. In Fig. 2 D, the points for the B10.BR TCL-3–B10.A APC are 37 ng/ml of IL-10 after the ninth restimulation and 45 ng/ml of IL-10 after the tenth restimulation. Data presented are average of duplicate wells for each sample. (E) At the ninth restimulation, TCL 3 described for A–D were restimulated for 24 h with peptide and APC of the same (indicated) origin. Supernatant fluids depleted of IL-4 were analyzed for IL-2 levels, using the growth factor dependent cell line HT-2. Data presented are means and standard deviations of four wells for each sample (n = 4).
Expression of the Th1, high TNF-α phenotype only by B10.BR TCL serially restimulated with B10.BR APC correlates well with the encephalitogenicity of this TCL in adoptive transfer of EAE (Table 2). These results indicate that B10.A and F1 APC prevent the development of the Th1, high TNF-α encephalitogenic cytokine phenotype in response to MBP.
IFN-γ and TNF-α Production by Established Th1 B10.BR TCL can Be Downregulated by a Single Restimulation with B10.A APC or in the Presence of 2-h Supernatants from Activated B10.A Cultures.
To determine whether a single restimulation with B10.A APC can alter the production of IFN-γ and TNF-α by established B10.BR-derived Th1s, two independently derived Th1 MBP-specific B10.BR TCL (TCL 3 and TCL 4, both of which had been maintained with syngeneic B10.BR APC) were restimulated overnight with B10.BR or B10.A APC before harvesting supernatants for cytokine assays. As shown in Fig. 3, A–C (top bars), IFN-γ and TNF-α levels were downregulated by 40–60% when B10.A, instead of B10.BR, APC were used for the single restimulation. Similar downregulation of IFN-γ and TNF-α was observed when B10.A (but not B10.BR) T cell–depleted splenic APC were used to restimulate B10.BR TCL (Fig. 3 A, bottom bars). Irrespective of the genetic source of APC, IL-2 levels and proliferative responses remained high and IL-4 was not induced (not shown).
Figure 3.

Downregulation of B10.BR TCL IFN-γ and TNF-α production by B10.A APC or by 2-h supernatant from activated B10.A APC. (A–C) Independent experiments with MBP-specific B10.BR TCL 3 (A and B) and TCL 4. TCL were restimulated for 24 h in the presence of B10.BR or B10.A splenic APC, or splenic APC depleted of T cells (A, lower bars). In some cultures, B10.BR TCL were restimulated with B10.BR APC and MBP peptide in the presence of 2-h supernatant fluids from cultures of B10.BR or B10.A TCL stimulated with syngeneic APC and MBP peptide (B, bottom bars; C, middle bars), or in the presence of 2-h supernatants from B10.BR or B10.A spleen cells stimulated with LPS (C, bottom bars). Data presented are means and standard deviations of triplicate wells for each sample (n = 3). All experiments were repeated at least three times with similar results.
To determine whether the inhibitory activity of B10.A APC on IFN-γ and TNF-α levels was mediated by a soluble factor, the two B10.BR TCL were restimulated with mMBP Ac1-16 and B10.BR splenic APC in the presence of supernatant fluids from a culture of B10.A TCL stimulated with B10.A splenic APC and mMBP Ac1-16 for 2 h. The 2-h time point was chosen because most cytokines induced by activation of T cells and APC are not detectable at this time point, except TNF-α and -β (60, 61; and our unpublished data). The B10.A culture-derived 2-h supernatant downregulated IFN-γ and TNF-α production by at least 50% relative to control cultures with 2-h supernatants from B10.BR T cells and APC plus peptide (Fig. 3 B, bottom bars, and C, middle bars), comparable to the effects of B10.A APC themselves. Similar results were obtained with B10.BR TCL 4 stimulated with B10.BR splenic APC and MBP peptide in the presence of supernatants from freshlyisolated B10.A and B10.BR spleen cells activated for 2 h with LPS (Fig. 3 C, bottom bars). Thus, downregulation of IFN-γ and TNF-α production seems to be mediated by a soluble factor secreted by B10.A splenic APC within the first 2 h after activation by antigen-stimulated T cells or by LPS. However, a single restimulation of B10.BR TCL with B10.A splenic APC or in the presence of 2-h supernatant from B10.A cultures did not induce any IL-4 expression in B10.BR T cells (see below). B10.A TCL remained Th2, low TNF-α producing cells irrespective of the genetic source of APC or 2 h supernatant (not shown and see below).
The B10.A-derived Soluble Cytokine Regulatory Factor Functions when Ak-expressing L Cell Fibroblasts Are Used as APC.
To begin to clarify the target of action of the B10.A-derived regulatory factor, B10.BR or B10.A T cells (TCL 4) were restimulated twice with mMBP Ac1-16 presented by nonprofessional APC (Ak-expressing L cells which do not produce splenic APC-derived cytokines), in the presence of 2-h supernatant from B10.A cultures. In either normal medium or in the 2-h B10.BR supernatant, B10.BR T cells produced high levels of IFN-γ and TNF-α, and B10.A T cells produced high levels of IL-4 with little or no TNF-α and IFN-γ. However, the levels of IFN-γ and TNF-α produced by B10.BR TCL were reduced in the presence of 2-h supernatant from B10.A cultures, similar to the effects seen when professional splenic APC were used (Fig. 4). This result indicates that B10.A-derived factor does not require the presence of splenic APC for its action and probably acts directly on the responding T cells.
Figure 4.

Supernatant from B10.A T cells activated with B10.A APC for 2 h downregulates IFN-γ and TNF-α production by B10.BR Th1 cells when Ak-expressing L cell fibroblasts are used as APC. B10.BR (top bars) or B10.A (bottom bars) TCL 4 were activated with mMBP Ac1-16 and Ak-expressing L cell fibroblasts as APC for two consecutive restimulations in either normal medium or in 2-h supernatants from B10.BR or B10.A T cells activated with syngeneic splenic APC. Culture supernatants were harvested 24 h after restimulation and IFN-γ, TNF-α, and IL-4 were measured as above. Data presented are means and standard deviations of triplicate wells for each sample (n = 3). Similar results were obtained in two additional experiments.
The B10.A-derived Factor Acts Independently of Known Th1/Th2 Regulatory Cytokines in Downregulating B10.BR Production of IFN-γ and TNF-α.
To assess whether downregulation of IFN-γ and TNF-α by B10.A APC is mediated by or depends on any of the known Th1/Th2 regulators, 2-h culture supernatants derived from B10.BR or B10.A TCL/APC cultures were depleted of IL-4, IL-10, IL-12p40, IL-13, TNF-α, IFN-γ, and TGF-β and tested for their effects on IFN-γ and TNF-α production by B10.BR TCL 4 restimulated with B10.BR APC and mMBP peptide. Cytokine depletion did not diminish the downregulatory activity of the B10.A culture supernatant (Fig. 5, A and B). Serial dilutions of B10.A and B10.BR 2-h culture supernatants (Fig. 5 C) demonstrated that the downregulatory activity of B10.A-derived factor is rapidly lost by dilution, indicating that any depletion of the downregulatory cytokine would have been detected. Essentially identical results were obtained when the anti-cytokine antibodies were also used at half the concentrations (not shown).
Figure 5.

Downregulation of IFN-γ and TNF-α levels is not affected when 2-h B10.A culture supernatant is depleted of known Th1/Th2 cytokine regulators. (A and B) B10.BR or B10.A T cells (TCL 4) were restimulated with mMBP Ac1-16 and syngeneic splenic APC for 2 h. Neutralizing antibodies specific for mouse IL-4, IL-10, TNF-α, IFN-γ, TGF-β, IL-13, and IL-12p40 were added to these culture supernatants for 30 min, and antibody–cytokine complexes were removed using protein G–coated agarose beads. Cytokine depleted 2-h supernatants were then used as media in which B10.BR TCL 4 were restimulated with mMBP Ac1-16 and B10.BR splenic APC for 24 h. IFN-γ (A) and TNF-α (B) levels were measured as above. Lower levels of TNF-α after treatment with anti– TNF-α antibody and protein G beads were due to the presence of residual anti–TNF-α antibody, which has low affinity for protein G in medium with serum. Neutralizing antibodies were used at concentrations equal to (not shown) and twice what were recommended by the manufacturers for complete neutralization of cytokine levels produced by high expressing cells, giving similar results. Data presented are means and standard deviations of triplicate wells for each sample (n = 3). (C) B10.BR TCL 4 cells were restimulated with B10.BR splenic APC and mMBP Ac1-16 in 2-h culture supernatants from B10.A or B10.BR TCL 4, used either undiluted or at serial 1:2 dilutions in RPMI, 10% FCS. TNF-α levels are means and standard deviations of triplicate wells for each sample (n = 3).
As summarized in Fig. 6, blocking or adding IL-4 and IL-12 (alone or in combinations) did not reduce the downregulatory effects of B10.A APC on TNF-α levels, although there were effects on the absolute levels of TNF-α production. In contrast, adding IFN-γ significantly reduced the downregulation by B10.A APC. For IFN-γ production, adding TNF-α did not prevent the downregulation of IFN-γ by B10.A APC, and adding IL-4 did not synergize with this downregulation (Fig. 6 B). As expected because of their pro-Th1 effects, addition of IL-12 alone, or in combination with anti-IL-4, did increase the overall IFN-γ levels and reduced the downregulation of IFN-γ by B10.A APC.
Figure 6.

Modulation of downregulatory activity by addition or neutralization of known cytokine regulators during restimulation of B10.BR T cells with either B10.BR or B10.A splenic APC. B10.BR T cells (TCL 4) were restimulated with mMBP Ac1-16 and either B10.BR or B10.A splenic APC for 24 h in the absence or presence of mouse recombinant IL-4, IFN-γ, TNF-α, and IL-12 alone or combination with neutralizing antibody specific for mouse IL-12, or mouse IL-4. Cytokine levels resulting from each treatment are expressed as a percentage of control (B10.BR TCL, B10.BR APC, and peptide with no cytokine or antibody additions). Data presented are means and standard deviations of triplicate wells for each sample (n = 3). At least two independent experiments were performed for each treatment, yielding similar results.
In the experiments shown in Figs. 5 and 6, none of the cytokines or anti-cytokine antibodies induced detectable levels of IL-4 production by the B10.BR TCL (not shown). Overall the results shown in Figs. 5 and 6 suggest that the B10.A-derived factor acts independently of known cytokine regulators, and that strong pro-Th1 conditions can overcome its downregulatory effect on TNF-α and IFN-γ.
B10.A APC Downregulate IFN-γ and TNF-α Production by a Th1 Clone with Different Antigen and MHC Restriction Specificities.
The cytokines produced by the B10.BRderived, KLH-specific, Ek-restricted Th1 clone BR E7 (62) were assayed after two consecutive restimulations with B10.BR or B10.A splenic APC. As shown in Fig. 7, presentation of KLH by B10.A APC results in the progressive reduction of IFN-γ and TNF-α levels produced by this Th1 clone. Thus, the B10.A APC-derived factor acts independently of the specificity of the Th1 and is able to downregulate IFN-γ and TNF-α production by a fully committed clonal Th1 population.
Figure 7.

Downregulatory effects of B10.A splenic APC on IFN-γ and TNF-α production by a B10.BR-derived, KLH-specific, Ek-restricted T cell clone. The BR E7 T cell clone was activated with KLH and either B10.BR or B10.A splenic APC for two consecutive restimulations. Culture supernatants were harvested and assayed for TNF-α (A) and IFN-γ (B) 24 h after stimulation. Data presented are means and standard deviations of triplicate wells for each sample (n = 3). Similar results were ovserved in at least two additional experiments.
Discussion
This paper presents evidence for a genetic difference(s) between inbred strains B10.A and B10.BR that act(s) in APC to control Th1/Th2 cytokine profiles and the encephalogenicity of MBP-Ac1-16–specific T cells. The regulatory effects are manifested both at the time of commitment to Th1/Th2 phenotypes as B10.A APC block the development of IL-2, IFN-γ, and TNF-α production, and in the production of IFN-γ and TNF-α by committed Th1s as a single restimulation with B10.A-derived APC or in the presence of B10.A-derived supernatant downregulates the production of these cytokines by established Th1 B10.BR cells. The downregulation of IFN-γ and TNF-α production by a B10.BR-derived, KLH-specific, Ek-restricted T cell clone when restimulated by KLH presented by B10.A APC demonstrates that the regulatory effects of B10.Aderived factor (a) are not limited to responses of MBP-specific, Ak-restricted Th1s, and (b) can act by affecting individual clonal populations of mature Th1s rather than by expanding uncommitted precursors or Th0 cells. The induction of comparable proliferative and IL-2 responses in Th1 B10.BR TCL by MBP peptides presented by either B10.A or B10.BR APC indicates that B10.A APC do not inhibit the activation of these T cells, but downregulate production of IFN-γ and TNF-α. The target of the factor appears to be the responding T cells, as the downregulatory effects of the B10.A supernatants are observed when MBP peptide is presented to B10.BR T cells by nonprofessional APC, Ak-expressing L cell fibroblasts. B10.A TCL maintain a Th2, low TNF-α cytokine profile irrespective of the genetic source of the APC or 2-h supernatant, suggesting that they had become committed in vivo, perhaps due to the pro-Th2 effects of their own APC. The Th2-inducing phenotype of B10.A is dominant in (B10.A × B10.BR) F1 cells.
The identity of the APC cell type(s) within the irradiated B10.A splenocyte population that is responsible for the downregulation of IFN-γ and TNF-α production has not yet been determined. Splenic T cells do not seem to be required for the downregulation. Preliminary results indicate that B10.A-derived splenic macrophages have downregulatory activity, while splenic B cells do not (not shown). While the results to date indicate that the downregulation of IFN-γ and TNF-α production by B10.BR Th1s is mediated by B10.A APC, we have not ruled out the possibility that activated B10.A T cells may also produce the downregulatory factor.
The B10.A-derived soluble factor that modulates Th cytokine production appears to be novel. The results of adding or blocking cytokines implicated in regulating Th1/ Th2 cytokine production indicate that the downregulatory effects of this factor probably do not act through or require IL-4, IL-10, IL-13, IL-12p40, TGF-β, IFN-γ, or TNF-α. The B10.A-derived factor also acts independently of prostaglandin E2, an APC-derived molecule known to inhibit production of IFN-γ by activated CD4+ T cells (63, 64), indicated by the failure of indomethacin to block downregulation of IFN-γ and TNF-α production by B10.A APC (not shown). The activity of this factor in downregulating IFN-γ production also differs from that of IL-4 in that IL-4, but not the factor, blocks IFN-γ induction by IL-12 through its inhibition of signaling through the IL-12 receptor (21).
The finding that the downregulatory effects of the B10.Aderived factor on Th1 IFN-γ production is overcome by exogenous IFN-γ and by IL-12, especially in combination with anti–IL-4, suggests that the in vivo effects of the B10.A-derived factor should be most evident in the absence of strong pro-Th1 conditions. This may occur during initial antigen priming, especially when T cell responses are weak, e.g., with poor antigens such as the MBP Ac1-16 peptide, and perhaps more generally with self antigens. The apparent direct effect of B10.A APC and of the secreted cytokine regulatory factor on IFN-γ production by mature Th1s, even when IL-4 has been depleted, suggests that inhibition of IFN-γ production may be the primary mechanism by which the B10.A-derived factor skews initial Th1/ Th2 subset determination. When T cells are strongly activated, or where significant levels of IL-12 and/or IFN-γ are already present, the B10.A-derived factor may not downregulate IFN-γ production.
This may help to explain why the factor's anti-Th1 regulatory effects in B10.A mice may not have been observed in earlier studies of Leishmania susceptibility in inbred strains, which showed B10.A to be resistant (44), or in in vitro studies of Th1/Th2 commitment of T cells from B10.A TCR transgenic mice (19). Unlike IFN-γ, downregulation of TNF-α is maintained in the presence of exogenous IL-12, implying IFN-γ and TNF-α are regulated independently.
The basis for the difference between B10.A and B10.BR APC in downregulatory factor production remains to be determined. The gene for the factor itself could be polymorphic, resulting in differences in the levels or activity of the factor produced. Alternatively, the polymorphism could be in gene(s) that indirectly affect factor activity by controlling its synthesis, modification, or secretion. Initial analysis of the genetics downregulatory activity have yielded surprising results. We anticipated that the gene would be H-2linked, as B10.A and B10.BR are H-2 congenic strains on the C57BL/10 background; the H-2k haplotype of B10.BR was derived from C57BR and the H-2a recombinant haplotype of B10.A (k haplotype in the H-2K-Ea region, d haplotype telomeric to Ea) was derived from strain A/WySn (65, 66). However, preliminary tests of (B10.A × B10.BR) F2 progeny and (F1 × B10.BR) backcross progeny, in which their splenic APC were assayed for the ability to downregulate TNF-α production by MBP-specific B10.BR TCL, suggest that the polymorphic gene(s) controlling factor production is (are) not linked to H-2. If confirmed, this would indicate that strains B10.A and B10.BR also differ by a non-H-2 locus or loci. Splenic APC from A strain subline A/WySn and from the A strain background H-2 congenic A.TL downregulate IFN-γ and TNF-α production by B10.BR TCL in response to MBP Ac1-16. Thus, the production of the downregulatory factor by B10.A appears to be due to a contaminating non-H-2 gene(s) derived from the A/WySn donor of the H-2a haplotype, despite the 10 backcrosses to the C57BL/10 inbred partner during the initial derivation of B10.A (65).
The genetic and phenotypic properties of the B10.Aderived factor clearly distinguish it from the other major polymorphic gene previously shown to regulate Th1/Th2 cytokine production. Described initially in the Leishmania system, this polymorphic gene regulates resistance (associated with Th1 responses) to L. major (reviewed in 47), and more recently has been shown to regulate cytokine responses to other antigens (44, 49, 67). The Leishmania resistance gene acts in T cells to regulate their Th1/Th2 cytokine phenotypes (67, 47), whereas the results reported here show that the B10.A-derived factor acts in the APC used for T cell activation. Also, for Leishmania, most inbred strains, including B10.A and A strains, have the resistant (Th1) phenotype (44). Finally, in the Leishmania system, the resistant (Th1) phenotype is largely dominant in heterozygotes (47, 68), whereas for the B10.A-derived factor discussed here, the Th2-promoting phenotype is dominant or co-dominant in (B10.A × B10.BR) F1 mice.
While the effects of the B10.A-derived factor have been characterized in only a few mouse strains, this gene may represent a previously identified or a new member of a growing number of polymorphic genes known to influence susceptibility to autoimmune diseases (48). Additional studies are underway to characterize more fully the genetics and mechanisms of action of this novel cytokine regulatory gene. Identification of the soluble cytokine regulatory factor controlled by the B10.A-derived gene, its cellular origin, and its mode of action, should contribute to deciphering the mechanisms regulating Th1s and Th2s and their pathogenicity in autoimmune, inflammatory, and allergic disorders.
Acknowledgments
The authors thank Cathy Carswell-Crumpton for flow cytometry, Hugh McDevitt, Beth Moore, and Stacey Edelman for helpful discussion, and Mike Conboy for comments on the manuscript and helpful discussion.
Footnotes
This work was supported by Multiple Sclerosis Society grant PP0454 to P.P. Jones and National Institutes of Health grants AI19512 to P.P. Jones, AI24571 to R.H. DeKruyff, and AI26322 to D.T. Umetsu. I. Conboy was supported by NIH training grants GM07276 and HD07249.
1 Abbreviations used in this paper: EAE, experimental autoimmune encephalomyelitis; LAk, L cell fibroblast expressing Ak class II proteins; MBP, myelin basic protein; mMBP, mouse MBP; TCL, T cell line.
References
- 1.Mosmann TR, Coffman RL. Two types of mouse helper T-cell clone: implications for immune regulation. Immunol Today. 1987;8:223–226. doi: 10.1016/0167-5699(87)90171-X. [DOI] [PubMed] [Google Scholar]
- 2.Umetsu DT, Jabara HH, DeKruyff RH, Abbas AK, Abrams JS, Geha RS. Functional heterogeneity among human inducer T cell clones. J Immunol. 1988;140:4211–4216. [PubMed] [Google Scholar]
- 3.Mosmann TR, Sad S. The expanding universe of T-cell subsets: Th1, Th2 and more. Immunol Today. 1996;17:138–146. doi: 10.1016/0167-5699(96)80606-2. [DOI] [PubMed] [Google Scholar]
- 4.Mosmann TR, Coffman RL. Heterogeneity of cytokine secretion patterns and function of helper T cells. Adv Immunol. 1989;46:111–147. doi: 10.1016/s0065-2776(08)60652-5. [DOI] [PubMed] [Google Scholar]
- 5.Parronchi P, Macchia D, Piccinni M-P, Biswas P, Simonelli C, Magi E, Rocci M, Ansari AA, Romagnani S. Allergen- and bacterial antigen-specific T-cell clones established from atopic donors show a different profile of cytokine production. Proc Natl Acad Sci USA. 1991;88:4538–4542. doi: 10.1073/pnas.88.10.4538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Durham SR, Ying S, Varney VA, Jacobson MR, Sudderick RM, Mackay IS, Kay AB, Hamid QA. Cytokine messenger RNA expression for IL-3, IL-4, IL-5, and granulocyte/macrophage-colony–stimulating factor in the nasal mucosa after local allergen provocation: relationship to tissue eosinophilia. J Immunol. 1992;148:2390–2394. [PubMed] [Google Scholar]
- 7.Ohmen JD, Hanifin J, Nickoloff BJ, Rea TH, Wyzykowski R, Kim J, Jullien D, McHugh T, Nassif AS, Chan SC, et al. Overexpression of IL-10 in atopic dermatitis: contrasting cytokine patterns with delayed-type hypersensitivity reactions. J Immunol. 1995;154:1956–1963. [PubMed] [Google Scholar]
- 8.Liblau RS, Singer SM, McDevitt HO. Th1 and Th2 CD4+ T cells in the pathogenesis of organ-specific autoimmune diseases. Immunol Today. 1995;16:34–38. doi: 10.1016/0167-5699(95)80068-9. [DOI] [PubMed] [Google Scholar]
- 9.Seder RA, Gazzinelli R, Sher A, Paul WE. Interleukin 12 acts directly on CD4+ T cells to enhance priming for interferon γ production and diminishes interleukin 4 inhibition of such priming. Proc Natl Acad Sci USA. 1993;90:10188–10192. doi: 10.1073/pnas.90.21.10188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu CY, Demeure C, Kiniwa M, Gately M, Delespesse G. IL-12 induces the production of IFN-γ by neonatal human CD4 T cells. J Immunol. 1993;151:1938–1949. [PubMed] [Google Scholar]
- 11.Manetti R, Parronchi P, Giudizi MG, Piccinni MP, Maggi E, Trinchieri G, Romagnani S. Natural killer cell stimulatory factor interleukin 12 (IL-12) induces T helper type 1 (Th1)-specific immune responses and inhibits the development of IL-4–producing Th cells. J Exp Med. 1993;177:1199–1204. doi: 10.1084/jem.177.4.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Manetti R, Gerosa F, Guidici MG, Biagotti R, Parronchi P, Piccinni M-P, Sampognato S, Maggi E, Romagnani S, Trinchieri G. Interleukin 12 induces stable priming for interferon γ (IFN-γ) production during differentiation of human T helper (Th) cells and transient IFN-γ production in established Th2 cell clones. J Exp Med. 1994;179:1273–1283. doi: 10.1084/jem.179.4.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murphy EE, Terres G, Macatonia SE, Hsieh C-S, Mattison J, Lanier L, Wysocka M, Trinchieri G, Murphy K, O'Garra A. B7 and interleukin 12 cooperate for proliferation and interferon γ production by mouse T helper cell 1 clones that are unresponsive to B7 costimulation. J Exp Med. 1994;180:223–231. doi: 10.1084/jem.180.1.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kubin M, Kamoun M, Trinchieri G. Interleukin 12 synergizes with B7/CD28 interaction in inducing efficient proliferation and cytokine production of human T cells. J Exp Med. 1994;180:211–222. doi: 10.1084/jem.180.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Flesch IEA, Hess JH, Huang S, Arguet M, Rothe J, Bluethmann H, Kaufmann SHE. Early interleukin 12 production by macrophages in response to mycobacterial infection depends on interferon γ and tumor necrosis factor α. J Exp Med. 1995;181:1615–1621. doi: 10.1084/jem.181.5.1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fiorentino DF, Bond MW, Mosmann TR. Two types of mouse helper T cell. IV. Th2 clones secrete a factor that inhibits cytokine production by Th1 clones. J Exp Med. 1989;170:2081–2095. doi: 10.1084/jem.170.6.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fiorentino DF, Zlotnik A, Vieira P, Mosmann TR, Howard M, Moore KW, O'Garra A. IL-10 acts on the antigen-presenting cell to inhibit cytokine production by Th1 cells. J Immunol. 1991;146:3444–3451. [PubMed] [Google Scholar]
- 18.Hsieh G-S, Heimberger AB, Gold JS, O'Garra A, Murphy K. Differential regulation of T helper phenotype development by IL-4 and IL-10 in an αβ-transgenic system. Proc Natl Acad Sci USA. 1992;89:6065–6069. doi: 10.1073/pnas.89.13.6065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seder RA, Paul WE, Davis MM, Groth BF-S. The presence of interleukin 4 during in vitro priming determines the lymphokine-producing potential of CD4+T cells from T cell receptor transgenic mice. J Exp Med. 1992;176:1091–1098. doi: 10.1084/jem.176.4.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mocci S, Coffman RL. Induction of a Th2 population from a polarized Leishmania-specific Th1 population by in vitro culture with IL-4. J Immunol. 1995;154:3779–3787. [PubMed] [Google Scholar]
- 21.Szabo SJ, Jacobson NG, Dighe AS, Gubier U, Murphy KM. Developmental commitment to the Th2 lineage by extinction of IL-12 signaling. Immunity. 1995;2:665–675. doi: 10.1016/1074-7613(95)90011-x. [DOI] [PubMed] [Google Scholar]
- 22.de Waal Malefyt, R., C.G. Fidgor, R. Huijbens, S. MohanPeterson, B. Bennett, J. Culpepper, W. Dang, G. Zurawski, and J.E. de Vries. Effects of IL-13 on phenotype, cytokine production, and cytotoxic function of human monocytes. Comparison with IL-4 and modulation by IFN-gamma or IL-10. J Immunol. 1993;151:6370–6381. [PubMed] [Google Scholar]
- 23.Doherty TM, Kastelein R, Menon S, Andrade S, Coffman RL. Modulation of murine macrophage function by IL-13. J Immunol. 1993;151:7151–7160. [PubMed] [Google Scholar]
- 24.Cosentino G, Soprana E, Thienes CP, Siccardi AG, Viale G, Vercelli D. IL-13 downregulates CD14 expression and TNF-alpha secretion in normal human monocytes. J Immunol. 1995;155:3145–3151. [PubMed] [Google Scholar]
- 25.Mattner F, Fischer S, Guckes S, Jin S, Kaulen H, Schmitt E, Rude E, Germann T. The interleukin12 subunit p40 specifically inhibits effects of the interleukin12 heterodimer. Eur J Immunol. 1993;23:2202–2208. doi: 10.1002/eji.1830230923. [DOI] [PubMed] [Google Scholar]
- 26.Gillessen S, Carvajal D, Ling P, Podlaski FJ, Stremly DL, Familletti PC, Gubler U, Presky D, Stern AS, Gately MK. Mouse interleukin-12 (IL-12) p40 homodimer: a potent IL-12 antagonist. Eur J Immunol. 1995;25:200–206. doi: 10.1002/eji.1830250133. [DOI] [PubMed] [Google Scholar]
- 27.Chen Y, Kuchroo VK, Inobe J, Hafler DA, Weiner HL. Regulatory T cell clones induced by oral tolerance: suppression of autoimmune encephalomyelitis. Science (Wash DC) 1994;265:1237–1240. doi: 10.1126/science.7520605. [DOI] [PubMed] [Google Scholar]
- 28.Swain S, Huston LG, Tonkonogy S, Weinberg A. Transforming growth factor–beta and IL-4 cause helper T cell precursors to develop into distinct effector helper cells that differ in lymphokine secretion pattern and cell surface phenotype. J Immunol. 1991;147:2991–3000. [PubMed] [Google Scholar]
- 29.Nagelkerken L, Gollob KJ, Tielemans M, Coffman RL. Role of transforming growth factor–β in the preferential induction of T helper cells of type 1 by staphylococcal enterotoxin B. Eur J Immunol. 1993;23:2306–2310. doi: 10.1002/eji.1830230938. [DOI] [PubMed] [Google Scholar]
- 30.Hoehn P, Goedert S, Germann T, Koelsch S, Jin S, Palm N, Ruede E, Schmitt E. Opposing effects of TGF-β2on the Th1 cell develpment of naive CD4+ T cells isolated from different mouse strains. J Immunol. 1995;155:3788–3793. [PubMed] [Google Scholar]
- 31.Murray JS, Madri J, Tite J, Carding SR, Bottomly K. MHC control of CD4+T cell subset activation. J Exp Med. 1989;170:2135–2140. doi: 10.1084/jem.170.6.2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Constant S, Pfeiffer C, Woodard A, Pasqualini T, Bottomly K. Extent of T cell receptor ligation can determine the functional differentiation of naive CD4+T cells. J Exp Med. 1995;182:1591–1596. doi: 10.1084/jem.182.5.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aubry JP, Pochon S, Graber P, Jansen KU, Bonnefoy JY. CD21 is a ligand for CD23 and regulates IgE production. Nature (Lond) 1992;358:505–507. doi: 10.1038/358505a0. [DOI] [PubMed] [Google Scholar]
- 34.Weinberg AD, Wallin JJ, Jones RE, Sullivan TJ, Bourdette DN, Vandenbark AA, Offner H. Target organ–specific up-regulation of the MRC OX-40 marker and selective production of Th1 lymphokine mRNA by encephalitogenic T helper cells isolated from the spinal cords of rats with experimental autoimmune encephalitomyelitis. J Immunol. 1994;152:4712–4721. [PubMed] [Google Scholar]
- 35.Del Prete G, Maggi E, Pizzolo G, Romagnani S. CD30, Th2 cytokines and HIV infection: a complex and fascinating link. Immunol Today. 1995;16:76–80. doi: 10.1016/0167-5699(95)80092-1. [DOI] [PubMed] [Google Scholar]
- 36.Elson LH, Nutman TB, Metcalfe DD, Prussin C. Flow cytometric analysis for cytokine production identifies T helper 1, T helper 2, and T helper 0 cells within human CD4+CD27− lymphocyte population. J Immunol. 1995;154:4294–4301. [PubMed] [Google Scholar]
- 37.Kuchroo VK, Das MP, Brown JA, Ranger AM, Zamvil SS, Sobel RA, Weiner HL, Nabavi N, Glimcher LH. B7-1 and B7-2 costimulatory molecules activate differentially the Th1/Th2 developmental pathways: application to autoimmune disease theory. Cell. 1995;80:707–718. doi: 10.1016/0092-8674(95)90349-6. [DOI] [PubMed] [Google Scholar]
- 38.Kamanaka M, Yu P, Yasui T, Yoshida K, Kawabe T, Horii T, Kishimoto T, Kikutani H. Protective role of CD40 in Leishmania majorinfection at two distinct phases of cell-mediated immunity. Immunity. 1996;4:275–281. doi: 10.1016/s1074-7613(00)80435-5. [DOI] [PubMed] [Google Scholar]
- 39.Campbell K, Ovendale PJ, Kennedy MK, Fanslow WC, Reed SG, Maliszewski CR. CD40 ligand is required for protective cell-mediated immunity to Leishmania major . Immunity. 1996;4:283–289. doi: 10.1016/s1074-7613(00)80436-7. [DOI] [PubMed] [Google Scholar]
- 40.Soong L, Xu J-C, Grewal IS, Kima P, Sun J, Longley BJ, Jr, Ruddle NH, McMahon-Pratt D, Flavell RA. Disruption of CD40-CD40 ligand interactions results in an enhanced susceptibility to Leishmania amazonensis infection. Immunity. 1996;4:263–273. doi: 10.1016/s1074-7613(00)80434-3. [DOI] [PubMed] [Google Scholar]
- 41.Secrist H, DeKruyff RH, Umetsu DT. Interleukin 4 production by CD4+T cells from allergic individuals is modulated by antigen concentration and antigen-presenting cell type. J Exp Med. 1995;181:1081–1089. doi: 10.1084/jem.181.3.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Macatonia S, Hosken NA, Litton M, Vieira P, Hsieh C-S, Culpepper JA, Wysocka M, Trinchieri G, Murphy KM, O'Garra A. Dendritic cells produce IL-12 and direct the development of Th1 cells from naive CD4+ T cells. J Immunol. 1995;154:5071–5079. [PubMed] [Google Scholar]
- 43.Stockinger B, Zal T, Zal A, Gray D. B cells solicit their own help from T cells. J Exp Med. 1996;183:891–899. doi: 10.1084/jem.183.3.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Howard JG, Hale C, Chan-Liew WL. Immunological regulation of experimental cutaneous leishmaniasis. I. Immunogenetic aspects of susceptibility to Leishmania tropicain mice. Parasite Immunol (Oxf) 1980;2:303–314. doi: 10.1111/j.1365-3024.1980.tb00061.x. [DOI] [PubMed] [Google Scholar]
- 45.Fritz RB, Skeen MJ, Chou C-HJ, Garcia M, Egorov IK. Major histocompatibility complex–linked control of the murine immune response to myelin basic protein. J Immunol. 1985;134:2328–2332. [PubMed] [Google Scholar]
- 46.Fritz RB, Mcfarlin DE. Encephalitogenic epitopes of myelin basic protein. Chem Immunol. 1989;46:101–125. [PubMed] [Google Scholar]
- 47.Reiner SL, Locksley RM. The regulation of immunity to Leishmania major . Annu Rev Immunol. 1995;12:151–177. doi: 10.1146/annurev.iy.13.040195.001055. [DOI] [PubMed] [Google Scholar]
- 48.Vyse TJ, Todd JA. Genetic analysis of autoimmune disease. Cell. 1996;85:311–318. doi: 10.1016/s0092-8674(00)81110-1. [DOI] [PubMed] [Google Scholar]
- 49.DeKruyff RH, Fang Y, Umetsu DT. IL-4 synthesis by in vivo primed keyhole limpet hemocyanin-specific CD4+ T cells. Influence of antigen concentration and antigen-presenting cell type. J Immunol. 1992;149:3468–3476. [PubMed] [Google Scholar]
- 50.Sher A, Coffman RL. Regulation of immunity to parasites by T cells and T cell-derived cytokines. Annu Rev Immunol. 1992;10:385–409. doi: 10.1146/annurev.iy.10.040192.002125. [DOI] [PubMed] [Google Scholar]
- 51.Guler ML, Gorham JD, Hsieh C-S, Mackey AJ, Steen RG, Dietrich WF, Murphy KM. Genetic susceptibility to Leishmania: IL-12 responsiveness in Th1 cell development. Science (Wash DC) 1996;271:984–987. doi: 10.1126/science.271.5251.984. [DOI] [PubMed] [Google Scholar]
- 52.Powell MB, Mitchell D, Lederman J, Buhmeier J, Zamvil S, Graham G, Ruddle NH, Steinman L. Lymphotoxin and tumor necrosis factor-α production by myelin basic protein-specific T cell clones correlates with encephalitogenicity. Int Immunol. 1990;2:539–544. doi: 10.1093/intimm/2.6.539. [DOI] [PubMed] [Google Scholar]
- 53.Ruddle NH, Bergman CM, McGrath KM, Lingenheld EG, Grunnet ML, Padula SJ, Clark RB. An antibody to lymphotoxin and tumor necrosis factor prevents transfer of experimental allergic encephalomyelitis. J Exp Med. 1990;172:1193–1200. doi: 10.1084/jem.172.4.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van der Veen RC, Stohlman SA. Encephalitogenic Th1 cells are inhibited by Th2 cells with related peptide specificity: relative roles of interleukin IL-4 and IL-10. J Neuroimmunol. 1993;48:213–220. doi: 10.1016/0165-5728(93)90194-4. [DOI] [PubMed] [Google Scholar]
- 55.Olsson T. Critical influences of the cytokine orchestration on the outcome of myelin antigen-specific T-cell autoimmunity in experimental autoimmune encephalomyelitis and multiple sclerosis. Immunol Rev. 1995;144:245–268. doi: 10.1111/j.1600-065x.1995.tb00072.x. [DOI] [PubMed] [Google Scholar]
- 56.Baron JL, Madri JA, Ruddle NH, Hashim G, Janeway CA., Jr Surface expression of α4 integrin by CD4+T cells is required for their entry into brain parenchyma. J Exp Med. 1993;177:57–68. doi: 10.1084/jem.177.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Branch DR, Shan A, Guilbert LJ. A specific and reliable bioassay for the detection of femtomolar levels of human and murine tumor necrosis factors. J Immunol Methods. 1991;143:251–261. doi: 10.1016/0022-1759(91)90050-p. [DOI] [PubMed] [Google Scholar]
- 58.Tate KM, Lee C, Edelman S, Carswell-Crumpton C, Liblau R, Jones PP. Interactions among polymorphic and conserved residues in MHC class II proteins affect MHC–peptide conformation and T cell recognition. Intl Immunol. 1995;7:747–761. doi: 10.1093/intimm/7.5.747. [DOI] [PubMed] [Google Scholar]
- 59.Zamvil S, Nelson P, Trotter J, Mitchell D, Knobler R, Fritz R, Steinman L. T-cell clones specific for myelin basic protein induce chronic relapsing paralysis and demyelination. Nature (Lond) 1985;317:355–358. doi: 10.1038/317355a0. [DOI] [PubMed] [Google Scholar]
- 60.Openshaw P, Murphy EE, Hosken NA, Maino V, Davis K, Murphy K, O'Garra A. Heterogeneity of intracellular cytokine synthesis at the single-cell level in polarized T helper 1 and T helper 2 populations. J Exp Med. 1995;182:1357–1367. doi: 10.1084/jem.182.5.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Millet I, Ruddle NH. Differential regulation of lymphotoxin (LT), lymphotoxin-β (LT-β), and TNF-α in murine T cell clones activated through the TCR. J Immunol. 1994;152:4336–4346. [PubMed] [Google Scholar]
- 62.DeKruyff RH, Ju S, Mosmann T, Hunt AJ, Umetsu DT. Induction of antigen specific responses in primed and unprimed B cells: functional heterogeneity among Th1 and Th2 T cell clones. J Immunol. 1989;142:2575–2582. [PubMed] [Google Scholar]
- 63.Katamura K, Shintaku N, Yamauchi Y, Fukui T, Ohshima Y, Mayumi M, Furusho K. Prostaglandin E2at priming of naive CD4+ T cells inhibits acquisition of ability to produce IFN-γ and IL-2, but not IL-4 and IL-5. J Immunol. 1995;155:4604–4612. [PubMed] [Google Scholar]
- 64.Hilkens CMU, Snijders A, Vermeulen H, van der Meide PH, Wierenga EA, Kapsenberg ML. Accessory cell-derived IL-12 and prostaglandin E2determine the IFN-γ level of activated human CD4+ T cells. J Immunol. 1996;156:1722–1727. [PubMed] [Google Scholar]
- 65.Stimpfling JH, Reichert AE. Strain C57BL/ 10ScSn and its congenic resistant sublines. Transplant Proc. 1970;2:39–47. [PubMed] [Google Scholar]
- 66.Klein, J. 1989. Congenic and segregating inbred strains. In Genetic Variants and Strains of the Laboratory Mouse. M.F. Lyon and A.G. Searle, editors. Oxford University Press, New York. 797–825.
- 67.Hsieh C-S, Macatonia SE, O'Garra A, Murphy KM. T cell helper background determines default T helper phenotype in vitro. J Exp Med. 1995;181:713–721. doi: 10.1084/jem.181.2.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mitchell GF. Murine cutaneous leishmaniasis: resistance in reconstituted nude mice and several F1 hybrids infected with Leishmania Tropica Major.1983. J Immunogenet (Oxf) 1983;10:395–412. doi: 10.1111/j.1744-313x.1983.tb00351.x. [DOI] [PubMed] [Google Scholar]