

Abstract
Transfer of vSAG7, the endogenous superantigen encoded in the Mtv7 locus, from MHC class II− to MHC class II+ cells has been suggested to occur both in vivo and in vitro. This transfer usually leads to the activation and deletion of T cells expressing responsive Vβs. However, there is no direct molecular evidence for such a transfer. We have developed an in vitro system which confirms this property of vSAGs. vSAG7 was transfected into a class II− murine fibroblastic line. Coculture of these cells with class II+ cells and murine T cell hybridomas expressing the specific Vβs led to high levels of IL-2 production which was specifically inhibited by vSAG7- and MHC class II–specific mAbs. Moreover, injection of vSAG7+ class II− cells in mice led to expansion of Vβ6+ CD4+ cells. We show that this transfer activity is paracrine but does not require cell-to-cell contact. Indeed, vSAG7 was transferred across semi-permeable membranes. Transfer can occur both from class II− and class II+ cells, indicating that MHC class II does not sequester vSAG7. Finally, competition experiments using bacterial toxins with well defined binding sites showed that the transferred vSAG7 fragment binds to the α1 domain of HLA-DR.
Superantigens (SAGs)1 are a family of proteins which can induce the stimulation, followed in vivo by the deletion of a subset of T cells sharing particular TCR Vβ segments. These SAGs can be either integrated in the host genome (endogenous superantigens) or can be produced by an increasing array of pathogens (exogenous superantigens). These include microorganisms such as bacteria (staphylococci [1], streptococci [2, 3]), or viruses (MMTVs [4, 5], rabies virus [6]). The fact that they are conserved in such a wide array of pathogens indicates that they might play a pivotal role in the pathogenic process. Indeed, in the case of mouse mammary tumor virus (MMTV) encoded superantigens, it has been clearly demonstrated that viral infection is directly associated with the expansion of cells carrying the Vβ elements responding to the vSAGs (7–9). Moreover, the presence of Vβ6+ cells which are responsive to the rabies virus SAG is required to induce paralysis of rabies-infected mice (6, 10).
Endogenous superantigens are encoded in the mammalian genome by retroviruses (MMTVs) that have integrated in the germline host DNA. Most of these integrated proviruses have lost their ability to form virions, but continue to express viral proteins. More than 30 sites of integration (Mtv) for distinct MMTV on different chromosomes are known, with most of the common murine strains possessing two to eight of these loci [11, 12]. The 3′ LTR of Mtvs contains an open reading frame encoding the viral superantigen (vSAG) (13–15), which has been characterized as a type II transmembrane protein (16) with five glycosylation sites and three furin-like cleavage sites (17).
The primary sequence of different vSAGs is highly conserved except in the COOH terminus region (18, 19), which imparts Vβ specificity (20, 21). When vSAG7, the 3′ LTR product of Mtv7 is presented at the cell surface by MHC class II molecules, T cells expressing TCR Vβ 6, 7, 8.1, or 9 chains are triggered to proliferate in vitro or are deleted in vivo (4, 5, 22, 23).
Different studies have demonstrated that vSAG presentation is not strictly restricted to particular alleles of class II. However, some murine alleles of MHC class II do present vSAGs better than others, and the hierarchy was established as: I-E >I-A >I-Aq (24–27). As demonstrated by Labrecque et al. (28), human DR alleles can also present vSAGs to human PBMCs. Moreover, human class II alleles and isotypes show a hierarchy in vSAG presentation (29).
Several reports have suggested that vSAGs can be intercellularly transferred from class II− cells that express vSAGs to class II+ cells (30–33): first, it was shown that the response of a T cell clone to minor lymphocyte stimulatory (Mls) required the presence of B cells and of splenic adherent cells (SAC) (30), suggesting that SAC cells present an Mls fragment transferred from B cells. However, these experiments failed to demonstrate whether spleen cells provide a costimulatory signal or if they stimulate the T cell clone after the transfer of vSAG7 from B cells. It was then shown that F1→ parent chimeras between mtv7+ I-E− mice from the H-2q haplotype (which do not delete Vβ6 cells) and mtv7− I-Ed mice leads to the elimination of Vβ6+ cells (31). Lastly, injection of mtv7− mice with CD8+ cells from mtv7+ mice leads to the activation and expansion of the mtv7 responsive Vβ6+ cells; it is noteworthy that the magnitude of the activation of Vβ6+ cells was comparable after the injection of CD8+ cells which are class II− and B cells which express high levels of class II, suggesting the transfer of vSAG from CD8+ cells to class II+ cells (32, 33).
These in vivo studies have provided strong arguments for the transfer of vSAGs from cells which do not express class II (or which express class II isotypes that fail to present vSAG7) to class II+ cells proficient in vSAG presentation. However, it has never been possible to exclude that undetected expression or passive acquisition of MHC class II by T cells rather than vSAG7 transfer is responsible for the observed Vβ-specific expansions. In this report we have developed an in vitro system to confirm at the molecular level if vSAG7 transfer occurs and to get insights at the mechanisms leading to the intercellular transfer of vSAGs.
Materials and Methods
Mice.
5-wk-old CBA/CaJ female mice were purchased from Jackson Laboratories (Bar Harbor, ME).
Culture Media.
PBMCs were cultured in RPMI 1640 supplemented with 5% FCS, 10 μM β-mercaptoethanol, and 20 μg/ml gentamycin (all GIBCO BRL, Gaithersburg, MD). Fibroblasts were cultured in DMEM supplemented with 10% calf serum, 10−2 M Hepes (all GIBCO BRL), and 20 μg/ml gentamycin.
T cell hybridomas were grown in DMEM supplemented with 5% FCS, 10 mM Hepes, 10 μM β-mercaptoethanol, 20 μg/ml gentamycin, 4 mM Dextrose (Sigma Chemical Co., St. Louis, MO), essential and nonessential amino acids (GIBCO BRL), 1 mM Sodium pyruvate (GIBCO BRL), and 10 mM Sodium Bicarbonate (Sigma).
Cell Lines.
We used DAP DR1 cells (DAP cells transfected with MHC class II DR1 α and β chains) (34), DAP vSAG7 cells (DAP cells transfected with vSAG7 gene) and 3B2, called DAP DR1 vSAG7 cells thereafter (DAP cells transfected both with DR1 and vSAG7 genes [28]). A panel of T cell hybridomas, Kmls 13.11, Kmls 12.6 (kindly provided by Drs. J. Kappler and P. Marrack, National Jewish Hospital and Howard Hughes Institute, Denver, CO), RG17 (kindly provided by Dr. B. Huber, Tufts University, Boston, MA), and KR3 (kindly provided by Dr. O. Kanagawa, Washington University, St. Louis, MO) was used in these experiments. Kmls 13.11, Kmls 12.6, and RG17 express Vβ6. KR3 expresses Vβ8.1.
Monoclonal Antibodies.
The mAb specific for the murine Vβ3 (KJ25) was purified and biotinylated using standard procedures (35). The mAb directed against Vβ6 (RR4-7) was used as a supernatant. The CD4 specific mAb (GK1-5) was purified and FITC-conjugated using standard procedures (35). FITC mAb directed against human Vβ12 and Vβ13 were purchased from Immunotech (Marseille, France). OT145, a mAb directed against Vβ6.7, is a kind gift or Dr. D. Posnett (The New York Hospital, Cornell Medical Center, New York). PE-conjugated anti-CD4 (leu-3a) mAb was purchased from Becton Dickinson (Mountain View, CA).
FITC-conjugated goat anti–mouse Ig antibodies and biotinylated goat anti–rat Ig antibodies were, respectively, purchased from Caltag (San Fransisco, CA) and Vector Laboratories (Burlingame, CA).
XD5.117, an IgG1 mAb specific for the human class II molecule was used as a supernatant. HUT78, a mAb specific for human Vβ23, was purified in the laboratory and used as an isotypic control for the mAb XD5.117. Purified mAb directed against vSAG7 (6E1) was kindly provided by Dr. H. Acha-Orbea (University of Lausanne, Switzerland) (36).
Transfections.
DAP cells were transfected using the Calcium Phosphate precipitation technique (37) with the vSAG7 gene cloned in the expression vector pHβ-Apr1-neo (38). G418 resistant clones were grown and screened for the expression of vSAG7 by Northern blot analysis.
Northern Blot Analysis.
RNA was isolated using RNAzol B (Cinna/Biotecx Laboratories, Friendswood, TX). 20 μg of RNA were fractionated on 1.2% formaldehyde-agarose gels (39), transferred onto nylon membranes (Amersham Corporation, Oakville, Ontario, Canada) and hybridized at 42°C in 50% formamide with vSAG7 (EcoRI–BglII fragment of 0.9 kb) (29) or actin (PstI fragment of 1.1 kb) (40) probes labeled using a random priming kit (Pharmacia, Uppsala, Sweden). Blots were washed at 65°C in 5× SSC, 1× SSC and finally in 0.1× SSC solutions, and exposed on Kodak XAR-5 film for 24 h.
Functional Assays.
60 × 104 DAP DR1 cells were cocultured with 60 × 104 T cell hybridomas and various concentrations of DAP vSAG7 for 24 h in 250 μl of DMEM supplemented with 10% calf serum. Supernatants were harvested, and IL-2 production was measured using the CTLL hexoaminidase assay (41).
Transfer experiments were carried out using transwells (Nunc, Naperville, IL) with 0.2-μm pores. 60 × 103 DAP DR1 and 60 × 104 hybridoma cells were added in the lower compartment, while various amounts of DAP vSAG7 cells were added in the upper compartment.
PBMC Stimulation.
Blood was obtained from different healthy donors, and PBMCs were purified using ficoll-hypaque gradients (Pharmacia) as previously described (28). One million PBMCs in 1 ml of RPMI 10% FCS were incubated for 7 d in 24well plates (Falcon, Becton Dickinson, Plymouth, UK) in the presence of either DAP vSAG7 cells treated with mitomycin C (100 μg/ml, 1 h at 37°C) or for 4 d in the presence of the bacterial superantigen Staphylococcal enterotoxin B (SEB) (Toxin technologies, Sarasota, FL). Viral or bacterial superantigens were either put in the same chamber as PBMCs, or were separated from PBMCs by a transwell (Costar, Cambridge, MA) with 0.4μm pores. Cells were then harvested and tested by flow cytometry for Vβ expression.
Cytofluorimetry.
For human TcR Vβ repertoire studies, 2–5 × 105 cells were stained with anti-Vβ antibodies at previously defined optimal titers for 20 min at 4°C in the dark and washed in PBS containing 2% FCS. When necessary cells were then incubated with FITC-conjugated goat anti–mouse mAb, and 10% normal mouse serum (Jackson Laboratories) in PBS. After washing, cells were finally incubated with PE- or PerCP-conjugated antiCD4 for 20 min in the dark. For murine TcR Vβ repertoire studies, cells were first incubated with biotinylated anti-Vβ mAbs. In a second step, they were incubated with FITC-conjugated antiCD4, and finally, with PE-conjugated streptavidin. For RR4-7, two additional steps (incubation with biotinylated goat anti–rat and then with normal rat serum 10% in PBS) were made before adding FITC-conjugated anti-CD4. Acquisition and analysis of cells were carried out using a FACScan® and the Lysis II software (Becton Dickinson, Mountain View, CA). For each analysis, a minimum of 1 × 104 live cells gated by forward and side scatter were analyzed.
Results
vSAG7 Is Transferred In Vitro and Stimulates T Cells.
The vSAG7 gene was transfected into the MHC class II− DAP3 murine fibroblastic line. Seven clones of G418-resistant cells were analyzed by Northern Blot. Equal loading of all wells was confirmed by comparable levels of actin mRNA in all clones tested (Fig. 1 a). When compared to other clones, clones 17 and 18 showed significantly higher levels of vSAG7 mRNA (Fig. 1 b). Clone 2 also expressed vSAG7 mRNA, although at lower levels. To verify the capacity of MHC class II− vSAG7+ cells to transfer vSAGs, we developed a transfer assay in which equal numbers (6 × 104) of DAP DR1 cells and the Vβ6+ vSAG7-responsive Kmls 13.11 hybridomas were cocultured for 16 to 20 h together with various amounts of the DAP vSAG7 cells. T cell stimulation was assessed by measuring IL-2 present in the supernatants. The three vSAG7+ clones (clones 2, 17, and 18) were able to induce a dose dependent T cell activation in several independent experiments. A representative experiment is shown in Fig. 1 c where increases in the production of IL-2 ranged from 10- to 20-fold as compared to cocultures of DAP vSAG7 cells and untransfected DAP cells. The latter indicated the absolute prerequisite for MHC class II molecules in order to obtain vSAG7 presentation. Levels of IL-2 production were directly correlated with the number of cells expressing vSAG7 or with the levels of vSAG7 expressed by the class II− fibroblasts. Indeed, little or no stimulation was observed with less than 2 × 103 DAP vSAG7 cells, whereas maximal response was obtained with 5 × 104 DAP vSAG7 cells per well.
Figure 1.

In vitro transfer of vSAG7 from class II− cells to class II+ cells. (a and b) Expression levels of vSAG7 and actin mRNA in different clones of DAP cells transfected with vSAG7. RNA was extracted from cloned DAP vSAG7 transfectants. 10 μg of RNA were run on a 1.2% agarose-formaldehyde gel and transferred on a nylon membrane. Membranes were then hybridized with either actin (a) or vSAG7 (b) radiolabeled probes and exposed for 24 h. Each track represents an individual clone. (c) Murine fibroblasts transfected with vSAG7 can stimulate T cell hybridomas in the presence of DAP DR1 cells. 6 × 104 DAP DR1 cells and 6 × 104 Kmls 13.11 cells were incubated overnight with either 5 × 104 DAP cells or various amounts of DAP vSAG7 cells. Supernatants were then harvested and tested for IL-2 activity. (d and e) Levels of vSAG7 expression by DAP vSAG7 clone 18 and DAP DR1 vSAG7 are comparable. RNA was extracted from DAP vSAG7 clone 18 and DAP DR1 vSAG7 clone 3B2, run on a 1.2% agarose-formaldehyde gel and hybridized with actin (d) or vSAG7 ( f ) radiolabeled probes. ( f ) Levels of stimulation in the transfer assay (DAP vSAG7 + DAP DR1) and in direct presentation (DAP DR1 vSAG7) are comparable. 6 × 104 Kmls 13.11 cells were cocultured in 250 μl either with 6 × 104 DAP DR1 cells and 5 × 104 DAP vSAG7 clone 18 cells, or with 6 × 104 DAP DR1 vSAG7 cells. Supernatants were then harvested and tested for IL-2 production.
Controls were performed to eliminate the possibility that this activity could be caused by the fusion of DAP DR1 with DAP vSAG7 cells. A flow cytometric assay was developed in which two different fibroblastic lines expressing distinct surface markers were cocultured for the duration of the above described functional assay (16 to 20 h). Results indicate that the percentage of fused cells was below 1% and fusion could thus not account for the superantigenic activity (data not shown). Our experiments thus indicate that a fragment of the vSAG7 protein carrying superantigenic activity is transferred intercellularly. Additional experiments were performed to compare the superantigenic activity in the transfer assay and in the direct presentation assay. Results illustrated in Fig. 1 (d and e) show that DAP vSAG7 and DAP DR1 vSAG7 express comparable levels of vSAG7 mRNA. The levels of stimulation induced by vSAG7 in transfer assays or in direct presentation assays are similar (Fig. 1, d–f ), indicating that transfer is an efficient process.
A similar strategy was used to monitor the transfer capacity of the exogenous vSAG GR. Results indicate that transfer of vSAG GR can occur from DAP DR1 vSAG GR to CH12 cells and lead to the stimulation of KOX15, a Vβ15-expressing hybridoma (data not shown).
Transfer Is Inhibited by Anti-vSAG7 and Anti-Class II MAbs.
To further demonstrate the requirement for vSAG7 in order to obtain T cell stimulation we used the vSAG7specific mAb 6E1 to inhibit stimulation in the transfer assay. Results illustrated in Fig. 2 a clearly show that this mAb inhibits vSAG7 activity in a dose-dependent manner, as previously demonstrated for presentation of endogenously expressed vSAG7. As a control of specificity, we show that mAb 6E1 fails to inhibit SEB presentation (data not shown).
Figure 2.
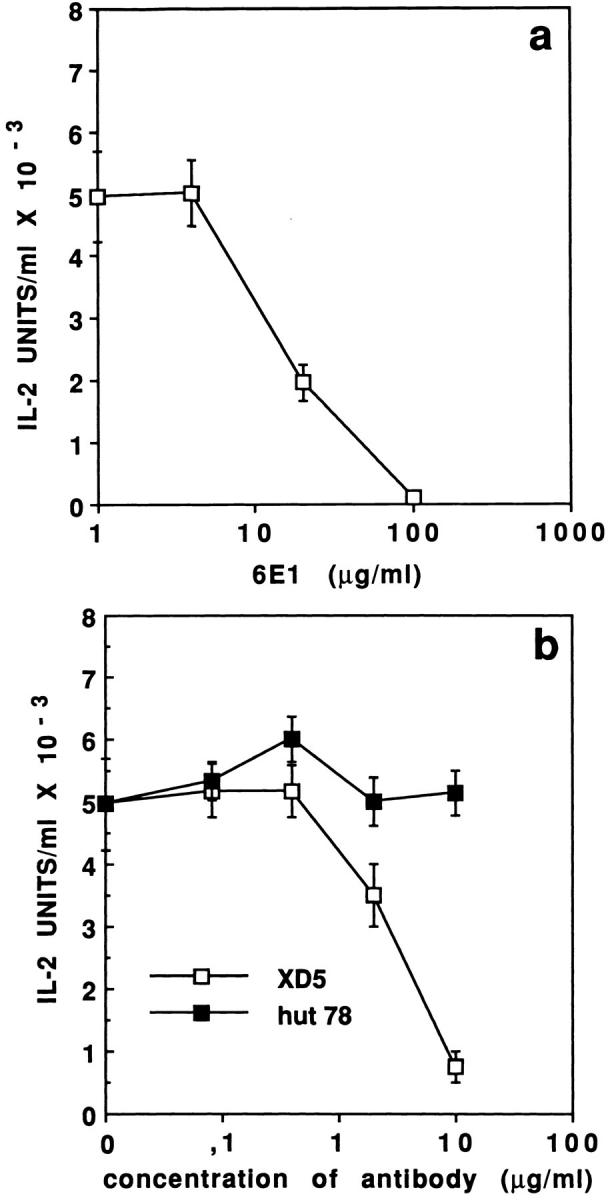
(a) vSAG7 Transfer can be inhibited by 6E1, a vSAG7 specific monoclonal antibody. Various concentrations of the mAb 6E1 were added in the transfer assay (6 × 104 Kmls 13.11 cells, 6 × 104 DAP DR1 cells and 5 × 104 DAP vSAG7 cells in 250 μl). After an overnight culture, supernatants were harvested and tested for IL-2 production. (b) Specific inhibition of vSAG7 transfer by an MHC class II specific mAb. Various concentrations of the mAbs XD5.117 (HLA DR) or HUT78 (human TCR Vβ23) were added in the transfer assay. After an overnight culture, supernatants were harvested and tested for IL-2 production.
Inhibition experiments carried out with the MHC class II DR–specific mAb XD5.117 further confirmed the requirement for DR expression in order to stimulate T cells in the transfer assay (Fig. 2 b). Indeed, the XD5.117 mAb inhibited vSAG7 stimulation in a dose-dependent manner, further confirming the similarity between the transfer of vSAG7 and the presentation of endogenous vSAGs. Altogether, these experiments confirmed that vSAG7 can be expressed at the surface of MHC class II− cells and is directly transferred to MHC class II molecules expressed at the surface of DAP DR1 cells.
Transfer of vSAG7 Stimulates T Cells Bearing Distinct TCRs and Occurs between Cell Lines of Different Origin.
vSAG7 not only reacts with Vβ6-expressing T cells, but also Vβ8.1expressing T cells. A panel of Vβ6- (Kmls 13.11, Kmls 12.6 and RG17)- or Vβ8.1 (KR3)-expressing hybridomas was used to demonstrate that transfer of vSAG7 was capable of stimulating T cells expressing different vSAG7responding TCRs. All these hybridomas were stimulated at comparable levels by vSAG7 both in the transfer assay and in direct presentation by DAP DR1 vSAG7 cells (Fig. 3 a).
Figure 3.
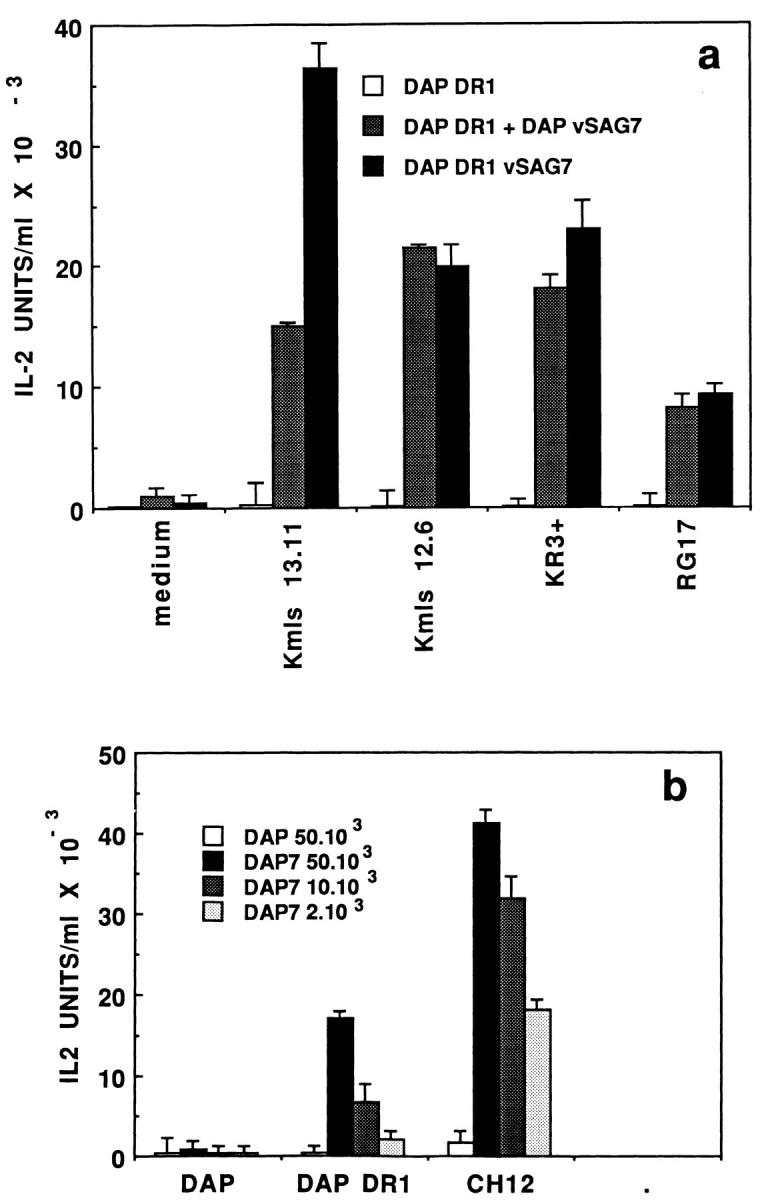
(a) A panel of vSAG7-expressing hybridomas expressing Vβ6 or Vβ8.1 respond to the transferred vSAG7. 6 × 104 T cell hybridomas cells Kmls 13.11 (Vβ6), Kmls 12.6 (Vβ6), RG17 (Vβ6), and KR3 (Vβ8.1) were tested in direct presentation assays (overnight coculture with 6 × 104 DAP DR1 vSAG7 cells) or in transfer assays (overnight coculture with 6 × 104 DAP DR1 cells and 6 × 104 DAP vSAG7 cells) for IL-2 production. (b) Presentation of the transferred vSAG7 is not restricted to DAP DR1 cells. 6 × 104 DAP DR1 cells or 6 × 104 CH12 B lymphoma cells were cocultured overnight with 5 × 104 DAP vSAG7 cells and 6 × 104 Kmls 13.11 cells. Supernatants were then harvested and tested for IL-2 activity.
In order to provide evidence that transfer of vSAG7 is not dependent on particular properties of DAP DR1 cells, we used CH12 cells, a murine B cell hybridoma expressing both I-Ek and I-Ak. CH12 cells did not stimulate Kmls 13.11 cells when cocultured with untransfected DAP cells, as indicated in Fig. 3 b. However, when CH12 cells were cocultured with DAP vSAG7 cells, efficient stimulation of Kmls 13.11 hybridoma occurred. Levels of T cell stimulation were directly correlated to the number of DAP vSAG7 cells added to the coculture and ranged in the same order of magnitude than those obtained using DAP DR1 cells (Fig. 3 b).
In Vivo Transfer of vSAG7.
To further demonstrate that the transfer activity bears physiological relevance, experiments were set up to verify if transfer of vSAG7 from DAP vSAG7 occurs in vivo. For this purpose, mice were injected with 3 × 106 DAP vSAG7 in the hind footpad. Five days following injection, draining lymph node cells were analyzed by flow cytometry for TcR Vβ6 expression. A representative experiment is shown in Fig. 4. In this experiment, the percentage of Vβ6-expressing cells among the CD4+ cells increases from 11% (mice injected with DAP DR1 cells) in control mice to 25% in mice injected with DAP vSAG7 cells. This enhancement is comparable to the one observed after injection of DAP DR1 vSAG7 cells. As previously shown following injection of B cells or CD8+ cells expressing vSAG7, we did not observe any expansion of Vβ6+ CD8+ cells.
Figure 4.

DAP vSAG7 induces Vβ6-specific expansion in vivo. 3 × 106 DAP vSAG7 cells in 30 μl of PBS were injected in the hind footpad. 5 d later, lymph nodes were taken and CD4+ lymphocytes were analyzed by flow cytometry for Vβ3 and Vβ6 expression. Live cells were gated by forward and side scatter. Percentages of Vβ6-expressing cells among CD4+ cells were respectively 11, 25, and 23%, respectively, after DAP DR1, DAP vSAG7, and DAP DR1 vSAG7 injection. This experiment was repeated four times and led to similar results.
These results demonstrate that the fragment carrying the superantigenic activity is thus transferred from class II− DAP vSAG7 to MHC class II+ cells in lymph nodes.
Ex Vivo Transfer of vSAG7 to Human Lymphocytes Stimulates Vβ12+ Cells.
Labrecque et al. (28) have shown that DAP DR1 vSAG7 cells are able to induce the proliferation of human PBMCs. We show here that class II− DAP vSAG7 cells are also able to stimulate PBMCs in a Vβ-restricted manner. DAP vSAG7 (2 × 105) were cocultured with PBMCs (106) for 10 d. PBMCs were then harvested and tested by cytofluorimetry for Vβ expression among CD4+ blasts. A representative experiment is shown in Fig. 5 and demonstrates a three- to fourfold increase in the percentage of Vβ12+ CD4+ blasts, as compared to PBMCs cocultured with untransfected DAP cells. The percentage of Vβ6.7 cells, which are not responsive to vSAG7, remained unchanged.
Figure 5.

DAP vSAG7 can induce a Vβ-specific response of ex vivo derived human PBMCs. 1.5 × 105 mitomycin treated DAP vSAG7 cells were added to 1 × 106 fresh PBMCs in a total volume of 1 ml. After 7 d of culture, cells were harvested and the percentage of CD4+ blasts expressing Vβ6.7 and Vβ12 was assayed using flow cytometry. Blasts were gated by forward and side scatter. Results are illustrated as dot plots, with CD4 on the x axis and Vβ on the y axis. This experiment was performed twice and led to similar results.
Characterization of the Intercellular Transfer of vSAG7.
Whereas the above results undoubtedly showed that transfer of vSAGs is possible, they did not address the requirement for cellular interactions in this process. We thus performed transfer assays in which DAP vSAG7 cells were separated from DAP DR1 cells and Kmls 13.11 hybridomas by a semi-permeable membrane. No stimulation could be observed under these conditions (Fig. 6 a) even with concentrations of DAP vSAG7 cells as high as 2 × 106 per ml. Conversely, SEB, even at low concentrations (1.5 ng/ml), was able to cross this membrane and stimulate the Vβ6 hybridoma. These results raised the hypothesis that transfer is mediated by an insoluble or unstable fragment. However, we were not able to exclude the possibility that a small proportion of vSAG7 molecules are able to cross the membrane.
Figure 6.
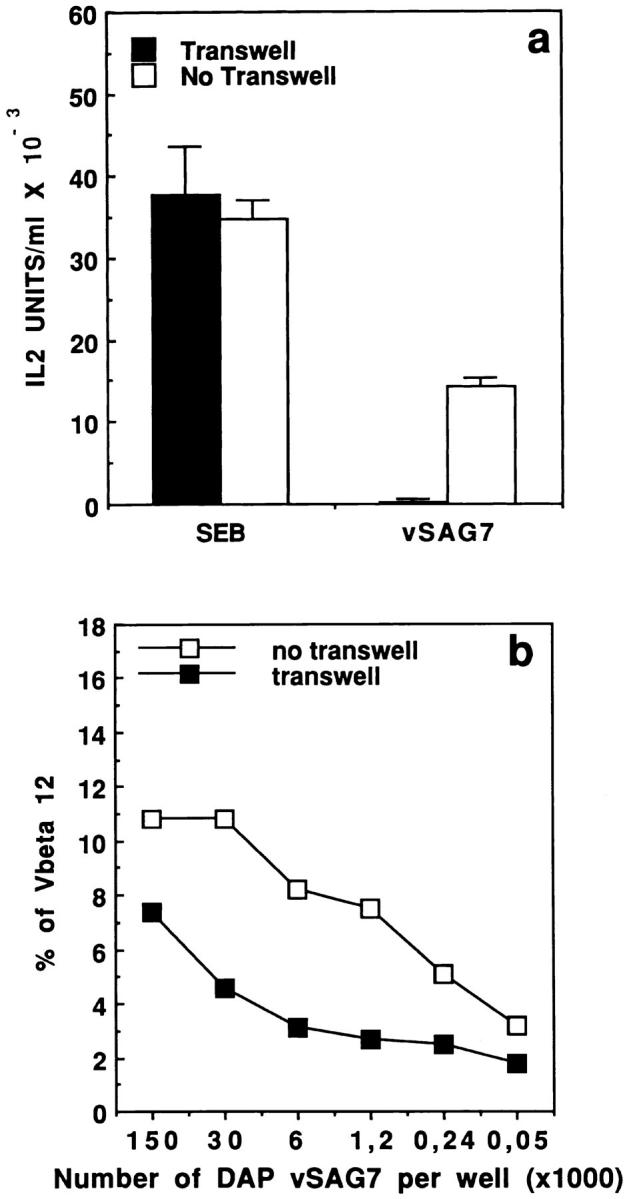
(a) Transfer of vSAG7 requires cell proximity to stimulate murine T cell hybridoma. 5 × 104 DAP vSAG7 cells or 75 pg SEB were disposed in the upper chamber while 6 × 104 DAP DR1 cells and 6 × 104 Kmls 13.11 cells were disposed in the bottom chamber, separated by a 0.2-μm porous membrane. As a control, 5 × 104 DAP vSAG7 cells or SEB were disposed with the other cells in absence of compartmentalization. Assays were then performed as previously described. (b) The transferred fragment of vSAG7 can cross the membrane and stimulate human PBMCs. Different concentrations of DAP vSAG7 were disposed in the upper chamber while 106 human PBMCs were disposed in the bottom chamber. Percentage of CD4+ blasts expressing Vβ12 or Vβ6.7 was measured by flow cytometry after 7 d of culture. As a control, DAP vSAG7 cells and human PBMCs were disposed in absence of compartmentalization. Results are illustrated as dot plots with Vβ in x axis and CD4 in y axis.
For this purpose, we tried to identify a more sensitive readout and compared stimulation of human PBMCs to stimulation of T cell hybridomas (Korman, A., personal communication). Titration experiments involving coculture of PBMCs with decreasing numbers of DAP vSAG7 cells indicated that stimulation of PBMCs required as few as 250 DAP vSAG7 cells (Fig. 6 b), while the stimulation of T cell hybridomas was repeatedly shown to require at least 2 × 103 DAP vSAG7 cells (Fig. 1 c). The use of human PBMCs thus provides a readout which is 10-fold more sensitive than that obtained using T cell hybridomas.
In vitro assays were then established in which DAP vSAG7 cells were separated from PBMCs by a 0.4-μm semipermeable membrane. A threefold enrichment in CD4+ blasts expressing Vβ12 was observed by flow cytometry (Fig. 6 b). However, this enrichment was only detected when high amounts (>3 × 104) of DAP vSAG7 cells were used. This result clearly indicates that the fragment of vSAG7 carrying the superantigenic activity is able to cross the semi-permeable membrane. Our results also show that at least 100-fold more DAP vSAG7 cells are required when cells are separated by a transwell as compared to direct coculture of DAP vSAG7 cells and PBMCs, in order to obtain the same levels of Vβ12 expansion. It is thus possible to estimate that only ∼1% of the soluble vSAG7 molecules are able to efficiently cross membranes. In comparison, SEB was shown to freely cross the membrane, as the SEB-induced stimulation of PBMCs is not altered by this compartmentalization, even at the lowest concentrations of bacterial toxin (data not shown).
Similar experiments were performed using DAP DR1 vSAG7 cells in the upper compartment. Our results (Fig. 7) show that the transferred fragment of vSAG7 crosses the membrane and stimulates efficiently Vβ12+ PBMCs. These results were obtained using two different DAP DR1 vSAG7 clones, 3B2 and 3A5. The results obtained using DAP vSAG7 or DAP DR1 vSAG7 (Figs. 6 b and 7) were very similar: in both cases a small and comparable proportion of vSAG molecules were able to cross the membrane and stimulate PBMCs. These results indicate that the MHC class II molecules expressed by DAP DR1 cells are not interfering with the transfer of vSAG7 molecules.
Figure 7.

vSAG7 can be transferred despite the presence of MHC class II molecules on donor cells. Various concentrations of DAP DR1 vSAG7 cells clone 3B2 or 3A5 were disposed in the upper compartment. The experiment was then performed as described in Fig. 6 b.
vSAG7 Interacts with the HLA-DR α Chain.
Bacterial toxins bind to well identified sites on MHC class II. To provide a molecular characterization of the class II site which is involved in the interaction with vSAG7, we have performed inhibition experiments of both vSAG7 in direct and transfer presentation assays using bacterial toxins. We show in Fig. 8 that the DAP DR1 vSAG7-induced stimulation of Kmls 13.11 is not inhibited by addition of SEA, even at the highest concentration tested (50 μg/ml). On the other hand, intercellular transfer of vSAG7 is significantly affected by the simultaneous incubation of cells with high concentrations of SEA (Fig. 8). This inhibition of vSAG7dependent T cell stimulation was directly correlated with the amount of SEA. Addition of increasing concentrations of SEA led to up to 95% inhibition of T cell stimulation. These experiments allowed us to suggest that the molecular interactions of the whole vSAG7 or of the transferred fragment with MHC class II are different.
Figure 8.

vSAG7 transfer is inhibited by SEA. 6 × 104 DAP DR1 cells, 6 × 104 Kmls 13.11 cells and 5 × 104 DAP vSAG7 cells were cocultured overnight in presence of different concentrations of SEA. IL-2 released in supernatants was measured 24 h later.
SEA binds to the α and β chains of MHC class II molecules (41a). We were thus interested to determine if the binding sites for the bacterial toxin were also involved in the binding of vSAG7. To address this question, we used a mutant of SEA, SEA F47, which has lost its ability to bind MHC class II through the α chain but is still able to bind with high affinity to the β chain of MHC class II (41a, 42). We show here (Fig. 9) that SEA F47 partially blocks the activity of vSAG7 in the transfer assay, as compared to the 20-fold inhibition induced by SEA wt. Indeed, the addition of high concentrations of SEA F47 decreases T cell stimulation by less than a twofold (∼40% of inhibition). We concluded from these studies that vSAG7 interacts mainly with MHC class II through its α chain. This result was further confirmed by the fact that high concentrations of TSST-1, a toxin known to bind exclusively to the α chain of MHC class II (43, 44), also exerted a partial (∼60%) but reproducible inhibition of vSAG7 presentation in the transfer system (Fig. 9).
Figure 9.

vSAG7 binds to the DR α chain. To standard conditions of transfer assay were added various concentrations of either SEA, SEA F47, or TSST-1. IL-2 was measured in supernatants as previously described.
Discussion
In this report we provide in vitro and in vivo evidence indicating that a soluble form of vSAG7 can activate primary T cells of human or murine origin. This transfer is absolutely dependent on the presence of class II+ cells, and is specific for the vSAG, as shown by inhibition experiments. Our demonstration that this transfer can occur across a semipermeable membrane clearly rules out the possibility that fusion between class II+ and class II− cells is responsible for this effect.
Moreover, the use of the semi-permeable membrane provided conclusive evidence that cell-to-cell contact is not required for intercellular transfer of vSAG7. In these conditions, however, transfer is not very efficient and involves only a small proportion (∼1%) of vSAG7 molecules. Indeed, titration curves of vSAG7 in the presence or in the absence of a semi-permeable membrane shows that comparable Vβ specific expansion requires 100-fold more vSAG7 cells in the presence of a semi-permeable membrane. The fact that vSAG7 molecules can only cross membranes with low efficiency suggests that vSAG7 is weakly soluble, and that most of the transferable molecules remain uncovalently bound at the cellular membrane to its NH2-terminal part (45). These features leads us to conclude that vSAG7 transfer does not require strict cell-to-cell contact but rather cellular proximity. It can thus be considered as a paracrine phenomenon. These results also provide convincing evidences that vSAG7 molecules can reach the cell surface in the absence of MHC class II molecules and are biologically active.
Titration experiments involving DAP DR1 vSAG7+ cells in the presence or in the absence of a semi-permeable membrane indicate that DR molecules do not sequester vSAGs. Indeed, the Vβ skewing obtained using DAP DR1 vSAG7 and DAP vSAG7 shows that vSAG7 can be transferred efficiently from both cells. This suggests that vSAG molecules are not sequestered by MHC class II at the surface of DAP DR1 vSAG7 cells. Alternatively, vSAGs could be loosely associated to class II enabling the dissociation of the transferred fragment and subsequent binding to another class II+ cell. It is possible that the transferred fragment has never been associated with class II molecules. The presence of three cleavage sites for furin-like proteases could lead to the dissociation of a COOH-terminal fragment, a situation which is highly analogous to the one observed with the HIV gp120, which readily dissociates from the membrane gp41, and remains anchored at the cell surface (46).
Class II− CD8+ T cells can delete vSAG-responsive T cells more efficiently than class II+ B cells (32), although these cells express comparable levels of vSAG7 mRNA (47). This paradox could have been attributed to the sequestration of vSAGs by MHC class II+ molecules on the surface of B cells. Our demonstration that both DR1+ and DR1− vSAG7+ cells are equally efficient in inducing the deletion of Vβ6-expressing T cells indicates that the reported difference between CD8+ and B cells is not due to their differential expression of class II and subsequent sequestration of vSAG7 by MHC class II, but rather to other cellular characteristics such as adhesion molecules, ability of being activated in different conditions or half life after injection.
We have then shown that SEA can compete with the transferred vSAG while it has no effect on the presentation of vSAG7 endogenously expressed by DAP DR1+ cells. Two models can be proposed to explain this discrepancy. First, it is possible that the transferred fragment of vSAG7 displays a lower affinity for class II MHC molecules than the full-length vSAG7 molecule. These differences could be attributed to additional contact points between the fulllength and the transferred fragment. Alternatively, when endogenously expressed, vSAG7 could induce a conformational modification of the vSAG7–class II complex generating a tighter association between the two molecules.
Although considerable progress has been made in the understanding of TCR:vSAG interaction, little is available concerning the sites involved in the binding of vSAGs to MHC class II. However, different results indicate that the β chain of MHC class II is involved in the binding of vSAG7 (29, 48). Whereas SEA molecules efficiently abrogate vSAG7 transfer to DR1 molecules, a mutated SEA which fails to interact with the α chain is much less efficient in exerting this inhibitory effect. This result suggests that vSAG7 interacts with the DR α chain. Partial inhibition could be due to the residual binding of SEA F47 to the DR α chain. Indeed our own results have suggested that SEA also binds to the second loop of the DR α chain which includes residue DR α 39 (41a). Binding of vSAG7 to the DR α chain was further confirmed using TSST-1, which predominantly binds to the DR α chain (43, 44) and can also compete (60% of inhibition) with the transferred fragment of vSAG7. These results do not exclude however that vSAGs could also interact with the β chain of DR, as suggested by the hierarchy in vSAG presentation by DR alleles, which differ only through their β chain. (29)
vSAGs contain furin-like cleavage sites and are naturally cleaved. However, the cleaved fragment remains bound to the rest of the protein through non covalent bonds (45). These observations raise the hypothesis that the soluble molecule involved in the transfer phenomenon is a fragment of vSAG7 obtained by cleavage of vSAG7 at one of the three furin-like cleavage sites. The precise nature of this transferred fragment remains to be determined. In this context it is important to note that these cleavage sites have been conserved in most of the known sequences of exogenous and endogenous MMTVs. Such a degree of conservation would favor an important role for these cleavage sites. It is well established that the virus is strictly dependent for its replication on its capacity to stimulate a large pool of T cells (7, 9). Transfer of the vSAG fragment from infected cells to other MHC class II+ cells would favor T cell activation, upregulate viral production, and hence lead to viral dissemination in the host.
On the other hand, transfer of vSAGs could also provide an advantage to the host; Moore et al. (49) have demonstrated that the expression of some endogenous vSAGs in thymic class II+ cells is low or null, and that the bulk of vSAGs is expressed in lymphocytes of the T lineage at different stages of maturation. The efficient paracrine transfer of vSAGs from MHC class II− vSAG+ thymocytes (47) to class II+ vSAG− dendritic cells or macrophages would provide a mechanism leading to the deletion of vSAG-responsive CD4+ or CD8+ cells. Transfer of vSAGs could then provide an alternative way of thymic deletion of thymocytes expressing responsive Vβs, this deletion being critical in order to protect mice from exogenous infection by MMTVs sharing the same Vβ specificity (7) in the first weeks after birth.
It is also quite likely that this transfer could play a role in peripheral deletion. CD8+ T cells express high levels of vSAGs (32, 47) which are upregulated upon T cell activation (15). This could lead to an increased transfer of vSAGs to adjacent class II+ cells and hence to the deletion of activated CD8+ T cells. Different reports demonstrate that vSAGs can also be expressed in cells of non-immune origin, such as lung cells, brain cells (47) or intestinal epithelial cells (50). A role of vSAG reservoir could be attributed to these cells, which could hence be involved in the deletion of activated T cells infiltrating these organs.
Finally, studies concerning binding of vSAGs to MHC class II, its intracellular transport, and cofactors involved in transfer, that remain unaddressed, should be made possible using the above described transfer assays.
Acknowledgments
We would like to thank Pascal Lavoie and Dr. Michel Braun for critical review of the manuscript, and H. McGrath for excellent technical support.
Footnotes
This work was supported by grants RG-544/95 from Human Frontier Science Project, MT-10055 from Medical Research Council of Canada, and 007273 from National Cancer Institute of Canada, attributed to R.P. Sékaly. R.P. Sékaly holds an MRC scientist award. F. Denis and J. Thibodeau are supported by fellowships from National Health Research and Development Program. M. Delcourt has a French government Assistant Moniteur Normalien fellowship.
1 Abbreviations used in this paper: MMTV, mouse mammary tumor virus; SAC, splenic adherent cell; SAG, superantigens; SEA SEB, staphylococcal enterotoxia A/B; vSAG, viral superantigen.
References
- 1.Kappler JW, Kotzin B, Herron L, Gelfand EW, Bigler RD, Boylston A, Carrel S, Posnett DN, Choi Y, Marrack P. V beta-specific stimulation of human T cells by staphylococcal toxins. Science (Wash DC) 1989;244:811–813. doi: 10.1126/science.2524876. [DOI] [PubMed] [Google Scholar]
- 2.Tomai MA, Aelion JA, Dockter ME, Majumdar G, Spinella DG, Kotb M. T cell receptor V gene usage by human T cells stimulated with the superantigen streptococcal M protein. J Exp Med. 1991;174:285–292. doi: 10.1084/jem.174.1.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tomai M, Kotb M, Majumdar G, Beachey EH. Superantigenicity of streptococcal M protein. J Exp Med. 1990;172:359–362. doi: 10.1084/jem.172.1.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.MacDonald HR, Schneider R, Lees RK, Howe RC, Acha-Orbea H, Festenstein H, Zinkernagel RM, Hengartner H. T-cell receptor V beta use predicts reactivity and tolerance to Mlsa-encoded antigens. Nature (Lond) 1988;332:40–45. doi: 10.1038/332040a0. [DOI] [PubMed] [Google Scholar]
- 5.Kappler JW, Staerz U, White J, Marrack PC. Self-tolerance eliminates T cells specific for Mls-modified products of the major histocompatibility complex. Nature (Lond) 1988;332:35–40. doi: 10.1038/332035a0. [DOI] [PubMed] [Google Scholar]
- 6.Lafon M, Lafage M, Martinez-Arends A, Ramirez R, Vuillier F, Charron D, Lotteau V, Scott-Algara D. Evidence for a viral superantigen in humans. Nature (Lond) 1992;358:507–510. doi: 10.1038/358507a0. [DOI] [PubMed] [Google Scholar]
- 7.Held W, Waanders GA, Shakhov AN, Scarpellino L, Acha-Orbea H, MacDonald HR. Superantigeninduced immune stimulation amplifies mouse mammary tumor virus infection and allows virus transmission. Cell. 1993;74:529–540. doi: 10.1016/0092-8674(93)80054-i. [DOI] [PubMed] [Google Scholar]
- 8.Golovkina TV, Chervonsky A, Dudley JP, Ross SR. Transgenic mouse mammary tumor virus superantigen expression prevents viral infection. Cell. 1992;69:637–645. doi: 10.1016/0092-8674(92)90227-4. [DOI] [PubMed] [Google Scholar]
- 9.Golovkina TV, Dudley JP, Jaffe AB, Ross SR. Mouse mammary tumor viruses with functional superantigen genes are selected during in vivo infection. Proc Natl Acad Sci USA. 1995;92:4828–4832. doi: 10.1073/pnas.92.11.4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Astoul E, Lafage M, Lafon M. Rabies superantigen as a V-beta T-dependent adjuvant. J Exp Med. 1996;183:1623–1631. doi: 10.1084/jem.183.4.1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kozak C, Peters G, Pauley R, Morris V, Michalides R, Dudley J, Green M, Davisson M, Prakash O, Vaidya A. A standardized nomenclature for endogenous mouse mammary tumor viruses. J Virol. 1987;61:1651–1654. doi: 10.1128/jvi.61.5.1651-1654.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Simpson E. T cell repertoire selection by mouse mammary tumour viruses. Eur J Immunogenetics. 1993;20:137–149. doi: 10.1111/j.1744-313x.1993.tb00104.x. [DOI] [PubMed] [Google Scholar]
- 13.Choi Y, Kappler JW, Marrack P. A superantigen encoded in the open reading frame of the 3′ long terminal repeat of mouse mammary tumour virus. Nature (Lond) 1991;350:203–209. doi: 10.1038/350203a0. [DOI] [PubMed] [Google Scholar]
- 14.Acha-Orbea H, Shakhov AN, Scarpellino L, Kolb E, Muller V, Vessaz-Shaw A, Fuchs R, Blochlinger K, Rollini P, Billotte J. Clonal deletion of Vβ14-bearing T cells in mice transgenic for mammary tumour virus. Nature (Lond) 1991;350:207–217. doi: 10.1038/350207a0. [DOI] [PubMed] [Google Scholar]
- 15.Beutner U, Frankel WN, Cote MS, Coffin JM, Huber BT. Mls-1 is encoded by the long terminal repeat open reading frame of the mouse mammary tumor provirus Mtv-7. Proc Natl Acad Sci USA. 1992;89:5432–5436. doi: 10.1073/pnas.89.12.5432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Korman AJ, Bourgarel P, Meo T, Rieckhof GE. The mouse mammary tumour virus long terminal repeat encodes a type II transmembrane glycoprotein. EMBO (Eur Mol Biol Organ) J. 1992;11:1901–1905. doi: 10.1002/j.1460-2075.1992.tb05242.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Choi Y, Marrack P, Kappler JW. Structural analysis of a mouse mammary tumor virus superantigen. J Exp Med. 1992;175:847–852. doi: 10.1084/jem.175.3.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yazdanbakhsh K, Park CG, Winslow GM, Choi Y. Direct evidence for the role of COOH terminus of mouse mammary tumor virus superantigen in determining T cell receptor V beta specificity. J Exp Med. 1993;178:737–741. doi: 10.1084/jem.178.2.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Held W, Shakhov AN, Waanders G, Scarpellino L, Luethy R, Kraehenbuhl JP, MacDonald HR, AchaOrbea H. An exogenous mouse mammary tumor virus with properties of Mls- 1a (Mtv-7) J Exp Med. 1992;175:16231633. doi: 10.1084/jem.175.6.1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rudy CK, Kraus E, Palmer E, Huber BT. Mls-1-like superantigen in the MA/MyJ mouse is encoded by a new mammary tumor provirus that is distinct from Mtv-7. J Exp Med. 1992;175:1613–1621. doi: 10.1084/jem.175.6.1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pullen AM, Choi Y, Kushnir E, Kappler JW, Marrack PC. The open reading frames in the 3′ long terminal repeats of several mouse mammary tumor virus integrants encode V beta 3- specific superantigens. J Exp Med. 1992;175:41–47. doi: 10.1084/jem.175.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Happ MP, Woodland DL, Palmer E. A third T-cell receptor beta-chain variable region gene encodes reactivity to Mls-1a gene products. Proc Natl Acad Sci USA. 1989;86:6293–6296. doi: 10.1073/pnas.86.16.6293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marrack P, Blackman M, Kushnir E, Kappler J. The toxicity of staphylococcal enterotoxin B in mice is mediated by T cells. J Exp Med. 1990;171:455–464. doi: 10.1084/jem.171.2.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Katz ME, Janeway CA., Jr The immunobiology of T cell responses to Mls-locus-disparate stimulator cells. II. Effects of Mls-locus-disparate stimulator cells on cloned, protein antigen-specific, Ia-restricted T cell lines. J Immunol. 1985;134:2064–2070. [PubMed] [Google Scholar]
- 25.MacDonald HR, Glasebrook AL, Schneider R, Lees RK, Pircher H, Pedrazzini T, Kanagawa O, Nicolas JF, Howe RC, Zinkernagel RM. T-cell reactivity and tolerance to Mlsa-encoded antigens. Immunol Rev. 1989;107:89–108. doi: 10.1111/j.1600-065x.1989.tb00004.x. [DOI] [PubMed] [Google Scholar]
- 26.Bill J, Kanagawa O, Woodland DL, Palmer E. The MHC molecule I-E is necessary but not sufficient for the clonal deletion of V beta 11-bearing T cells. J Exp Med. 1989;169:1405–1419. doi: 10.1084/jem.169.4.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Okada CY, Weissman IL. Relative V beta transcript levels in thymus and peripheral lymphoid tissues from various mouse strains. Inverse correlation of I-E and Mls expression with relative abundance of several V beta transcripts in peripheral lymphoid tissues. J Exp Med. 1989;169:1703–1719. doi: 10.1084/jem.169.5.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Labrecque N, McGrath H, Subramanyam M, Huber BT, Sekaly RP. Human T cells respond to mouse mammary tumor virus-encoded superantigen: V beta restriction and conserved evolutionary features. J Exp Med. 1993;177:1735–1743. doi: 10.1084/jem.177.6.1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Subramanyam M, McLellan B, Labrecque N, Sekaly RP, Huber BT. Presentation of the Mls-1 superantigen by human HLA class II molecules to murine T cells. J Immunol. 1993;151:2538–2545. [PubMed] [Google Scholar]
- 30.DeKruyff RH, Ju ST, Laning J, Cantor H, Dorf ME. Activation requirements of cloned inducer T cells. III. Need for two stimulator cells in the response of a cloned line to Mls determinants. J Immunol. 1986;137:1109–1114. [PubMed] [Google Scholar]
- 31.Speiser DE, Schneider R, Hengartner H, MacDonald HR. Clonal deletion of self-reactive T cells in irradiation bone marrow chimeras and neonatally tolerant mice. Evidence for intercellular transfer of Mlsa. J Exp Med. 1989;170:595–600. doi: 10.1084/jem.170.2.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Webb SR, Sprent J. Induction of neonatal tolerance to Mlsa antigens by CD8+T cells. Science (Wash DC) 1990;248:1643–1648. doi: 10.1126/science.1973003. [DOI] [PubMed] [Google Scholar]
- 33.Webb S, Morris C, Sprent J. Extrathymic tolerance of mature T cells: clonal elimination as a consequence of immunity. Cell. 1990;63:1249–1256. doi: 10.1016/0092-8674(90)90420-j. [DOI] [PubMed] [Google Scholar]
- 34.Jacobson S, Sekaly RP, Jacobson CL, McFarland HF, Long EO. HLA class II-restricted presentation of cytoplasmic measles virus antigens to cytotoxic T cells. J Virol. 1989;63:1756–1762. doi: 10.1128/jvi.63.4.1756-1762.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ausubel, F.M., R. Brent, R.E. Kingston, D.D. Moore, J.G. Seidman, J.A. Smith, and K. Struhl. 1995. Current protocols in immunology. In Molecular Cloning. Wiley Interscience, editor. 201–203.
- 36.Acha-Orbea H, Scarpellino L, Shakhov AN, Held W, MacDonald HR. Inhibition of mouse mammary tumor virus-induced T cell responses in vivo by antibodies to an open reading frame protein. J Exp Med. 1992;176:1769–1772. doi: 10.1084/jem.176.6.1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Graham FL, Van der Eb AJ. Transformation of rat cells by DNA of human adenovirus 5. Virology. 1973;54:536–539. doi: 10.1016/0042-6822(73)90163-3. [DOI] [PubMed] [Google Scholar]
- 38.Gunning P, Leavitt J, Muscat G, Ng SY, Kedes L. A human beta-actin expression vector system directs high-level accumulation of antisense transcripts. Proc Natl Acad Sci USA. 1987;84:4831–4835. doi: 10.1073/pnas.84.14.4831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maniatis, T., E.F. Fritsch, and J. Sambrook. 1982. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 40.Bond JF, Farmer SR. Regulation of tubulin and actin mRNA production in rat brain: expression of a new beta-tubulin mRNA with development. Mol & Cell Biol. 1983;3:1333–1342. doi: 10.1128/mcb.3.8.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Landegren U. Measurement of cell numbers by means of the endogenous enzyme hexosaminidase. Applications to detection of lymphokines and cell surface antigens. J Immunol Methods. 1984;67:379–388. doi: 10.1016/0022-1759(84)90477-0. [DOI] [PubMed] [Google Scholar]
- 41a.Thibodeau, J., M. Dohlsten, I. Cloutier, P.M. Lavoie, P. Bjork, F. Michel, C. Léveillé, W. Mourad, I. Kalland, and R.P. Sékaly. Molecular characterization and role in T cell activation of staphylococcal enterotoxin A binding to HLADRα chain. J. Immunol. In press. [PubMed]
- 42.Abrahmsen L, Dohlsten M, Segren S, Bjork P, Jonsson E, Kalland T. Characterization of two distinct MHC class II binding sites in the superantigen staphylococcal enterotoxin A. EMBO (Eur Mol Biol Organ) J. 1995;14:2978–2986. doi: 10.1002/j.1460-2075.1995.tb07300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim J, Urban RG, Strominger JL, Wiley DC. Toxic shock syndrome toxin-1 complexed with a class II major histocompatibility molecule HLA-DR1. Science (Wash DC) 1994;266:1870–1874. doi: 10.1126/science.7997880. [DOI] [PubMed] [Google Scholar]
- 44.Thibodeau J, Cloutier I, Lavoie PM, Labrecque N, Mourad W, Jardetzky T, Sekaly RP. Subsets of HLA-DR1 molecules defined by SEB and TSST-1 binding. Science (Wash DC) 1994;266:1874–1878. doi: 10.1126/science.7997881. [DOI] [PubMed] [Google Scholar]
- 45.Winslow GM, Scherer MT, Kappler JW, Marrack P. Detection and biochemical characterization of the mouse mammary tumor virus 7 superantigen (Mls-1a) Cell. 1992;71:719–730. doi: 10.1016/0092-8674(92)90549-r. [DOI] [PubMed] [Google Scholar]
- 46.McCune JM, Rabin LB, Feinberg MB, Lieberman M, Kosek JC, Reyes GR, Weissman IL. Endoproteolytic cleavage of gp160 is required for the activation of human immunodeficiency virus. Cell. 1988;53:55–67. doi: 10.1016/0092-8674(88)90487-4. [DOI] [PubMed] [Google Scholar]
- 47.Jarvis CD, Germain RN, Hager GL, Damschroder M, Matis LA. Tissue-specific expression of messenger RNAs encoding endogenous viral superantigens. J Immunol. 1994;152:1032–1038. [PubMed] [Google Scholar]
- 48.Torres BA, Griggs ND, Johnson HM. Bacterial and retroviral superantigens share a common binding region on class II MHC antigens. Nature (Lond) 1993;364:152–154. doi: 10.1038/364152a0. [DOI] [PubMed] [Google Scholar]
- 49.Moore NC, Anderson G, McLoughlin DE, Owen JJ, Jenkinson EJ. Differential expression of Mtv loci in MHC class II-positive thymic stromal cells. J Immunol. 1994;152:4826–4831. [PubMed] [Google Scholar]
- 50.Kaiserlian D, Vidal K, MacDonald HR, Grosjean I. Mouse intestinal epithelial cells express the self superantigen Mls1a. Eur J Immunol. 1993;23:2717–2720. doi: 10.1002/eji.1830231053. [DOI] [PubMed] [Google Scholar]