

Abstract
NF-κB is an important transcription factor required for T cell proliferation and other immunological functions. The NF-κB1 gene encodes a 105-kD protein that is the precursor of the p50 component of NF-κB. Previously, we and others have demonstrated that NF-κB regulates the NF-κB1 gene. In this manuscript we have investigated the molecular mechanisms by which T cell lines stimulated with phorbol 12-myristate 13-acetate (PMA) and phytohemagglutin (PHA) display significantly higher levels of NF-κB1 encoding transcripts than cells stimulated with tumor necrosis factor-α, despite the fact that both stimuli activate NF-κB. Characterization of the NF-κB1 promoter identified an Egr-1 site which was found to be essential for both the PMA/ PHA-mediated induction as well as the synergistic activation observed after the expression of the RelA subunit of NF-κB and Egr-1. Furthermore, Egr-1 induction was required for endogenous NF-κB1 gene expression, since PMA/PHA-stimulated T cell lines expressing antisense Egr-1 RNA were inhibited in their ability to upregulate NF-κB1 transcription. Our studies indicate that transcriptional synergy mediated by activation of both Egr-1 and NF-κB may have important ramifications in T cell development by upregulating NF-κB1 gene expression.
Tcell activation results in a rapid activation of NF-κB (1, 2) which appears to play an important role in T cell proliferation, partly because of the involvement of this transcription factor in the upregulation of the IL-2 and cognate receptor (IL-2R) genes (3). NF-κB was originally described to be composed of heterodimeric subunits of p50 and p65 proteins (1, 2) and is typically found in the cytoplasm of cells as an inactive precursor complex with an inhibitor protein, IκB (4). The NF-κB1 gene product p105 is processed into the DNA-binding subunit of p50 through a mechanism that requires serine phosphorylation (5) and the induction of an ATP-dependent ubiquitin–proteasome pathway (6, 7). Cellular activation leads to an increase in NF-κB1 transcripts, p105 processing, and subsequent p50 accumulation in both fibroblasts and in T cells (5, 8).
Although little is known about the regulation of NFκB1 in T lymphocytes, its expression has been demonstrated to be essential for proliferation after costimulation through CD2 or CD28 receptors (9). Recently, it has been demonstrated that lymphocytes derived from p50−/−-deficient mice display defects in immune responses (10). In addition Sha et al. (10) noted that T cells derived from these animals were unable to effectively proliferate in response to TCR and CD28 costimulation. Although disruption of the NF-κB1 gene did not result in lethality, possibly because NF-κB2/p52 could substitute for p50, these studies strongly suggest that the p50 DNA-binding component of NF-κB is critical for T cell proliferation.
Analysis of the NF-κB1 promoter revealed several NF-κB DNA-binding sites within the upstream regulatory region which are capable of binding p50 homodimers, p50/p65, or p50/c-Rel heterodimers and, in part, account for the ability of NF-κB to regulate NF-κB1 gene expression (11, 12). Although the NF-κB1 promoter has been shown to require NF-κB for activity, it is presently unclear whether NF-κB needs other transcription factors to mediate a transcriptional response. This is especially important because on many responsive promoters NF-κB alone is insufficient at activating transcription (2). T cell stimulation mediated by the addition of either PMA and PHA or TNF-α results in an increase in NF-κB1 expression (13). Both stimuli activate NF-κB nuclear translocation and DNA-binding within 15 to 30 min (14); however, phorbol esters in combination with mitogens result in a greater accumulation of NF-κB1 transcripts (11, 13). Our research demonstrates that full PMA/PHA-mediated activation of the NF-κB1 promoter not only required the NF-κB transcription factor, but also the activation of the early growth response gene product (Egr-1)1. Egr-1, also known as NGFI-A, Zif268, Krox-24, and Tis 8, is transiently induced during the activation of T cells from the G0 to G1 phase in response to various cellular stimuli (15), has been recently demonstrated to be required for T cell proliferation, and is known to regulate transcription of several genes (15–17). Our data establishes Egr-1 and RelA as important transcription factors capable of synergistically transactivating the NF-κB1 promoter.
Materials and Methods
Cell Culture and Transient Transfection Analysis.
CEM cells were cultured in RPMI 1640 medium containing 10% FCS and antibiotics. Cells were stimulated by supplementing their growth medium with either PMA and PHA (Sigma Chemical Co., St. Louis, MO) to a final concentration of 50 ng/ml and 5 μg/ml, respectively, or TNF-α (10 ng/ml; Promega Corp., Madison, WI). Transient transfections by electroporation (11) and luciferase or chloramphenicol acetyl-transferase assays were performed as previously described (11, 18, 19).
Plasmid Constructs and Site-directed Mutagenesis.
Both the NFκB1 promoter constructs, p50HS- and p50SS-CAT, were previously described (11). The SpS-, HiS-, and AS-CAT constructs were generated by digesting the p50HS-CAT reporter with HindIII, and SphI, HinfI, or ApoI, respectively. The SSLUC reporter plasmid was generated by cloning the StuI/SmaI fragment from p50HS-CAT into the pGL2 basic reporter containing the luciferase gene (Promega Corp.). The StuI/SmaI fragment of the NF-κB1 promoter was subjected to site-directed mutagenesis using a two-staged PCR (20). The mNLUC construct, containing the disrupted 289-bp NF-κB site, was made using a 5′ primer (5′-ATGTAACTGAGACACGCTTAAATGGAATA-3′) and a 3′ primer (5′-AGGCCATCAGCGCCGCGCCATGGCCGCA-3′) in the first round of amplification and two internal primers (5′-AGGCGCTTCCTGGAAGCTTGAATACCGGCTCCAG-3′ and 5′-CTGGAGCCGGTATTCAAGCTTCCAGGAAGCGCCT-3′) which mutated the κB element to a HindIII site. The NF-κB1 promoter sequences containing a site-directed mutation within the Egr-1 DNA binding site (mELUC) was created using the same 5′ and 3′ primers described above plus two internal primers (5′-GCGCACGCAGCGAATTCGGGAGGTAGGGTC-3′) and (5′-GACCCTACCTCCCGAATTCGCTGCGTGCGC-3′) which destroyed both the SP1/Egr-1 motif and created a unique EcoRI site. The mNELUC construct was created by mixing the mutant Egr-1 oligos described above and using the mNF-κB fragment as a template. The PCR amplified NF-κB1 promoter fragments were cloned into the SSLUC construct by replacing the wild-type PvuII/SmaI fragment with the corresponding site-directed mutant promoter sequences. Nucleotide sequences of mN, mE, and mNELUC constructs were confirmed by sequence analysis.
Northern Blot and Electrophoresis Mobility Shift Analysis.
Total cellular RNA was isolated from CEM cells using the RNeasy total RNA kit (Qiagen, Inc., Chatsworth, CA). Northern blot analysis was performed as previously described (11). Nuclear extracts were isolated and electrophoresis mobility shift analyses (EMSAs) were performed as previously described (11, 18). Oligonucleotides corresponding to the NF-κB1 promoter Egr-1 DNA binding site (5′-TCGACTCCCGCCCCCGCTGCG-3′ and 5′-TCGACGCAGCGGGGGCGGGAG-3′) were radiolabeled using α-[32P]dCTP and the Klenow fragment of DNA polymerase I. For antibody super shift assays and competition experiments, nuclear extracts were preincubated (10 min, room temperature) with either 1 μg of antiserum or with a nonradioactive double stranded oligonucleotide before the addition of the radiolabeled gel shift probe. The mutant Egr-1 probe (annealed 5′-TCGACTCCCGAATTCGCTGCG-3′ and 5′-TCGACGCAGCGAATTCGGGAG-3′) was used in competition assays. The Egr-1–specific antibody (SC No. 189) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
Generation of Stable Cell Lines.
The full-length human Egr-1 cDNA was cloned into the EcoR1 site of the CMV-5 expression vector in the antisense orientation. CEM cells were cotransfected with either CMV-AsEgr-1 or with the CMV-5 control vector in the presence of pcDNA-3.1 Neo (Invitrogen, San Diego, CA). Transfected cells were grown in complete RPMI-1640 medium supplemented with 1.5 mg/ml of Geneticin (GIBCO BRL, Gaithersburg, MD) and resistant clones were isolated. RNase protection assays were performed using the RPA II kit (Ambion, Inc., Austin, TX) to ensure that the CEM/AsEgr-1 cells were expressing the CMV-driven antisense human Egr-1 transcript.
Results and Discussion
Differential Expression of NF-κB1 mRNA After PMA/ PHA– or TNF-α–Stimulation.
To determine whether differential expression of NF-κB1 mRNA was observed after stimulation by PMA and PHA or TNF-α, Northern blot analysis was performed on total cellular RNA isolated from the human T cell line, CEM. As shown in Fig. 1 A, NFκB1 mRNA began to accumulate within 1 h of stimulation with PMA and PHA, and by 4 h a significant induction (10-fold) was observed in CEM cells. Although TNF-α–stimulated cells also displayed increases in NF-κB1 transcripts with similar kinetics, a substantially lower accumulation of mRNA was observed over the 4-h time course.
Figure 1.


NF-κB1 gene expression and promoter analysis in CEM cells after PMA/ PHA or TNF-α stimulation. (A) RNAs were isolated from CEM cells after addition of either PMA/PHA (50 ng and 5 μg/ml) or TNF-α (10 ng/ml) over the time course indicated. RNAs were detected using a 32Plabeled NF-κB1-specific probe. Transcripts encoding NF-κB1 were quantitated by densitometric scanning of autoradiograms and the fold accumulation of mRNAs were calculated after normalization to β-actin. (B) Schematic of the NF-κB1 promoter depicting the Egr-1 and NF-κB binding sites. CEM cells were transfected with either the HS-CAT reporter or with various deletion constructs (SS-, SpS-, HiS-, and AS-CAT). 18 h after transfection, cells were stimulated with either PMA/PHA (50 ng and 5 μg/ ml) or TNF-α (10 ng/ml) and CAT activity was analyzed. Transfection experiments were performed in triplicate and the mean fold induction and the standard deviations are shown.
Identification of the NF-κB1 Promoter Region Required for PMA/PHA-Responsive Upregulation.
To determine whether the increase in NF-κB1 mRNA, seen in Fig. 1 A, was due to transcriptional upregulation of the promoter and not due to PMA/PHA-induced message stabilization, CEM cells were transiently transfected with the p50HS-CAT NF-κB1 promoter construct and stimulated with either PMA/PHA or TNF-α for 24 h. As shown in Fig. 1 B, the NF-κB1 promoter (p50HS-CAT) was strongly activated in CEM cells after PMA/PHA stimulation, but only weakly after TNF-α addition, suggesting that PMA/PHA-mediated activation of the NF-κB1 promoter was through a transcription-dependent mechanism.
To localize the PMA/PHA-responsive region within the NF-κB1 promoter, a series of deletion promoter constructs were made. CEM cells were transiently transfected with each reporter construct and then either stimulated with PMA/PHA or with TNF-α. As shown in Fig. 1 B, no significant difference in stimulation was observed between the HS-, the SS-, or the SpS-CAT constructs. However, the HiS-CAT reporter, which lacked the −289 NF-κB site, displayed a significant decrease in NF-κB1 promoter activity after stimulation by either PMA/PHA or TNF-α. The AS-CAT reporter, lacking both the −103 and −11 κB sites, was greatly diminished in response to PMA/PHA– and TNF-α–mediated activation (Fig. 1 B). Collectively, this data indicates that the region between the SphI site (−425) and the ApoI site (−9) was critical for both inducers, but that PMA/PHA stimulation resulted in a higher fold activation than TNF-α. Based on the deletion analysis of the NF-κB1 promoter, these results suggest that both stimuli may use NF-κB, but that the PMA/PHA stimulation must activate additional transcription factors not induced by TNF-α.
The NF-κB1 Promoter Contains an Egr-1 Consensus Site and T Cells Stimulated with PMA and PHA, but not TNF-α, Display Egr-1 DNA Binding.
Computer analysis of the SphI/ApoI fragment of the NF-κB1 promoter identified potential DNA-binding motifs for five different transcription factors including AP-1, E2F, Egr-1, NF-κB, and SP1. Of these, NF-κB, Egr-1, and AP-1 are known to display increased transactivation potential after PMA stimulation (2, 15, 21). Since PMA/PHA and TNF-α both activated NF-κB, we excluded this transcription factor as the nuclear factor responsible for differentially regulating the NF-κB1 promoter. In addition, the putative AP-1 site located in the NF-κB1 promoter was not found to affect PMA-induced transactivation of the NF-κB1 promoter (12). To test whether extracts from PMA/PHA-stimulated cells would display Egr-1 binding, EMSAs were performed. As shown in Fig. 2 A, a 32P double stranded oligonucleotide probe corresponding to the 68-bp Egr-1 site within the NF-κB1 promoter bound a nuclear protein which was present in PMA/PHA-stimulated cell extracts (lane 2), but not in unstimulated cell extracts (lane 1). This DNA–protein complex could also be competed with an excess of wild-type unlabeled oligonucleotide (lane 3), but not by equal concentrations of a corresponding mutant Egr-1 site (lane 4). Moreover, this complex contained Egr-1 protein since incubation with an Egr-1–specific antibody completely supershifted the nuclear protein bound to the 32P-probe (lane 5). Pretreatment with cycloheximide, which has been previously demonstrated to block PMA-mediated increases in Egr-1 protein (15), prevented the detection of Egr-1–specific binding (lane 6). Although we do not effectively detect SP1 binding under our EMSA conditions (Fig. 2 A), we observed constitutive SP1 binding in nuclear extracts isolated from CEM cells using a modified binding protocol (data not shown). Since SP1 binding to the −68 site was constitutive and completely independent of cellular stimulation mediated by either PMA/ PHA or TNF-α (data not shown), we chose to focus on the inducible binding of the Egr-1 transcription factor.
Figure 2.


Identification of an Egr-1 DNA binding site within the NFκB1 promoter. (A) Nuclear extracts were isolated from CEM cells after a 2 h PMA/PHA stimulation in either the absence or presence of cycloheximide (50 μg/ml, 30 min pretreatment). Nuclear extracts (5 μg) were incubated with a 32P-labeled probe corresponding to the 68-bp Egr-1 site. Competition assays and antibody supershift experiments were performed by preincubating nuclear extracts (15 min) with either unlabeled mutant Egr-1 (mEgr-1) or wild-type Egr-1 oligonucleotide (50 ng total) before the addition of probe. Lane 1, unstimulated cells; lane 2, PMA/PHA for 2 h; lane 3, PMA/PHA + wild-type Egr-1 oligo; lane 4, PMA/PHA + mEgr-1 oligo; lane 5, PMA/PHA + Egr-1–specific antibody; lane 6, pretreatment with cycloheximide (30 min) + PMA/PHA for 2 h. SS indicates the position of the Egr-1 + antibody supershift complex; NS identifies nonspecific bands. (B) Nuclear proteins from CEM cells were isolated after the addition of either PMA/PHA or TNF-α over the indicated time course and EMSAs were performed using the 32P-labeled Egr-1 site. Note that the unbound 32P-labeled probe is not shown in both A and B.
To elucidate whether differential Egr-1 binding was observed in T cells after the addition of either PMA/PHA or TNF-α, nuclear proteins were isolated from stimulated CEM cells over a 4 h time course and EMSAs were performed using the NF-κB1 Egr-1 consensus site. After stimulation with PMA/PHA, Egr-1 binding begins to appear within 30 min and continues to increase at both 1 and 2 h with a slight decrease seen at 4 h (Fig. 2 B). Interestingly, no Egr-1 binding was observed in CEM cell extracts after TNF-α stimulation (Fig. 2 B). However, both PMA/ PHA– and TNF-α–stimulated CEM cells demonstrated NF-κB–specific binding after the addition of each inducer, confirming that CEM cells were capable of responding to TNF-α (data not shown). Identical results to those obtained in the CEM cell line were demonstrated in Jurkat T cells (data not shown). Collectively, these results indicate that the −68 site located in the NF-κB1 promoter is capable of binding the Egr-1 transcription factor and that, unlike PMA/PHA stimulation, TNF-α fails to stimulate Egr-1 binding in T cell lines.
Efficient PMA/PHA-Responsive Activation of the NF-κB1 Promoter Requires the Egr-1 DNA Binding Site.
To determine whether Egr-1 is required for PMA/PHA responsive activation of the NF-κB1 promoter, site-directed mutagenesis was performed. Since the NF-κB1 promoter is regulated directly by NF-κB (11, 12), we chose to mutate the −289 κB motif alone or in combination with the −68 Egr-1 binding site. The four luciferase NF-κB1 promoter constructs included (a) SSLUC, the wild-type construct, (b) mNLUC, which contained a mutated NF-κB site at position −289, (c) mELUC, which contained the disrupted 68-bp Egr-1 site, and (d ) mNELUC, which contained mutated NF-κB and Egr-1 sites. As shown in Fig. 3 A, site-directed mutagenesis of the Egr-1 binding element (mELUC) significantly diminished PMA/PHA-responsive activation of the NF-κB1 promoter, but had very little or no effect on TNF-α–mediated stimulation. In contrast, mutating the NF-κB −289 site alone had very little effect on either PMA/PHA– or TNF-α–induced activation (data not shown). These results in conjunction with the promoter deletion studies (shown in Fig. 1 B) would strongly suggest that other κB binding sites (namely −103 and −11) are important for transcriptional activation. Recently, McElhinny et al. (22) demonstrated that the −11 NF-κB site was required for HIV-mediated induction of NF-κB1 promoter in monocytes. Although the −289 motif is probably not the only functional NF-κB motif, site-directed mutagenesis of both the Egr-1 and NF-κB elements (mNELUC), resulted in a further decrease in PMA/PHA-responsive stimulation compared to the mELUC construct (Fig. 3 A). Taking into account that mutagenesis of the Egr-1 site in the NF-κB1 promoter significantly diminished PMA/PHA-responsive stimulation, these results indicate that the −68 Egr-1 site plays an important role in the activation of this gene in T lymphocytes.
Figure 3.


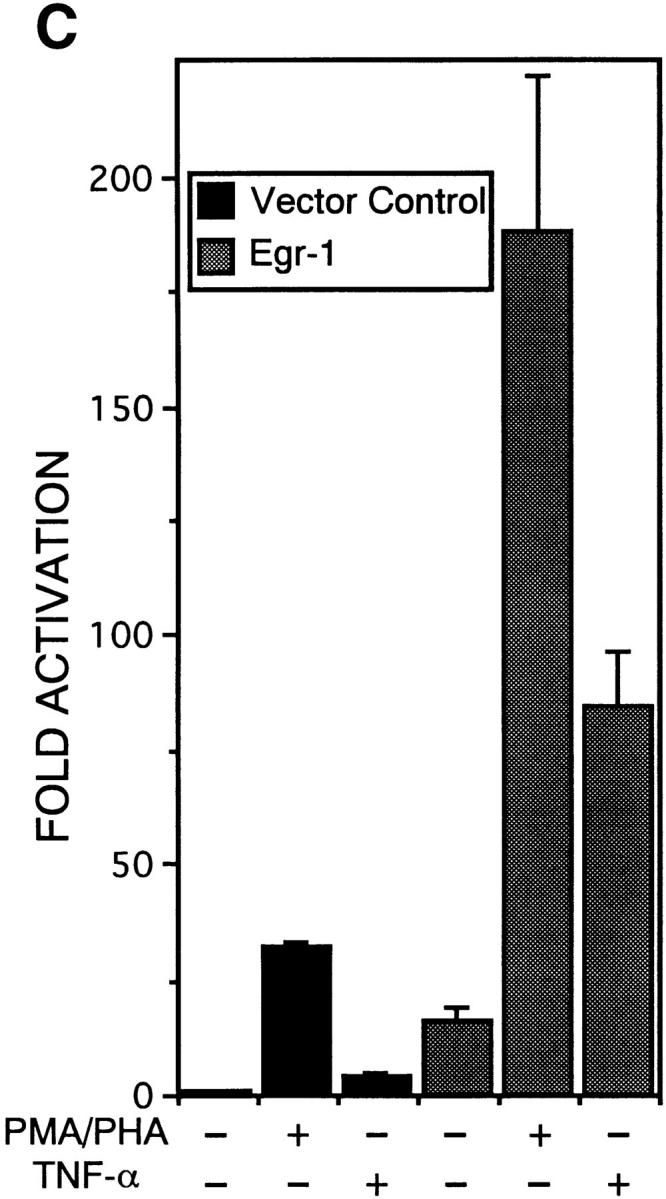
The Egr-1 DNA binding site is required for PMA/PHA–responsive activation and for transcriptional synergy mediated by RelA and Egr-1. (A) CEM cells were transfected with either the wild-type NF-κB1 promoter (SSLUC) or with each of the site-directed mutants including (a) mNLUC, which contained a mutated NF-κB site at position −289, (b) mELUC, which contained a disrupted 68-bp Egr-1 site, and (c) mNELUC, which contained mutated NF-κB and Egr-1 sites, and 18 h later cells were stimulated with either PMA/PHA or TNF-α. 24 h after stimulation, cell extracts were harvested and assayed for luciferase activity. (B) CEM cells were co-transfected with either the SSLUC construct or with mutant reporters (mN, ME, mNE; 5 μg each) and with various expression vectors (5 μg) including an empty vector control, RelA, Egr-1, or RelA plus Egr-1. Cells were harvested 48 h after transfection and luciferase activities were determined. (C) The SSLUC construct was transfected into CEM cells along with either the vector control or with an Egr-1 expression construct. 18 h after transfection, cells were either left untreated or stimulated with PMA/PHA or TNF-α. Cells were harvested 24 h after stimulation and extracts were analyzed for luciferase activity. All transfection assays were performed in triplicate in three independent experiments.
NF-κB and Egr-1 Transcription Factors Synergistically Transactivate the NF-κB1 Promoter.
To further explore the ability of each of these transcription factors to activate the NFκB1 promoter, the wild-type as well as the site-directed mutant reporters were cotransfected with the empty expression plasmid or with constructs encoding either RelA, the p65 subunit of NF-κB, Egr-1, or a combination of both. As shown in Fig. 3 B, the SSLUC reporter was strongly transactivated after cotransfection with the RelA encoding expression construct, but only weakly activated by the overexpression of Egr-1. Interestingly, transfection with both RelA and Egr-1 resulted in a potent synergistic transactivation of the NF-κB1 promoter. Although the −289 κB site was important for RelA-mediated transactivation, mutagenesis of this element did not affect activation by Egr-1, nor did it affect RelA/Egr-1 synergy (Fig. 3 B). This result would suggest that, in the presence of Egr-1 binding, other κB sites located within the NF-κB1 promoter are used for the synergistic response. Unlike the mNLUC reporter, mutagenesis of the Egr-1 site (mELUC) resulted in a significant reduction in RelA-, Egr1-, and RelA/Egr1mediated activation of the NF-κB1 promoter (Fig. 3 B). However, disruption of both the −289 and −68 sites (mNELUC) did not further diminish RelA/Egr-1–mediated synergy. This would suggest that overexpression of both of these transcription factors may indirectly lead to the activation of the NF-κB1 promoter by upregulating other trans-acting elements, or that Egr-1 (like NF-κB) recognizes other DNA-binding sites within the NF-κB1 promoter. Collectively, co-transfection studies strongly suggest that the Egr-1 DNA binding site is critical for both Egr1–mediated transactivation and for RelA/Egr-1-dependent synergy. Although the Rel homology domain of RelA has been shown to be responsible for protein–protein interactions with transcription factors outside of the NF-κB/Rel family (23–26), we were unable to demonstrate a physical interaction between RelA and Egr-1, nor were we able to generate synergistic transactivation mediated by these factors using either a 3×–NF-κB or 3×-Egr-1 CAT reporter (data not shown).
Egr-1 Complements TNF-α to Stimulate the NF-κB1 Promoter.
To determine whether the difference between PMA/ PHA– and TNF-α–stimulated NF-κB1 gene expression was due to the inability of TNF-α to activate Egr-1, CEM cells were transfected with the SSLUC reporter along with the empty vector control or with the Egr-1 expression construct and 24 h later cells were stimulated with PMA/PHA or TNF-α. As shown in Fig. 3 C, the fold increase between cells transfected with the vector control or with the Egr-1 expression construct was comparable to previous experiments, while PMA/PHA-stimulated cells (coexpressing Egr-1) displayed the highest level of NF-κB1 activation. Interestingly, cells transfected with Egr-1 and stimulated with TNF-α displayed a significant increase in promoter activity, compared to TNF-α–stimulated cells transfected with the vector control. The ability of Egr-1 to augment TNF-α–mediated activation of the NF-κB1 promoter was a direct effect since the transcriptional synergy was not observed with the mELUC construct containing the disrupted Egr-1 binding site (data not shown). These results demonstrate that the Egr-1 transcription factor can complement TNF-α–mediated stimulation of the NF-κB1 promoter. Furthermore, our data suggests that the inability of TNF-α to activate Egr-1 may account for the difference between PMA/PHA– and TNF-α–mediated NF-κB1 gene expression in T cells.
Endogenous NF-κB1 Gene Expression Is Affected in T Cells Expressing Antisense Egr-1 RNA.
To determine the importance of the Egr-1 transcription factor on endogenous NF-κB1 gene expression, we developed a CEM cell line which constitutively expressed antisense human Egr-1 RNA. Total RNAs were isolated from either CEM/AsEgr-1 cells (which express the human Egr-1 antisense RNA) or from CEM/CMV control cells (which harbor the empty expression vector) after the addition of either PMA/PHA or TNF-α over the time course indicated. As shown in Fig. 4 A, CEM/AsEgr-1 cells did not display significant increases in NF-κB1 transcripts until 4 h after PMA/PHA addition, while the control cells (CEM/CMV) responded within 1 to 2 h with similar kinetics as the parental CEM cell line (Fig. 1 A). Interestingly, TNF-α–stimulated CEM/ AsEgr-1 cells demonstrated a reduction in NF-κB1 transcripts 30 min after activation. Although TNF-α does not activate Egr-1, it would appear that under conditions in which endogenous Egr-1 levels are reduced (as with CEM/ AsEgr-1 cells), TNF-α stimulation represses NF-κB1 transcription (Fig. 4 A). Although the level of Egr-1 protein in the CEM/AsEgr-1 cells after PMA/PHA stimulation was significantly lower (fivefold) than protein levels observed in the control line, the antisense Egr-1 transcripts were not able to completely block PMA/PHA-mediated increases in Egr-1 protein (Fig. 4 B). Perhaps for this reason CEM/ AsEgr-1 cells are still able to display PMA/PHA-responsive upregulation of NF-κB1 transcripts but with slower kinetics.
Figure 4.


Egr-1 is required for endogenous NF-κB1 gene expression. (A) Total RNAs were isolated from CEM/CMV control cells or from CEM/AsEgr-1 cells stimulated with either PMA/PHA or TNF-α over a 4 h period. RNAs were detected using a 32P-labeled NF-κB1 specific probe. (B) Total proteins were isolated from CEM/CMV or CEM/AsEgr-1 cells after the addition of either PMA/PHA or TNF-α. Proteins were subjected to PAGE and analyzed using an Egr-1–specific antibody and enhanced chemiluminescent assay. NS, shows the presence of a nonspecific band and demonstrates that equal amounts of protein were loaded in each lane.
In this paper we have demonstrated that to achieve full activation of the NF-κB1 promoter, T cell signals require not only the activation of NF-κB, but also the Egr-1 transcription factor. Interestingly, it appears that the potent activation of NF-κB DNA binding activity by TNF-α is insufficient at inducing high levels of NF-κB1 transcription. This is consistent with the recent report that activation of NF-κB binding by TNF-α is insufficient at activating VCAM-1 transcription in some cells, but the co-activation of NF-κB and IRF-1 in endothelial cells functions to stimulate transcription (27, 28). Moreover, like NF-κB, Egr-1 is a relatively poor activator of NF-κB1 transcription. Thus, it is the combined action of NF-κB and Egr-1, both of which alone result in weak activation, that functions to synergistically stimulate the NF-κB1 promoter. A number of NFκBresponsive genes have been characterized and found to require Egr-1 DNA binding motifs for maximum transactivation. Such genes include TNF-α, IL-2, and ICAM-1 (17, 29, 30). In T cells, TNF-α does not fully upregulate its own promoter, presumably because of the inability of cytokines to activate Egr-1 (17). However, primary human fibroblasts and macrophage cell lines display dramatic increases in Egr-1 after TNF-α stimulation (15). Collectively, these studies would suggest that the synergistic potential mediated by the NFκB and Egr-1 transcription factors are not only restricted by the type of stimuli, but also may depend on the particular cell type. Thus, it appears that the functional interaction of NF-κB and Egr-1 in the context of a promoter is likely to mediate critical aspects of T cell signaling and potentially other important aspects of cellular growth control. The potent synergy between NF-κB and Egr-1 may explain, at least partially, the requirements for these transcription factors in T cell proliferation with NF-κB1 being a critical downstream target for Egr-1.
Acknowledgments
We would like to thank Dr. Vikas P. Sukhatme for providing the human Egr-1 cDNA. We would also like to thank S. Scott Drouin for technical assistance in the preparation of this manuscript.
Footnotes
This work was funded by the National Institute of Health (NIH) grant AI35098 awarded to A.S Baldwin and by the NIH postdoctoral fellowship grant 1F32-CA69790-01 awarded to M.W. Mayo.
1 Abbreviations used in this paper: Egr-1; early growth response gene product; EMSA, electrophoretic mobility shift assay.
P.C. Cogswell and M.W. Mayo contributed equally to the scientific merit and preparation of this manuscript.
References
- 1.Baeuerle PA, Henkel T. Function and activation of NF-κB in the immune system. Annu Rev Immunol. 1994;12:141–179. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- 2.Baldwin AS. The NF-κB and IκB proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–681. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 3.Baeuerle PA. The inducible transcription activator NF-κB: regulation by distinct protein subunits. Biochim Biophys Acta. 1991;1072:63–80. doi: 10.1016/0304-419x(91)90007-8. [DOI] [PubMed] [Google Scholar]
- 4.Baeuerle PA, Baltimore D. IκB: a specific inhibitor of the NF-κB transcription factor. Science (Wash DC) 1988;242:540–546. doi: 10.1126/science.3140380. [DOI] [PubMed] [Google Scholar]
- 5.MacKichan ML, Logeat F, Israel A. Phosphorylation of p105 PEST sequence via a redox-insensitive pathway up-regulates processing to p50 NF-κB. J Biol Chem. 1996;270:18347–18351. doi: 10.1074/jbc.271.11.6084. [DOI] [PubMed] [Google Scholar]
- 6.Palombella VJ, Rando OJ, Goldberg AL, Maniatis T. The ubiquitin-proteasome pathway is regulated for processing the NF-κB precursor protein and the activation of NF-κB. Cell. 1994;78:773–785. doi: 10.1016/s0092-8674(94)90482-0. [DOI] [PubMed] [Google Scholar]
- 7.Chen ZJ, Hagler J, Palombella VJ, Melandri F, Scherer D, Ballard D, Maniatis T. Signal-induced site-specific phosphorylation targets IkBα to the ubiquitinproteasome pathway. Genes Dev. 1995;9:1586–1597. doi: 10.1101/gad.9.13.1586. [DOI] [PubMed] [Google Scholar]
- 8.Rice NR, MacKichan ML, Israel A. The precursor of NF-κB p50 has IκB-like functions. Cell. 1992;71:243–253. doi: 10.1016/0092-8674(92)90353-e. [DOI] [PubMed] [Google Scholar]
- 9.Costello R, Cerdan C, Lipcey C, Algarte M, Martin Y, Baeuerle PA, Olive D, Imbert J. The role of NF-κB1 (p50/p105) gene expression in activation of human blood T-lymphocytes via CD2 and CD28 adhesion molecules. Cell Growth Differ. 1993;4:947–954. [PubMed] [Google Scholar]
- 10.Sha WC, Liou H-C, Tuomanen EI, Baltimore D. Targeted disruption of the p50 subunit of NF-κB leads to multifocal defects in immune responses. Cell. 1995;80:321–330. doi: 10.1016/0092-8674(95)90415-8. [DOI] [PubMed] [Google Scholar]
- 11.Cogswell PC, Scheinman RI, Baldwin AS. Promoter of the human NF-κB p50/p105 gene. J Immunol. 1993;150:2794–2804. [PubMed] [Google Scholar]
- 12.Ten RM, Paya CV, Israel N, LeBail O, Mattei M-G, Virelizier J-L, Kourilsky P, Israel A. The characterization of the promoter of the gene encoding the p50 subunit of NF-κB indicates that it participates in its own regulation. EMBO (Eur Mol Biol Organ) J. 1992;11:195–203. doi: 10.1002/j.1460-2075.1992.tb05042.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meyer R, Hatada H-P, Hohmann H-P, Haiker M, Bartsch C, Rotlisberger U, Lahm H-W, Schlaeger EJ, vanLoon APGM, Scheidereit C. Cloning of the DNA-binding subunit of human nuclear factor κB: the level of its mRNA is strongly regulated by phorbol ester or tumor necrosis factor α. Proc Natl Acad Sci USA. 1991;88:966–970. doi: 10.1073/pnas.88.3.966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hohmann H-P, Remmy R, Scheidereit C, vanLoon APGM. Maintenance of NF-κB activity is dependent on protein synthesis and the continuous presence of external stimuli. Mol Cell Biol. 1991;11:259–266. doi: 10.1128/mcb.11.1.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gashler A, Sukhatme VP. Early growth response protein 1 (Egr-1): prototype of a zinc-finger family of transcription factors. Prog Nucleic Acid Res Mol Biol. 1995;50:191–224. doi: 10.1016/s0079-6603(08)60815-6. [DOI] [PubMed] [Google Scholar]
- 16.Perez-Castillo A, Pipaon C, Garcia I, Alemany S. NGFI-A gene expression is necessary for T lymphocyte proliferation. J Biol Chem. 1993;268:19445–19450. [PubMed] [Google Scholar]
- 17.Kramer B, Meichle A, Hensel G, Charnay P, Kronke M. Characterization of an Krox-24/Egr-1-response element in the human tumor necrosis factor promoter. Biochim Biophys Acta. 1994;1219:413–421. doi: 10.1016/0167-4781(94)90066-3. [DOI] [PubMed] [Google Scholar]
- 18.Finco TS, Baldwin AS. κB site-dependent induction of gene expression by diverse inducers of NF-κB requires Raf-1. J Biol Chem. 1993;268:676–679. [PubMed] [Google Scholar]
- 19.Gilman MZ, Wilson RN, Weinberg RA. Multiple protein-binding sites in the 5′ flanking region regulate c-fosexpression. Mol Cell Biol. 1986;6:4305–4316. doi: 10.1128/mcb.6.12.4305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Higuchi R, Krummel G, Saiki R. A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res. 1988;16:7351–7367. doi: 10.1093/nar/16.15.7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bravo, R. 1990. Growth factor inducible genes in fibroblasts. In Growth Factors, Differentiation Factors and Cytokines. A. Habenich, editor. Springer-Verlag, Berlin. 324–343.
- 22.McElhinny JA, MacMorran WS, Bren GD, Ten RM, Israel A, Paya CV. Regulation of IκB and p105 in monocytes and macrophages persistently infected with human immunodeficiency virus. J Virol. 1995;69:1500–1509. doi: 10.1128/jvi.69.3.1500-1509.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stein B, Cogswell PC, Baldwin AS. Functional and physical association between NF-kappa B and C/EBP family members: a Rel domain-pZIP interaction. Mol Cell Biol. 1993;13:3964–3974. doi: 10.1128/mcb.13.7.3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Diehl J, Hannink M. Identification of a C/EBPRel complex in avian lymphoid cells. Mol Cell Biol. 1994;14:6635–6646. doi: 10.1128/mcb.14.10.6635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perkins N, Agranoff A, Pascal E, Nabel G. An interaction between the DNA binding domains of RelA (p65) and Sp1 mediates HIV gene activation. Mol Cell Biol. 1994;14:6570–6583. doi: 10.1128/mcb.14.10.6570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.John S, Reeves R, Lin J, Child R, Leiden J, Thompson C, Leonard W. Regulation of cell type specific IL-2 receptor α chain gene expression: potential role of physical interaction between Elf-1, HMG-I(Y) and NF-κB family proteins. Mol Cell Biol. 1995;15:1786–1796. doi: 10.1128/mcb.15.3.1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ahmad M, Marui N, Alexander R, Medford R. Cell type-specific transactivation of the VCAM-1 promoter through an NF-κB enhancer motif. J Biol Chem. 1995;270:8976–8983. doi: 10.1074/jbc.270.15.8976. [DOI] [PubMed] [Google Scholar]
- 28.Neish A, Read M, Thanos D, Pine R, Maniatis T, Collins T. Endothelial interferon regulatory factor 1 cooperates with NF-κB as a transcriptional activator of vascular cell adhesion molecule 1. Mol Cell Biol. 1995;15:2558–2569. doi: 10.1128/mcb.15.5.2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Skerka C, Decker EL, Zipfel PF. A regulatory element in the human interleukin 2 gene promoter is a binding site for the zinc finger proteins Sp1 and EGR-1. J Biol Chem. 1995;270:22500–22506. doi: 10.1074/jbc.270.38.22500. [DOI] [PubMed] [Google Scholar]
- 30.Maltzman JS, Carman JA, Monroe JG. Transcriptional regulation of the ICAM-1 gene in antigen receptor and phorbol ester stimulated B lymphocytes: role for transcriptional factor Egr-1. J Exp Med. 1996;183:1747–1759. doi: 10.1084/jem.183.4.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]