

Abstract
We have previously demonstrated that interleukin (IL)-10–deficient (IL-10 knockout [KO]) but not wild-type (WT) mice develop colitis after infection with Helicobacter hepaticus. Here, we show that infected recombination activating gene (RAG) KO mice develop intestinal inflammation after reconstitution with CD4+ T cells from IL-10 KO animals and that the cotransfer of CD4+ T cells from H. hepaticus–infected but not uninfected WT mice prevents this colitis. The disease-protective WT CD4+ cells are contained within the CD45RBlow fraction and unexpectedly were found in both the CD25+ and the CD25− subpopulations of these cells, their frequency being higher in the latter. The mechanism by which CD25+ and CD25− CD45RBlow CD4+ cells block colitis involves IL-10 and not transforming growth factor (TGF)-β, as treatment with anti–IL-10R but not anti–TGF-β monoclonal antibody abrogated their protective effect. In vitro, CD45RBlow CD4+ cells from infected WT mice were shown to produce IL-10 and suppress interferon-γ production by IL-10 KO CD4+ cells in an H. hepaticus antigen–specific manner. Together, our data support the concept that H. hepaticus infection results in the induction in WT mice of regulatory T cells that prevent bacteria-induced colitis. The induction of such cells in response to gut flora may be a mechanism protecting normal individuals against inflammatory bowel disease.
Keywords: inflammatory bowel disease, regulatory T cells, intestinal flora, CD25, immunoregulation
Introduction
Experimental models have established a crucial role for microbial flora in the induction of inflammatory bowel disease (IBD).*For example, several strains of immunocompromised mice and rats known to develop intestinal inflammation when housed in conventional animal facilities show minimal or no disease when reared under specific pathogen-free (SPF) or germfree (GF) conditions (1–6). A requirement for gut flora has also been demonstrated for the development of colitis in the well-established CD4/SCID transfer model of IBD in which intestinal inflammation is induced in T cell–deficient recipients by transfer of splenic CD45RBhi CD4+ cells from naive WT mice (7, 8). Thus, in this model, no inflammation is observed upon the transfer of CD45RBhi CD4+ cells to T cell–deficient mice raised under GF conditions (9) or with a reduced bacterial load (10). Moreover, the pathogenic CD4+ cells do not have to be primed by gut flora before cell transfer, as CD45RBhi cells from GF mice induce disease upon transfer to flora-containing SCID mice (11). Although the etiology of the two major chronic intestinal inflammatory diseases of man, Crohn's disease and ulcerative colitis, is largely unknown and believed to be multifactorial, several reports have also implicated a role for intestinal flora in the development of these diseases (12–14).
Considering the vast number of bacteria in the intestine, there must exist mechanisms by which immune responses to these microbes are controlled so that a state of chronic inflammation is prevented from developing in the gut. General mechanisms believed to play a role in protection against autoimmunity, such as T cell deletion, T cell anergy, and immunologic ignorance, could also potentially contribute to protection against unwanted immune responses in the gut. In addition, the presence in normal mice of T regulatory (Treg) cells able to prevent the development of intestinal inflammation has been demonstrated. Thus, the administration of CD45RBlow CD4+ cells to T cell–deficient mice along with the pathogenic CD45RBhi population leads to complete inhibition of colitis development (7, 15). These Treg cells block colitis though a mechanism involving TGF-β and IL-10 (16, 17), and recent reports have further established that the CD4+ Treg cells inhibiting disease are predominantly found within the CD25+ CD45RBlow population (18, 19).
We have previously demonstrated that SPF IL-10 KO mice experimentally infected with Helicobacter hepaticus develop severe inflammation in the cecum and colon, a disease associated with a Th1-type cytokine response by mesenteric lymph node (MLN) cells stimulated in vitro with soluble H. hepaticus Ag (SHelAg; references 20 and 21). In contrast, WT mice do not develop intestinal inflammation after inoculation with this bacterium and their MLN cells instead produce IL-10 in response to SHelAg (20). These results suggested that infected WT mice mount a disease-protective CD4+ T cell response to the bacterium, possibly a Treg type of response that prevents the development of colitis in these animals.
In this study, we have characterized the phenotype and mode of action of the CD4+ T cells in H. hepaticus–infected WT animals that protect against disease. Using a modification of the CD4/SCID IBD model, we induced colitis in H. hepaticus–infected RAG KO animals by the transfer of CD4+ T cells from infected colitic IL-10 KO mice and then analyzed the disease-protective effect of the cotransfer of various CD4+ cell populations from WT mice. We found that CD25− CD45RBlow and, to a much lesser extent, CD25+ CD45RBlow CD4+ cells from H. hepaticus–infected WT mice block colitis development and that this protection is dependent on IL-10. Importantly, similar populations of CD4+ cells derived from uninfected WT animals did not block disease development. The disease-protective cells are distinct from conventional Th2 lymphocytes and instead closely resemble the in vitro–generated CD4+ IL-10–producing Treg cells recently described (22–24). Taken together, our results suggest the involvement of bacteria-primed Treg cells in prevention of IBD in normal individuals.
Materials and Methods
Experimental Animals and Infections.
6–16-wk-old female SPF C57BL/10 IL-10 KO (backcrossed to the 10th generation), C57BL/10 (CD45.2+), C57BL/6.SJL (CD45.1+), C57BL/6 IL-4 KO (backcrossed to the 12th generation), C57BL/10 RAG-2 KO, and C57BL/10 RAG-2/IL-10 double KO (provided by T. Wynn, NIAID, NIH, Bethesda, MD) mice were obtained from Taconic Farms. The animals tested negative for antibodies to specific murine viruses and were free of Helicobacter species as assessed by PCR. Animals were housed in sterile microisolator cages with autoclaved bedding, food, and water at the animal facility at the NIAID in accordance with the Guide for the Care and Use of Laboratory Animals (25) under an animal study proposal approved by the NIAID Animal Care and Use Committee.
Mice were inoculated intragastrically with 0.5 ml of an H. hepaticus suspension (standard Frederick isolate 1A; references 26 and 27) prepared to a McFarland turbidity standard of 1.0 in PBS representing 2.45 × 109 CFU/ml. Age-matched uninfected animals were included as controls.
Bacterial Ag Preparation.
SHelAg was prepared from cultures of H. hepaticus as previously described (20, 21). SHelAg was boiled for 5 min, a procedure shown to have no effect on the ability of the Ag to activate T cells (unpublished data), and stored at −40°C until use.
Cell Purifications.
CD4+ cells were purified from the MLNs of naive or 5–21-wk infected WT, IL-10 KO, or IL-4 KO mice by negative selection using CD4 columns (R&D Systems). The CD4+ cells were generally >95% pure (range 91–98%) as analyzed by flow cytometry. For purification of CD45RBhigh, CD45RBlow, CD25+, and CD25− subpopulations of CD4+ cells, MLNs were stained with anti–CD4-PE (clone RM4-5) or anti–CD4-CyChrome (RM4-5), anti–CD45RB-FITC (16A), anti–CD25-biotin (7D4), and streptavidin-PE (all from BD Biosciences), and sorted on a FACSVantage™ SE or a FACStarPlus™ SE (Becton Dickinson). In some experiments, MLNs were passed over a CD4 column before staining and sorting. The sorted subpopulations of CD4+ cells were ≥99% pure. The CD45RBlow and CD45RBhi cells were collected after gating on 15–20% of the dimmest and ∼50% of the brightest CD4+ cells, respectively.
For APC, CD11c+ dendritic cells (DCs) were purified from the spleens of naive IL-10 KO mice using anti-CD11c MACS microbeads (Miltenyi Biotech). The CD11c+ DCs were ∼90% pure.
Cell Transfers to RAG KO Mice and In Vivo mAb Treatment.
MLN CD4+ cells or subpopulations thereof (3–4 × 105/mouse or as indicated) were transferred intravenously to naive or 2–4-d H. hepaticus–infected RAG KO mice. Mice receiving no cells were included as controls. 8–52 d after cell transfer, mice were killed and intestinal tissues were collected for histologic analysis.
In some experiments, mice were injected intraperitoneally 1 d after cell transfer and then each week for 4 wk with 2 mg anti–TGF-β (mAb 2G75A9, anti-TGF-β1,2,3), 1 mg anti–IL-10R (1B1.3a), or control mAb GL113 (anti–β-galactosidase) in 0.5 ml PBS. In a separate experiment, mice were given 2 mg anti–TGF-β twice a week for 2 wk. The anti–TGF-β mAb was purified from ascites by ion exchange chromatography. The anti–IL-10R and control mAb were purified from ascites by two sequential 50% ammonium sulfate precipitations.
Pathology and Immunohistochemistry.
Tissues were fixed in Bouin's fixative, embedded in paraffin, sectioned at 5 μm, and stained with hematoxylin and eosin (H&E). A longitudinal section of the entire cecum and a cross section of the ascending colon ∼1 cm from the cecum were made. Sections were evaluated in a blinded fashion by the same pathologist, A.W. Cheever, and an average score for the whole section was assigned based on a 0–4+ scoring system with emphasis on the number of infiltrating cells for the lamina propria, the submucosa, and the serosa (21). For the lamina propria and submucosa, a score of 1+ indicated the equivalent of a single layer of infiltrating cells throughout the section and 4+ indicated ≥4 cell layers. In addition, crypt abscesses and ulcers were each given a score from 0 to 4. A total score was calculated by adding the individual scores, meaning that the maximum total score was 20. Hyperplasia was evaluated as mucosal thickness using an ocular micrometer. Staining of colonic tissues for CD3+ T cells was performed as previously described (20, 21).
Cell Cultures and Cytokine Assays.
Cells were cultured in complete tissue culture medium (RPMI 1640 with 10% heat-inactivated FCS, 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM glutamine, 20 mM Hepes, 1 mM sodium pyruvate, 0.1 mM nonessential amino acids, and 50 μM 2-ME) and 5% CO2 at 37°C. CD4+ T cells (105/well) from infected IL-10 KO mice were cultured in 96-well round-bottomed plates (0.2 ml/well) with CD11c+ DC (4 × 104/well) from naive IL-10 KO animals as APC, 5 μg/ml SHelAg, and indicated numbers of CD45RBlow CD4+ cells from naive or infected WT mice for 72 h.
CD45RBlow CD4+ cells (2 × 105/well) from naive or infected WT mice were cultured in 96-well round-bottomed plates (0.2 ml/well) with CD11c+ DCs (4 × 104/well) from naive IL-10 KO animals as APC, 5 μg/ml SHelAg, and 20 U/ml human rIL-2 (Chiron Corp.) for 72 h. IL-2 was added to cultures as it has been shown to be necessary for revealing the responsiveness of some CD45RBlow populations (28).
Supernatants were analyzed for IFN-γ and IL-10 by ELISA using mAb from BD Biosciences.
Intracellular Cytokine Staining.
Analyses of intracellular cytokine expression were performed on cells from the same SHelAg-stimulated cultures used for cytokine secretion assays as previously described (29). In brief, cells were incubated for an additional 18 h in fresh medium containing 10 U/ml of rIL-2 added to replace the culture supernatant collected at 72 h. Thereafter, cells were stimulated with 1 μg/ml PMA and 1 ng/ml ionomycin for 4.5 h with the addition of 10 μg/ml brefeldin A (all from Sigma-Aldrich) during the last 2 h. Intracellular staining was then performed as previously described (29) using anti–IL-10–PE (JES5-16E3) and anti–IFN-γ–FITC (XMG1.2; both from BD Biosciences). Cell fluorescence was measured using a FACScan™ (Becton Dickinson) and the data were analyzed using Cell Quest™ software (Becton Dickinson).
Statistical Analysis.
Colitis scores were compared using the nonparametric Mann-Whitney U test and differences were considered statistically significant with P < 0.05.
Results
Transfer of CD4+ T Cells from IL-10 KO Mice Results in Colitis in H. hepaticus–infected but Not Uninfected RAG KO Recipients.
We have previously shown that SPF-reared IL-10 KO mice develop a Th1 cytokine-associated colitis after experimental infection with H. hepaticus (20, 21). In contrast, H. hepaticus–inoculated WT mice are free of disease and mount a T cell–dependent IL-10 response to the bacterium (20), suggesting the development of a disease-protective CD4+ T cell response. To characterize this protective T cell response in WT mice, we used a modification of the CD4/SCID transfer model of colitis involving experimental infection with bacteria in which disease is induced in H. hepaticus–infected RAG KO recipients by the transfer of CD4+ cells from infected colitic IL-10 KO mice. After inoculation with H. hepaticus, RAG KO mice on a C57BL/10 background showed no or minimal signs of intestinal inflammation when examined up to 34 wk after infection (Fig. 1, A and B , and not depicted). However, if 2–4-d infected RAG KO mice were reconstituted with MLN CD4+ cells from infected IL-10 KO animals, inflammation developed in the large intestine (colitis) including the cecum (typhlitis) within 1 wk, gradually increasing over a 4-wk period (Fig. 1 A). Similar results were observed when CD4+ cells from naive IL-10 KO mice were transferred to infected RAG KO recipients (unpublished data), a result we interpret as reflecting the sensitization of the naive transferred CD4+ cells by H. hepaticus in the infected recipients. The colitis observed in the RAG KO recipients was characterized by mucosal hyperplasia and inflammatory cell infiltrates in the lamina propria, submucosa, and serosa, as well as by the presence, in severe cases, of crypt abscesses and ulcers (Fig. 1 C). Importantly, when uninfected RAG KO mice were given IL-10 KO CD4+ cells, no inflammation was observed in the same time period (Fig. 1, A and D). These results demonstrate a critical role for both T cells and the Helicobacter organism in disease induction.
Figure 1.

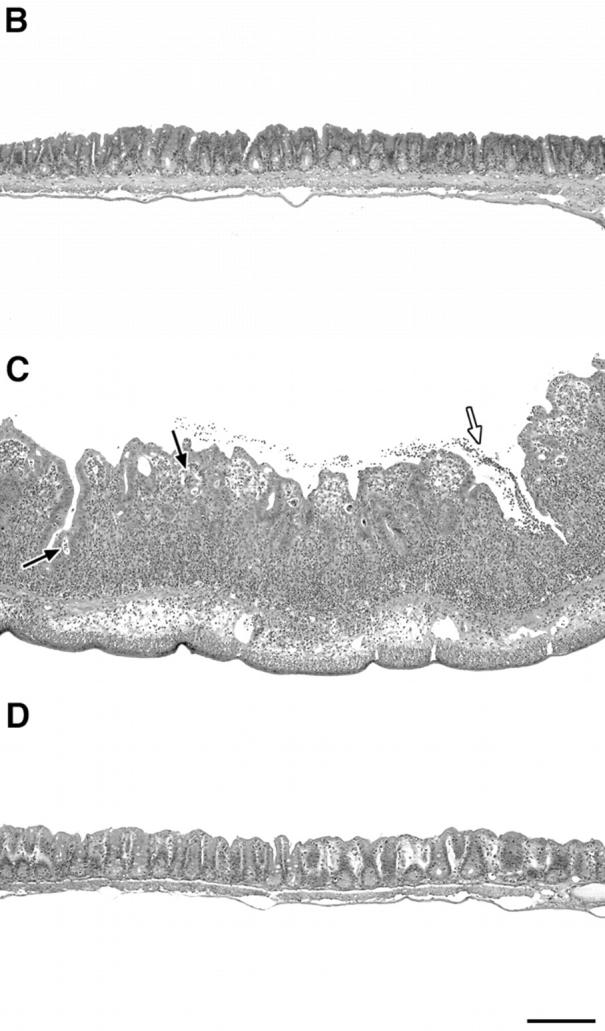
H. hepaticus–infected but not naive RAG KO mice develop intestinal inflammation after reconstitution with CD4+ T cells from IL-10 KO mice. (A) Uninfected or H. hepaticus–infected RAG KO mice were inoculated intravenously with 3 × 105 CD4+ cells from the MLNs of 11-wk infected IL-10 KO mice, and intestinal pathology was analyzed 8, 15, 22, and 29 d later (solid bars). Naive and infected RAG KO animals receiving no cells were analyzed in parallel (open bars). Bars represent mean cecal histology scores ± SD of three or four mice per group. *, P < 0.05 compared with infected mice receiving no cells. (B–D) Histology of ceca of representative sections from the mice shown in A analyzed 29 d after cell transfer: (B) infected RAG KO without cell transfer; (C) infected RAG KO receiving CD4+ cells from infected IL-10 KO mice; or (D) uninfected RAG KO receiving CD4+ cells from infected IL-10 KO mice. Crypt abscesses (solid arrows), ulcer (open arrow). H&E staining. Bar, 200 μm.
CD45RBlow CD4+ Cells from H. hepaticus–infected WT Mice Protect RAG KO Animals from Colitis Induced by IL-10 KO CD4+ Cells plus Bacterial Infection.
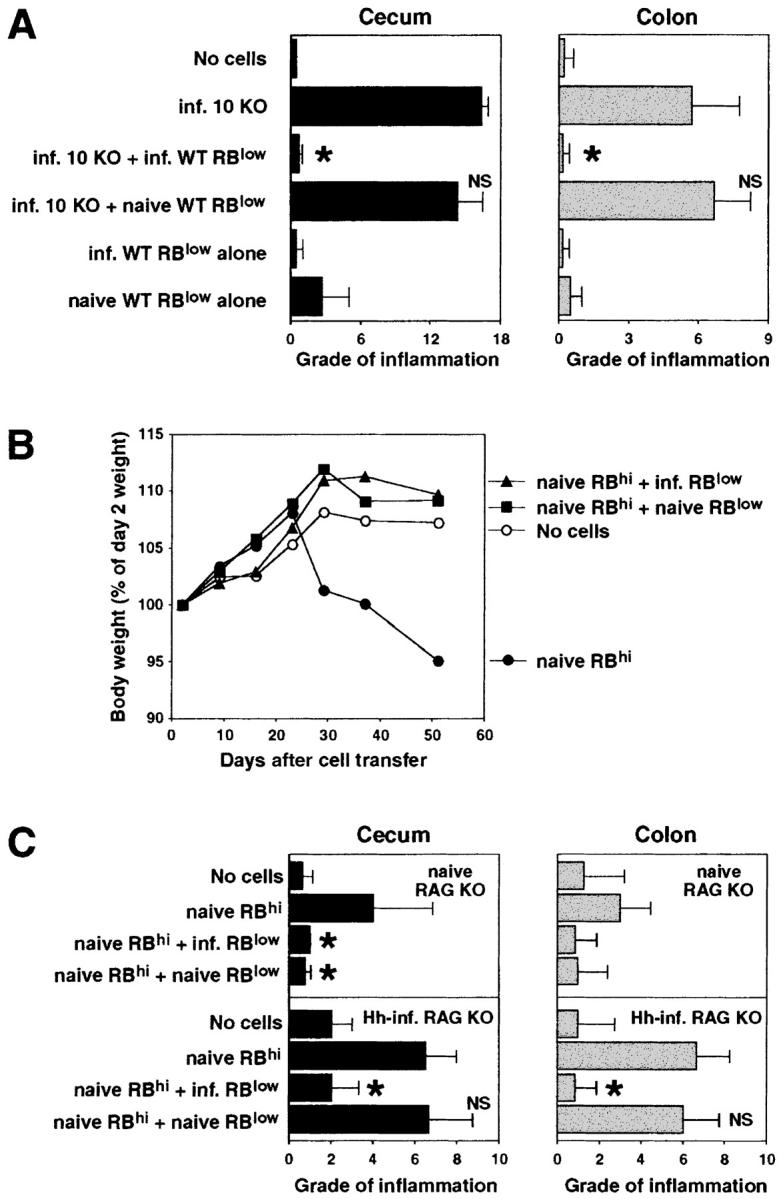
Next, we investigated whether we could protect the IL-10 KO T cell reconstituted, infected RAG KO mice from disease by cotransfer of WT cells. As shown in Fig. 2 A, whereas infected RAG KO hosts receiving IL-10 KO CD4+ cells alone developed colitis, animals receiving a 1:1 mixture of MLN CD4+ cells from IL-10 KO and infected WT mice did not develop intestinal inflammation. If the WT CD4+ cells originated from uninfected (naive) WT mice, however, no protection was observed (Fig. 2 A). In fact, the transfer of naive WT CD4+ cells alone induced colitis in the infected T cell–deficient mice, whereas CD4+ cells obtained from infected WT animals did not induce disease in these recipients. Whereas minimal inflammation was observed in infected T cell–deficient mice with no cell transfer (Fig. 3 A), infected RAG KO recipients of IL-10 KO CD4+ lymphocytes displayed large numbers of cells infiltrating the intestine (Fig. 3 B). Co-transfer of infected but not naive WT CD4+ cells blocked the accumulation of these inflammatory infiltrates (Fig. 3, C and D).
Figure 2.

CD45RBlow CD4+ cells from H. hepaticus–infected WT mice protect RAG KO animals from colitis induced by IL-10 KO CD4+ cells plus bacterial infection. (A) H. hepaticus–infected RAG KO mice (solid bars) were inoculated with CD4+ cells from infected IL-10 KO mice and CD4+ cells from naive or infected WT mice as indicated (4 × 105 of each population). Intestinal pathology was analyzed 4 wk later. Naive (open bar) and infected RAG KO animals receiving no cells were included as controls. Bars represent mean histology scores ± SD of three mice per group except for groups receiving WT cells alone, in which case, due to limited cell numbers, only two mice per group were used. Similar results were seen in ascending colon (although histology scores were lower) and when disease was induced by the transfer of CD4+ cells from naive IL-10 KO mice (not depicted). (B) Infected RAG KO mice were given either no cells or CD4+ cells from infected IL-10 KO mice and CD4+, CD45RBhi CD4+, or CD45RBlow CD4+ cells from infected WT mice as indicated (4 × 105 of each population). Pathology in the cecum (solid bars) and ascending colon (gray bars) were analyzed 4 wk later. Bars represent mean histology scores ± SD of six or seven mice per group pooled from two separate experiments except for the group receiving IL-10 KO CD4+ cells plus infected WT CD4+ cells (n = 3), which was included in only one of the experiments. Statistical significance was tested for groups receiving IL-10 KO cells. *, P < 0.05 compared with mice receiving IL-10 KO cells alone.
Figure 3.
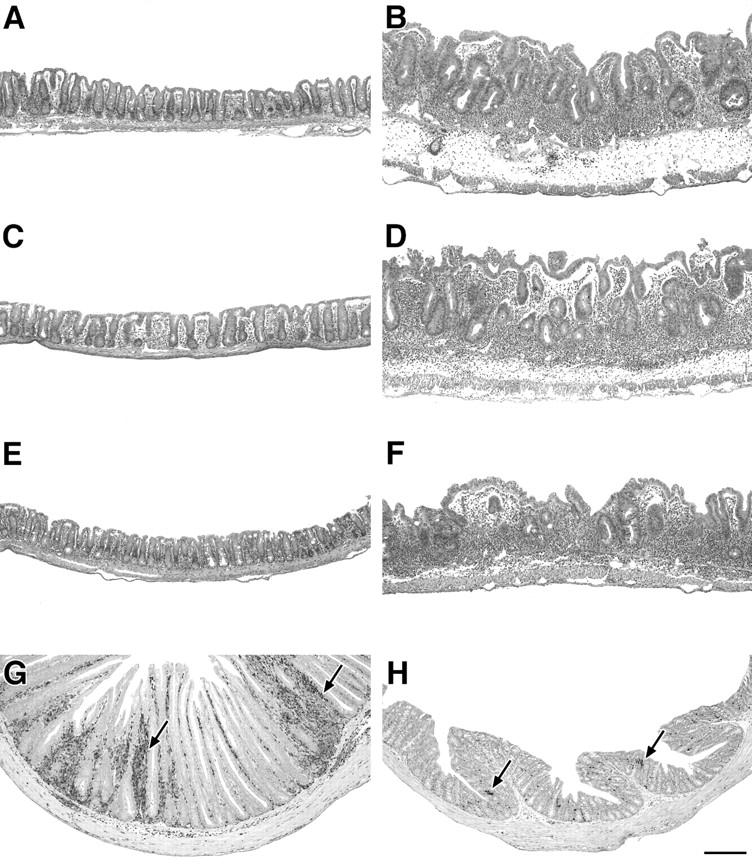
Intestinal pathology of H. hepaticus–infected RAG KO mice receiving CD4+ cells from IL-10 KO and WT mice. (A–F) Representative cecal sections of the infected RAG KO mice described in Fig. 2 A receiving (A) no cells, (B) infected IL-10 KO CD4+ cells, (C) infected IL-10 KO CD4+ cells plus infected WT CD4+ cells, (D) infected IL-10 KO CD4+ cells plus naive WT CD4+ cells, (E) infected WT CD4+ cells alone, and (F) naive WT CD4+ cells alone. H&E staining. (G and H) Immunohistochemical staining for CD3+ T cells (arrows) in colonic sections of infected RAG KO recipients receiving (G) infected IL-10 KO CD4+ cells alone and (H) infected IL-10 KO CD4+ cells plus infected WT CD45RBlow CD4+ cells. Bar, 200 μm.
The disease-protective effect of CD4+ cells from infected WT mice was not due to the clearance of H. hepaticus in the recipients. Although no accurate methods exist for the direct quantification of bacterial colonization of intestinal tissue by this pathogen, when analyzed by culture and H. hepaticus–specific PCR, fecal samples from animal groups receiving IL-10 KO CD4+ cells plus CD4+ cells from either infected or uninfected WT mice were both shown to be clearly positive for the organism (unpublished data).
To further characterize the disease-protective CD4+ cells, we purified by cell sorting the CD45RBhi and CD45RBlow CD4+ populations from infected WT mice and analyzed the ability of these cells to block colitis. Consistent with previous findings in the SCID IBD model (7, 15), CD45RBlow cells from infected WT mice completely protected the H. hepaticus–infected RAG KO recipients from colitis induced by IL-10 KO T cells (Fig. 2 B). The CD45RBhi population showed no disease-protective effect in the colon, and the small but significant reduction in pathology observed in the cecum was significantly less (P < 0.05) than that mediated by the CD45RBlow population.
Using cells from WT and IL-10 KO mice expressing different allotypes of CD45, we were able to track the donor origin of the CD4+ cells in the recipients. Although T cells in colonic sections from diseased RAG KO mice were mainly of IL-10 KO origin, comparable tissues of mice receiving IL-10 KO cells plus infected WT CD4+ CD45RBlow cells showed a marked reduction in the number of IL-10 KO cells with a simultaneous presence of scattered T cells of WT origin (unpublished data). The dramatic reduction in the numbers of T cells in the intestine of IL-10 KO CD4 cell recipients after cotransfer with infected WT CD45RBlow cells was even further evident when colonic sections were stained for CD3+ cells (Fig. 3, G and H). These data suggest that CD45RBlow CD4+ cells from infected WT mice regulate the recruitment and/or expansion of pathogenic IL-10 KO T cells in the intestine and are in accordance with findings by Annacker et al. (11), in which colitis in SCID mice transplanted with CD45RBhi and CD45RBlow cells expressing different CD45 allotypes was studied.
Both CD25+ and CD25− Subpopulations of CD45RBlow CD4+ Cells Protect against Bacteria-induced Colitis.
To further phenotype the disease-protective CD45RBlow CD4+ lymphocytes, we sorted CD25+ and CD25− cells from this population and asked whether these cells could inhibit colitis. Two recent reports have demonstrated that CD25+ CD45RBlow CD4+ cells from naive mice are superior to the CD25− subpopulation in blocking CD45RBhi-induced colitis in T cell–deficient mice (18, 19). Our experiments using IL-10 KO CD4+ cells plus H. hepaticus to induce disease indicate that the CD25+ and CD25− populations prevent disease to a similar degree when given at a 1:1 ratio with the pathogenic cells. However, at an IL-10 KO CD4+/WT CD45RBlow ratio of 3:1 or 9:1, the CD25− CD45RBlow cells are more effective in blocking intestinal inflammation than the CD25+ subpopulation (Fig. 4) . Thus, when given with 3 × 105 IL-10 KO CD4+ cells, 105 of the CD25− CD45RBlow WT cells were able to significantly suppress pathology in both the cecum and the colon, whereas the same number of cells of the CD25+ subpopulation conferred partial protection only in the cecum. Although disease protection by the CD25+ cells was completely lost at 3.3 × 104 cells, the CD25− cells still suppressed inflammation at this dosage (Fig. 4).
Figure 4.
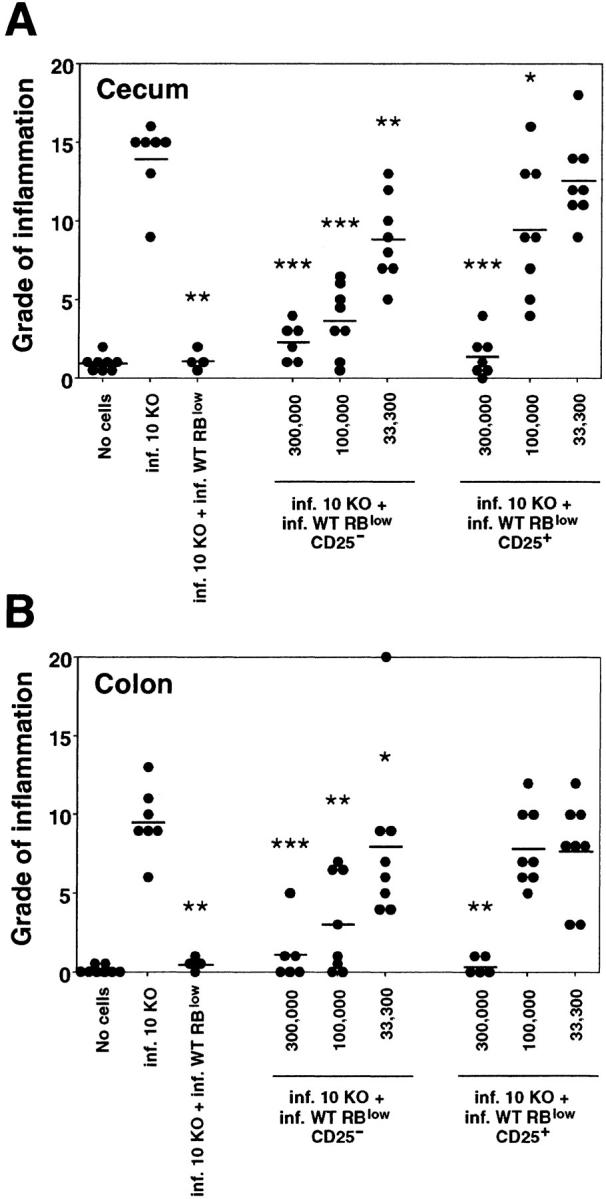
CD25− CD45RBlow as well as CD25+ CD45RBlow CD4+ cells from infected WT mice protect RAG KO mice against colitis. Infected RAG KO mice were given either no cells or 3 × 105 CD4+ cells from infected IL-10 KO mice either alone or with 3 × 105 CD45RBlow CD4+ cells, 3 × 105, 105, or 3.3 × 104 CD25− CD45RBlow, or CD25+ CD45RBlow CD4+ cells from infected WT mice as indicated. (A) Pathology in the cecum and (B) ascending colon was analyzed 4 wk later. •, an individual mouse; —, the average for each group. Data shown are pooled from two separate experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001 compared with infected mice receiving IL-10 KO cells alone.
CD25+ and CD25− Treg Cells from Infected WT Mice Block Colitis via an IL-10–dependent but TGF-β–independent Mechanism.
To analyze whether IL-10/TGF-β are important mediators in blocking H. hepaticus–induced colitis, infected RAG KO mice receiving pathogenic IL-10 KO CD4+ cells with disease-protective WT CD25+ or CD25− CD45RBlow CD4+ cells were treated with control mAb, anti–IL-10R, or anti–TGF-β at doses previously shown to abrogate protection in the SCID IBD model (16–18). As shown in Fig. 5 A, anti–IL-10R treatment completely abrogated the disease-protective effect of both the CD25+ and the CD25− subpopulations of the CD45RBlow CD4+ cells. In contrast, anti–TGF-β did not significantly affect the protection mediated by either population. These results were confirmed in a separate experiment in which protection by both CD25+ and CD25− CD45RBlow cells was largely unaffected by anti–TGF-β treatment, even after the amount of mAb was doubled to 4 mg/wk (Fig. 5 B). Moreover, in agreement with previously published results (16, 18), treatment with the same anti–TGF-β mAb reversed the protection of CD45RBhi reconstituted, uninfected RAG KO recipients mediated by CD45RBlow cells from the MLNs of naive WT animals (unpublished data).
Figure 5.

CD25− CD45RBlow and CD25+ CD45RBlow CD4+ cells from infected WT mice block colitis via an IL-10–dependent but TGF-β–independent mechanism. Infected RAG KO mice were given either no cells or CD4+ cells from infected IL-10 KO mice and CD25− CD45RBlow or CD25+ CD45RBlow CD4+ cells from infected WT mice (3 × 105 of each population) and treated with control mAb, anti–IL-10R (both at 1 mg/wk), or anti–TGF-β as indicated. (A) In the first experiment, mice were given 2 mg of anti–TGF-β per wk for 4 wk, (B) and in the second experiment, they were given 4 mg of anti–TGF-β per wk for 2 wk before analysis. •, the cecal histology score from an individual mouse; —, the average for each group. Similar results, although lower histology scores, were seen for the ascending colon where CD25− cell recipients treated with control or anti–TGF-β mAb displayed indistinguishable scores in B. In A, anti–IL-10R–treated groups had P values of <0.01 and <0.05 compared with control mAb–treated groups, whereas scores for mice given anti–TGF-β were not significantly different from those receiving control mAb. Similar results were seen in B except that it was not technically possible to perform Mann-Whitney U test statistics on recipients of CD25− cells due to identical scores of all mice receiving control mAb.
The possibility still existed that IL-10 derived from host non-T cells plays a role in disease protection mediated by infected WT cells in our model. Therefore, we performed cell transfers into double RAG/IL-10 KO mice. As demonstrated in single RAG KO recipients (Fig. 2 A), CD4+ cells from infected but not naive WT mice protected from colitis induced by IL-10 KO CD4+ cells plus H. hepaticus infection also in these IL-10–deficient RAG KO mice (unpublished data). This finding was supported by subsequent cell sorting experiments demonstrating that the disease-protective cells are found within the CD45RBlow CD4+ population and that their inhibitory effect is abrogated by treatment of the RAG/IL-10 KO recipients with anti–IL-10R mAb (Fig. 6 A).
Figure 6.

T cell–derived IL-10 mediates the disease-protective effect of WT CD45RBlow CD4+ cells. (A) Infected RAG/IL-10 double deficient mice were given either no cells or infected IL-10 KO CD4+ cells and infected WT CD4+ CD45RBlow cells as indicated (4 × 105 of each population) and then treated with control mAb or anti–IL-10R mAb for 4 wk. Bars represent mean cecal histology scores ± SD of three mice per group. (B) Infected RAG KO mice (solid bars) were inoculated with CD4+ cells from infected IL-10 KO, WT, and IL-4 KO mice as indicated (3 × 105 of each population) and then treated with control mAb or anti–IL-10R mAb for 4 wk. Naive (open bar) and infected RAG KO animals receiving no cells were included as controls. Bars represent mean cecal histology scores ± SD of three mice per group from one representative experiment out of two performed. Statistical significance was tested for groups receiving IL-10 KO cells. *, P < 0.05 compared with mice receiving infected IL-10 KO cells alone.
The disease-protective IL-10–secreting CD4+ cells were shown to develop independently of IL-4, as CD4+ cells isolated from H. hepaticus–infected IL-4 KO mice were as potent as CD4+ cells from infected WT mice in blocking disease (Fig. 6 B). Treatment with anti–IL-10R completely abolished this protective effect (Fig. 6 B), indicating that IL-4 is not required for either the development or the effector function of the disease-protective cells. Together, these results suggest that T cell–derived IL-10, but not TGF-β or IL-4, is important for protecting RAG KO mice against colitis induced by pathogenic cells plus H. hepaticus infection, and that the disease-protective cells are distinct from conventional Th2 cells.
Responsiveness of Disease-protective CD45RBlow CD4+ Treg Cells to Bacterial Ag.
Our results clearly show that CD4+ cells must be obtained from H. hepaticus–infected IL-10–competent mice to confer protection from colitis triggered by this bacterium. These data could imply that the infection per se results in a selective expansion of a particular Treg population (for example, CD45RBlow cells) and/or that the disease-protective cells specifically recognize and respond to Helicobacter Ag. To examine the first possibility, MLN CD4+ cells from naive and infected WT mice were analyzed for their CD45RB and CD25 expression. In multiple experiments using mice at various time points after infection, no difference was observed in CD45RB and CD25 expression of MLN CD4+ cells between naive and H. hepaticus–infected animals (unpublished data). In addition, CD45RBlow CD4+ cells from uninfected WT mice were unable to prevent intestinal inflammation in infected RAG KO mice reconstituted with IL-10 KO cells (Fig. 7 A). Similarly, CD25+ CD4+ cells from infected but not naive WT animals inhibited disease (unpublished data).
Figure 7.
CD45RBlow CD4+ cells from naive WT mice are unable to protect RAG KO recipients from colitis induced by pathogenic T cells plus H. hepaticus infection. (A) Infected RAG KO mice were given either no cells or CD4+ cells from infected IL-10 KO mice and CD45RBlow CD4+ cells from naive or infected WT mice (4 × 105 of each population), and tissues were analyzed 4 wk later. Bars represent mean cecal (solid bars) and colonic (gray bars) histology scores ± SD of three mice per group. (B and C) Uninfected or H. hepaticus–infected (Hh-inf.) RAG KO mice were given either no cells or CD45RBhi CD4+ cells from naive WT mice and CD45RBlow CD4+ cells from naive or infected WT mice (3 × 105 of each population). Body weights were measured weekly and tissues were analyzed after 4 and 7.5 wk for infected and uninfected recipients, respectively. (B) Results shown represent mean body weights (expressed as a percentage of the weight 2 d after cell transfer) of four mice per group from the uninfected RAG KO recipients. No difference in body weight was observed between the four groups of infected recipients over the 4-wk time period that these groups were being followed (not depicted). (C) Bars represent mean cecal (solid bars) and colonic (gray bars) histology scores ± SD of three or four mice per group. Statistical significance was tested for groups receiving pathogenic cells. *, P < 0.05 compared with mice receiving infected IL-10 KO CD4+ cells alone (A) or naive WT CD45RBhi CD4+ cells alone (C).
Additional evidence for Ag specificity came from experiments using naive WT CD45RBhi cells to induce disease in uninfected RAG KO recipients. Despite the inability of CD45RBlow CD4+ cells from naive WT mice to protect against Helicobacter-triggered colitis in IL-10 KO T cell reconstituted RAG KO mice, as previously reported (7, 15), these cells inhibited the wasting disease (Fig. 7 B) and colitis (Fig. 7 C, top) induced in uninfected RAG KO animals by the transfer of naive WT CD45RBhi CD4+ cells. However, if the recipients of the naive WT CD45RBhi cells were first infected with H. hepaticus, disease protection was again only observed when using WT CD45RBlow cells from H. hepaticus–primed mice (Fig. 7 C, bottom). These results suggest that the MLNs of H. hepaticus–infected but not naive WT mice contain Treg cells capable of protecting against Helicobacter-induced colitis.
To further address whether H. hepaticus–reactive Treg lymphocytes exist within the CD45RBlow CD4 population, we used an in vitro model in which the suppressive effect of Treg cells on a responder IL-10 KO CD4+ cell population was examined. Using this system, infected but not naive WT CD45RBlow CD4+ cells inhibited IFN-γ production by IL-10 KO CD4+ cells in an H. hepaticus Ag–specific manner (Fig. 8 A). In addition, the infected WT CD45RBlow CD4+ cells were shown to produce IL-10 after stimulation with SHelAg plus IL-2 as measured by both ELISA and intracellular cytokine staining, a response not detected from comparable cell populations from naive mice (Fig. 8, B and C). As expected, CD45RBlow cells from both naive and infected WT mice produced IL-10 in response to anti-CD3 plus IL-2 (Fig. 8 B). Taken together, our results argue that the mechanism by which CD4+ cells from infected WT mice block colitis involves the production of IL-10 from CD45RBlow CD4+ Treg cells induced by exposure to H. hepaticus.
Figure 8.
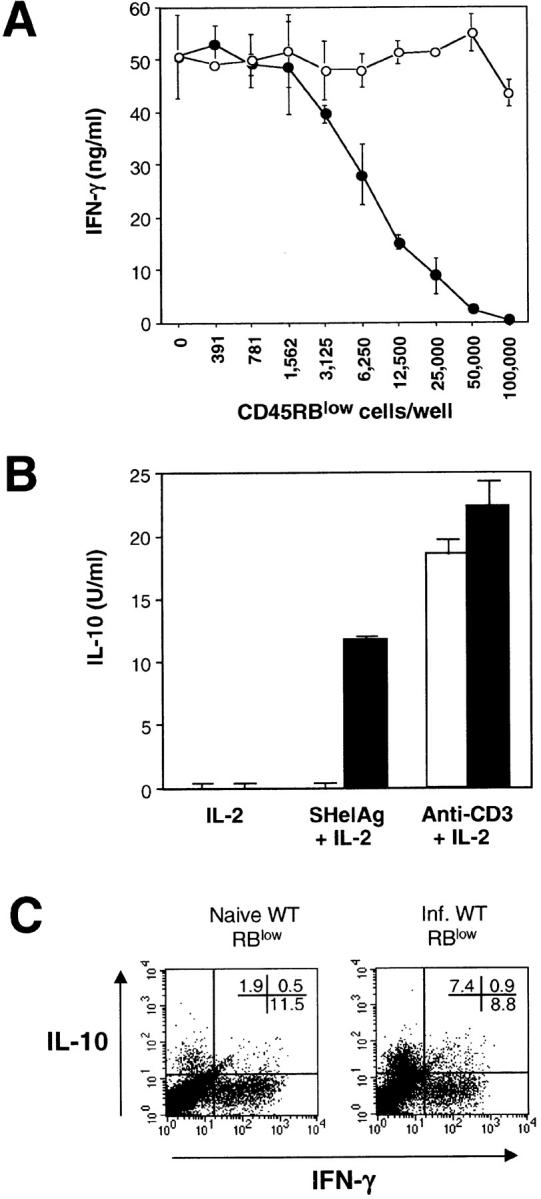
CD45RBlow CD4+ cells from infected WT mice inhibit IFN-γ production by IL-10 KO CD4+ cells and produce IL-10 after in vitro stimulation with SHelAg. (A) Responder CD4+ cells from infected IL-10 KO mice were cultured with APC, SHelAg, and indicated numbers of CD45RBlow CD4+ cells from naive (○) or infected (•) WT mice. IFN-γ was measured in 72-h supernatants. (B) CD45RBlow CD4+ cells from naive (□) or infected (▪) WT mice were cultured with IL-10–deficient APC and indicated Ag. IL-10 was measured in 72-h supernatants. No IFN-γ, IL-4, or IL-5 was detected in cultures stimulated with SHelAg plus IL-2. (C) CD45RBlow cells from naive or infected WT mice were cultured with IL-10–deficient APC and SHelAg plus IL-2. After removal of supernatants at 72 h, IL-2 was added for an additional 18 h before cells were stimulated with PMA plus ionomycin and analyzed for the expression of IL-10 and IFN-γ by intracellular staining. The FACS® dot plots shown are gated on CD4+ cells. In the same assay, the cells were negative for intracellular IL-4. The results shown in A–C are representative of two or three experiments performed.
Discussion
In this study, we show that H. hepaticus infection induces a population of CD4+ CD45RBlow Treg cells that inhibit the development of colitis triggered by this gram-negative bacterium. Based on their absence in uninfected animals and their restimulation in vitro by H. hepaticus Ag, we argue that these cells are directed against bacterial components and may be analogous to Treg cells that prevent the host from mounting pathologic responses to gut flora.
The concept that Treg cells are responsible for the prevention of IBD in normal hosts has emerged from a number of previous studies (7, 8, 30). Perhaps most pertinent are the findings of Powrie and co-workers (7, 15), who used a CD4/SCID transfer model to demonstrate the existence in normal mice of CD45RBlow CD4+ Treg cells that inhibit intestinal inflammation in CD45RBhi reconstituted SCID mice. Although the presence of bacterial flora was found to be essential for driving the CD45RBhi-induced pathogenic immune response (9, 10), the above studies did not address the Ag specificity of the disease-protective CD45RBlow Treg population. We have adapted the CD4/SCID model using H. hepaticus–infected T cell–deficient mice as recipients to examine this issue. The cells we found to protect from intestinal inflammation in this model differed from those described in the studies of Powrie and co-workers in that their frequency was higher in the CD25− compared with the CD25+ CD45RBlow CD4 subpopulation and their disease-protective function was blocked by anti–IL-10R but not anti–TGF-β treatment.
The original Treg populations described by Sakaguchi and co-workers (31, 32), and subsequently by other groups (18, 33–35), occur naturally in naive animals and express CD25 as a defining phenotypic marker. These conventional CD25+ cells are generated in the thymus (36, 37) and may require exposure to their target Ag to maintain their function as suppressors of autoimmune disease (38). Our observation that the cells that protect from H. hepaticus–induced colitis are enriched within the CD25− CD45RBlow subset and are not present in uninfected mice argues that such cells, rather than being endogenous, represent a memory population resulting from previous exposure to exogenous bacterial Ag. Although less suppressive than the CD25− CD45RBlow subset, the CD25+ subpopulation also possessed significant activity in our model. These disease inhibitory CD25+ CD45RBlow cells may differ from those described by Read et al. (18) and Annacker et al. (19), as no protection was observed when CD25+ cells from uninfected animals were tested in our model. One possibility is that the suppression obtained is due to recently activated IL-2R+ H. hepaticus–specific cells arising within the CD25+ CD45RBlow population and perhaps diluted by the presence of large numbers of conventional Treg cells.
Although disease protection in the SCID transfer model of IBD is abrogated by treatment with mAb against TGF-β, the H. hepaticus–induced Treg cells do not appear to block colitis through a mechanism involving this cytokine. Even though we have used an mAb different from that of Powrie and co-workers (16, 18), 30 μg/ml of this reagent was shown to efficiently neutralize at least 2.5 ng of TGF-β in vitro (unpublished data). Furthermore, we confirmed that the mAb we used abrogated CD45RBlow-mediated disease protection in the SCID IBD model. Finally, the same mAb has been shown by others to block TGF-β–mediated suppression of IBD in the trinitrobenzene sulfonic acid colitis model (39). Thus, we consider it highly unlikely that the difference in TGF-β dependency observed between the H. hepaticus–induced and the CD4/SCID colitis models is due to incomplete neutralization by the mAb used. Instead, as H. hepaticus induces a more rapid onset of, and a more severe colitis in, T cell reconstituted RAG KO mice than that of the SCID model (40), it is possible that a different type of Treg cell that preferentially secretes high amounts of IL-10 rather than TGF-β is required to prevent H. hepaticus–triggered disease. In this respect, our Treg cells more closely resemble the IL-10–producing Ag-induced Tr1 cells that develop in the presence of IL-10 (22, 23) and are thought to block colitis through the production of this cytokine (22), as well as the recently described IL-10–producing Treg cells shown to develop after in vitro Ag stimulation with vitamin D3 plus dexamethasone (24). Based on their lack of dependence on TGF-β, the H. hepaticus–induced regulatory cells appear to be distinct from the Th3 cells implicated in oral tolerance (41).
Perhaps the most intriguing finding of this study is that the Treg cells that protect from H. hepaticus–induced colitis appear to be primed by H. hepaticus Ag. This conclusion is based both on the requirement for H. hepaticus infection of the regulatory cell donors and on the ability of the disease-protective populations to produce IL-10 in response to specific Ag stimulation. The ability of cells from an infected donor to suppress intestinal inflammation triggered by the same pathogen was previously documented in a model of Toxoplasma gondii–induced ileitis (42). Although the phenotype of the protective intraepithelial lymphocytes was not determined, the cells involved appear to be distinct from those characterized here, as their disease inhibitory effect was abrogated by anti–TGF-β treatment. Moreover, in contrast to the situation in the H. hepaticus model, the T. gondii–infected donors from which the ileitis-preventing cells were derived are not protected against disease. To date, little information is available regarding the Ag specificity of Treg cells that protect from various pathogenic immune responses. Data from an elegant study by Seddon and Mason (38) on autoimmune thyroiditis have suggested that it is the presence of the peripheral autoantigen that maintains Treg cells in the periphery, as CD4+ cells from the spleen and LN of athyroid hosts were unable to protect against thyroiditis but did block the development of diabetes. Similarly, based on the findings reported here, it is tempting to speculate that the most efficient Treg cells protecting against bacteria-driven colitis require priming by bacterial Ag for their generation. One may infer from data by Singh et al. (43) that flora plays a role in the generation of disease-protective CD45RBlow cells also in the SCID IBD model, as at lower cell numbers, CD45RBlow cells from SPF-reared mice display a higher capacity to protect CD45RBhi reconstituted SCID mice from colitis compared with CD45RBlow cells isolated from GF animals. Thus, although Treg cells with no experience of bacterial Ag can exhibit protective activity, bacterial priming may result in much more effective disease suppression. Clearly, studies using Treg cells with defined specificities, e.g., Treg cell clones, are critical to formally establish the Ag specificity of the cells protecting from colitis in our model. However, such studies are technically difficult due to the poor proliferative capacity of Treg cells, and to date, our attempts to generate such clones have been unsuccessful.
The detailed mechanism by which Treg cells protect from colitis remains to be elucidated. Treg cells do not appear to prevent the initial activation of responder T cells, as normal up-regulation of early activation markers on the latter population is observed in the presence of Treg cells (44). Instead, the responder cells undergo cell cycle arrest at the G0/G1 stage (44). Moreover, in vivo studies have shown that the cotransfer of CD45RBhi and CD45RBlow cells limit the number of pathogenic CD45RBhi cells present in SCID or RAG KO recipients (11, 17, 19). Consistent with these findings, we observed reduced numbers of IL-10 KO T cells infiltrating the intestines of H. hepaticus–infected RAG KO recipients protected from colitis by the cotransfer with CD4+ cells from infected WT mice. The hypothesis that Treg cells limit the development of pathogenic Th1 cells is also supported by our failure to detect IFN-γ in SHelAg-stimulated total MLN cells (20), or CD45RBlow cultures (Fig. 8 B), from infected WT mice, even in the presence of neutralizing anti–IL-10 mAb (unpublished data). One explanation for this finding could be that Treg lymphocytes induce death of pathogenic cells perhaps through competition for growth-promoting cytokines. Another possible explanation for this impaired expansion of pathogenic cells is that disease-protective Treg cells, through their production of IL-10, prevent the activation of cells of the innate immune system (45, 46) such as DCs, as has been previously suggested in studies on the effects of anti-OX40L mAb treatment on colitis in the SCID IBD model (47). A role for Treg cell–produced IL-10 in this suppression of the innate immune response is supported by the observation of spontaneous enterocolitis in mice with a cell type–specific disruption of the Stat3 gene in macrophages and neutrophils that renders these cells IL-10 unresponsive (48).
A major question raised by this study is whether gut flora–reactive Treg cells, similar to those described here, prevent the appearance of IBD in humans. Evidence in support of this mechanism comes from studies demonstrating peripheral T cell tolerance in the intestine toward resident autologous gut flora, a state that is broken in active IBD (49) and appears to be mediated by bacterial Ag–reactive CD4+ T cells secreting IL-10 and TGF-β (50). Additional analysis should help establish whether the cells involved are phenotypically analogous to the Treg cells studied here. A major implication of these human and murine studies is that IBD may result from a defect in the development or function of bacteria-specific Treg cells. If so, treatments that augment the generation or function of such cells may offer an approach for therapeutic intervention in these intestinal inflammatory diseases.
Acknowledgments
We are grateful to R. Swafford, J. Clarke, and C. Eigsti for excellent cell sorting; S. Hieny for purification of anti–IL-10R and control mAb; R. Dreyfuss for help with photomicrographs; S. Jagannatha for assistance with statistical evaluation of our data; and G. Agyemang, J. Valez, and N. Gray for help with intragastric inoculations and body weight measurements. We also thank Drs. E. Shevach, W. Strober, C. Feng, and C. Piccirillo for their valuable discussions and for critical reading of the manuscript. Finally, we thank B. Marshall for editorial assistance.
Footnotes
Abbreviations used in this paper: DC, dendritic cell; GF, germfree; H&E, hematoxylin and eosin; IBD, inflammatory bowel disease; MLN, mesenteric lymph node; SHelAg, soluble Helicobacter hepaticus Ag; SPF, specific pathogen-free; Treg, T regulatory.
References
- 1.Sadlack, B., H. Merz, H. Schorle, A. Schimpl, A.C. Feller, and I. Horak. 1993. Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene. Cell. 75:253–261. [DOI] [PubMed] [Google Scholar]
- 2.Taurog, J.D., J.A. Richardson, J.T. Croft, W.A. Simmons, M. Zhou, J.L. Fernandez-Sueiro, E. Balish, and R.E. Hammer. 1994. The germfree state prevents development of gut and joint inflammatory disease in HLA-B27 transgenic rats. J. Exp. Med. 180:2359–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dianda, L., A.M. Hanby, N.A. Wright, A. Sebesteny, A.C. Hayday, and M.J. Owen. 1997. T cell receptor-αβ-deficient mice fail to develop colitis in the absence of a microbial environment. Am. J. Pathol. 150:91–97. [PMC free article] [PubMed] [Google Scholar]
- 4.Schultz, M., S.L. Tonkonogy, R.K. Sellon, C. Veltkamp, V.L. Godfrey, J. Kwon, W.B. Grenther, E. Balish, I. Horak, and R.B. Sartor. 1999. IL-2-deficient mice raised under germfree conditions develop delayed mild focal intestinal inflammation. Am. J. Physiol. 276:G1461–G1472. [DOI] [PubMed] [Google Scholar]
- 5.Berg, D.J., N. Davidson, R. Kühn, W. Müller, S. Menon, G. Holland, L. Thompson-Snipes, M.W. Leach, and D. Rennick. 1996. Enterocolitis and colon cancer in interleukin-10-deficient mice are associated with aberrant cytokine production and CD4+ TH1-like responses. J. Clin. Invest. 98:1010–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sellon, R.K., S. Tonkonogy, M. Schultz, L.A. Dieleman, W. Grenther, E. Balish, D.M. Rennick, and R.B. Sartor. 1998. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect. Immun. 66:5224–5231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Powrie, F., M.W. Leach, S. Mauze, L.B. Caddle, and R.L. Coffman. 1993. Phenotypically distinct subsets of CD4+ T cells induce or protect from chronic intestinal inflammation in C. B-17 scid mice. Int. Immunol. 5:1461–1471. [DOI] [PubMed] [Google Scholar]
- 8.Morrissey, P.J., K. Charrier, S. Braddy, D. Liggitt, and J.D. Watson. 1993. CD4+ T cells that express high levels of CD45RB induce wasting disease when transferred into congenic severe combined immunodeficient mice. Disease development is prevented by cotransfer of purified CD4+ T cells. J. Exp. Med. 178:237–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Powrie, F., S. Mauze, and R.L. Coffman. 1997. CD4+ T-cells in the regulation of inflammatory responses in the intestine. Res. Immunol. 148:576–581. [DOI] [PubMed] [Google Scholar]
- 10.Aranda, R., B.C. Sydora, P.L. McAllister, S.W. Binder, H.Y. Yang, S.R. Targan, and M. Kronenberg. 1997. Analysis of intestinal lymphocytes in mouse colitis mediated by transfer of CD4+, CD45RBhigh T cells to SCID recipients. J. Immunol. 158:3464–3473. [PubMed] [Google Scholar]
- 11.Annacker, O., O. Burlen-Defranoux, R. Pimenta-Araujo, A. Cumano, and A. Bandeira. 2000. Regulatory CD4 T cells control the size of the peripheral activated/memory CD4 T cell compartment. J. Immunol. 164:3573–3580. [DOI] [PubMed] [Google Scholar]
- 12.Sartor, R.B. 1997. Pathogenesis and immune mechanisms of chronic inflammatory bowel diseases. Am. J. Gastroenterol. 92:5S–11S. [PubMed] [Google Scholar]
- 13.Gionchetti, P., F. Rizzello, A. Venturi, F. Ugolini, M. Rossi, P. Brigidi, R. Johansson, A. Ferrieri, G. Poggioli, and M. Campieri. 1999. Review—antibiotic treatment in inflammatory bowel disease: rifaximin, a new possible approach. Eur. Rev. Med. Pharmacol. Sci. 3:27–30. [PubMed] [Google Scholar]
- 14.Campieri, M., and P. Gionchetti. 1999. Probiotics in inflammatory bowel disease: new insight to pathogenesis or a possible therapeutic alternative? Gastroenterology. 116:1246–1249. [DOI] [PubMed] [Google Scholar]
- 15.Powrie, F., R. Correa-Oliveira, S. Mauze, and R.L. Coffman. 1994. Regulatory interactions between CD45RBhigh and CD45RBlow CD4+ T cells are important for the balance between protective and pathogenic cell-mediated immunity. J. Exp. Med. 179:589–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Powrie, F., J. Carlino, M.W. Leach, S. Mauze, and R.L. Coffman. 1996. A critical role for transforming growth factor-β but not interleukin 4 in the suppression of T helper type 1–mediated colitis by CD45RBlow CD4+ T cells. J. Exp. Med. 183:2669–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Asseman, C., S. Mauze, M.W. Leach, R.L. Coffman, and F. Powrie. 1999. An essential role for interleukin 10 in the function of regulatory T cells that inhibit intestinal inflammation. J. Exp. Med. 190:995–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Read, S., V. Malmström, and F. Powrie. 2000. Cytotoxic T lymphocyte–associated antigen 4 plays an essential role in the function of CD25+CD4+ regulatory cells that control intestinal inflammation. J. Exp. Med. 192:295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Annacker, O., R. Pimenta-Araujo, O. Burlen-Defranoux, T.C. Barbosa, A. Cumano, and A. Bandeira. 2001. CD25+ CD4+ T cells regulate the expansion of peripheral CD4 T cells through the production of IL-10. J. Immunol. 166:3008–3018. [DOI] [PubMed] [Google Scholar]
- 20.Kullberg, M.C., J.M. Ward, P.L. Gorelick, P. Caspar, S. Hieny, A. Cheever, D. Jankovic, and A. Sher. 1998. Helicobacter hepaticus triggers colitis in specific-pathogen-free interleukin-10 (IL-10)-deficient mice through an IL-12- and gamma interferon-dependent mechanism. Infect. Immun. 66:5157–5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kullberg, M.C., A.G. Rothfuchs, D. Jankovic, P. Caspar, T.A. Wynn, P.L. Gorelick, A.W. Cheever, and A. Sher. 2001. Helicobacter hepaticus-induced colitis in interleukin-10-deficient mice: cytokine requirements for the induction and maintenance of intestinal inflammation. Infect. Immun. 69:4232–4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Groux, H., A. O'Garra, M. Bigler, M. Rouleau, S. Antonenko, J.E. de Vries, and M.G. Roncarolo. 1997. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature. 389:737–742. [DOI] [PubMed] [Google Scholar]
- 23.McGuirk, P., C. McCann, and K.H. Mills. 2002. Pathogen-specific T regulatory 1 cells induced in the respiratory tract by a bacterial molecule that stimulates interleukin 10 production by dendritic cells: a novel strategy for evasion of protective T helper type 1 responses by Bordetella pertussis. J. Exp. Med. 195:221–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barrat, F.J., D.J. Cua, A. Boonstra, D.F. Richards, C. Crain, H.F. Savelkoul, R. de Waal-Malefyt, R.L. Coffman, C.M. Hawrylowicz, and A. O'Garra. 2002. In vitro generation of interleukin 10–producing regulatory CD4+ T cells is induced by immunosuppressive drugs and inhibited by T helper type 1 (Th1)- and Th2-inducing cytokines. J. Exp. Med. 195:603–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.National Institutes of Health. 1996. Guide for the Care and Use of Laboratory Animals. National Institutes of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD.
- 26.Ward, J.M., J.G. Fox, M.R. Anver, D.C. Haines, C.V. George, M.J. Collins, Jr., P.L. Gorelick, K. Nagashima, M.A. Gonda, R.V. Gilden, et al. 1994. Chronic active hepatitis and associated liver tumors in mice caused by a persistent bacterial infection with a novel Helicobacter species. J. Natl. Cancer Inst. 86:1222–1227. [DOI] [PubMed] [Google Scholar]
- 27.Fox, J.G., F.E. Dewhirst, J.G. Tully, B.J. Paster, L. Yan, N.S. Taylor, M.J. Collins, Jr., P.L. Gorelick, and J.M. Ward. 1994. Helicobacter hepaticus sp. nov., a microaerophilic bacterium isolated from livers and intestinal mucosal scrapings from mice. J. Clin. Microbiol. 32:1238–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Read, S., S. Mauze, C. Asseman, A. Bean, R. Coffman, and F. Powrie. 1998. CD38+ CD45RBlow CD4+ T cells: a population of T cells with immune regulatory activities in vitro. Eur. J. Immunol. 28:3435–3447. [DOI] [PubMed] [Google Scholar]
- 29.Jankovic, D., M.C. Kullberg, N. Noben-Trauth, P. Caspar, W.E. Paul, and A. Sher. 2000. Single cell analysis reveals that IL-4 receptor/Stat6 signaling is not required for the in vivo or in vitro development of CD4+ lymphocytes with a Th2 cytokine profile. J. Immunol. 164:3047–3055. [DOI] [PubMed] [Google Scholar]
- 30.Powrie, F., and D. Mason. 1990. OX-22high CD4+ T cells induce wasting disease with multiple organ pathology: prevention by the OX-22low subset. J. Exp. Med. 172:1701–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sakaguchi, S., N. Sakaguchi, M. Asano, M. Itoh, and M. Toda. 1995. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor α-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 155:1151–1164. [PubMed] [Google Scholar]
- 32.Asano, M., M. Toda, N. Sakaguchi, and S. Sakaguchi. 1996. Autoimmune disease as a consequence of developmental abnormality of a T cell subpopulation. J. Exp. Med. 184:387–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suri-Payer, E., A.Z. Amar, A.M. Thornton, and E.M. Shevach. 1998. CD4+CD25+ T cells inhibit both the induction and effector function of autoreactive T cells and represent a unique lineage of immunoregulatory cells. J. Immunol. 160:1212–1218. [PubMed] [Google Scholar]
- 34.Salomon, B., D.J. Lenschow, L. Rhee, N. Ashourian, B. Singh, A. Sharpe, and J.A. Bluestone. 2000. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 12:431–440. [DOI] [PubMed] [Google Scholar]
- 35.Stephens, L.A., and D. Mason. 2000. CD25 is a marker for CD4+ thymocytes that prevent autoimmune diabetes in rats, but peripheral T cells with this function are found in both CD25+ and CD25− subpopulations. J. Immunol. 165:3105–3110. [DOI] [PubMed] [Google Scholar]
- 36.Jordan, M.S., A. Boesteanu, A.J. Reed, A.L. Petrone, A.E. Holenbeck, M.A. Lerman, A. Naji, and A.J. Caton. 2001. Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self-peptide. Nat. Immunol. 2:301–306. [DOI] [PubMed] [Google Scholar]
- 37.Bensinger, S.J., A. Bandeira, M.S. Jordan, A.J. Caton, and T.M. Laufer. 2001. Major histocompatibility complex class II–positive cortical epithelium mediates the selection of CD4+25+ immunoregulatory T cells. J. Exp. Med. 194:427–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seddon, B., and D. Mason. 1999. Peripheral autoantigen induces regulatory T cells that prevent autoimmunity. J. Exp. Med. 189:877–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fuss, I.J., M. Boirivant, B. Lacy, and W. Strober. 2002. The interrelated roles of TGF-β and IL-10 in the regulation of experimental colitis. J. Immunol. 168:900–908. [DOI] [PubMed] [Google Scholar]
- 40.Cahill, R.J., C.J. Foltz, J.G. Fox, C.A. Dangler, F. Powrie, and D.B. Schauer. 1997. Inflammatory bowel disease: an immunity-mediated condition triggered by bacterial infection with Helicobacter hepaticus. Infect. Immun. 65:3126–3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen, Y., V.K. Kuchroo, J. Inobe, D.A. Hafler, and H.L. Weiner. 1994. Regulatory T cell clones induced by oral tolerance: suppression of autoimmune encephalomyelitis. Science. 265:1237–1240. [DOI] [PubMed] [Google Scholar]
- 42.Buzoni-Gatel, D., H. Debbabi, F.J. Mennechet, V. Martin, A.C. Lepage, J.D. Schwartzman, and L.H. Kasper. 2001. Murine ileitis after intracellular parasite infection is controlled by TGF-β-producing intraepithelial lymphocytes. Gastroenterology. 120:914–924. [DOI] [PubMed] [Google Scholar]
- 43.Singh, B., S. Read, C. Asseman, V. Malmström, C. Mottet, L.A. Stephens, R. Stepankova, H. Tlaskalova, and F. Powrie. 2001. Control of intestinal inflammation by regulatory T cells. Immunol. Rev. 182:190–200. [DOI] [PubMed] [Google Scholar]
- 44.Thornton, A.M., and E.M. Shevach. 2000. Suppressor effector function of CD4+CD25+ immunoregulatory T cells is antigen nonspecific. J. Immunol. 164:183–190. [DOI] [PubMed] [Google Scholar]
- 45.Fiorentino, D.F., A. Zlotnik, P. Vieira, T.R. Mosmann, M. Howard, K.W. Moore, and A. O'Garra. 1991. IL-10 acts on the antigen-presenting cell to inhibit cytokine production by Th1 cells. J. Immunol. 146:3444–3451. [PubMed] [Google Scholar]
- 46.Ding, L., P.S. Linsley, L.Y. Huang, R.N. Germain, and E.M. Shevach. 1993. IL-10 inhibits macrophage costimulatory activity by selectively inhibiting the up-regulation of B7 expression. J. Immunol. 151:1224–1234. [PubMed] [Google Scholar]
- 47.Malmström, V., D. Shipton, B. Singh, A. Al-Shamkhani, M.J. Puklavec, A.N. Barclay, and F. Powrie. 2001. CD134L expression on dendritic cells in the mesenteric lymph nodes drives colitis in T cell-restored SCID mice. J. Immunol. 166:6972–6981. [DOI] [PubMed] [Google Scholar]
- 48.Takeda, K., B.E. Clausen, T. Kaisho, T. Tsujimura, N. Terada, I. Forster, and S. Akira. 1999. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity. 10:39–49. [DOI] [PubMed] [Google Scholar]
- 49.Duchmann, R., I. Kaiser, E. Hermann, W. Mayet, K. Ewe, and K.H. Meyer zum Buschenfelde. 1995. Tolerance exists towards resident intestinal flora but is broken in active inflammatory bowel disease (IBD). Clin. Exp. Immunol. 102:448–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Khoo, U.Y., I.E. Proctor, and A.J. Macpherson. 1997. CD4+ T cell down-regulation in human intestinal mucosa: evidence for intestinal tolerance to luminal bacterial antigens. J. Immunol. 158:3626–3634. [PubMed] [Google Scholar]