

Abstract
Nonreciprocal translocations and gene amplifications are commonly found in human tumors. Although little is known about the mechanisms leading to such aberrations, tissue culture models predict that they can arise from DNA breakage, followed by cycles of chromatid fusion, asymmetric mitotic breakage, and replication. Mice deficient in both a nonhomologous end joining (NHEJ) DNA repair protein and the p53 tumor suppressor develop lymphomas at an early age harboring amplification of an IgH/c-myc fusion. Here we report that these chromosomal rearrangements are initiated by a recombination activating gene (RAG)-induced DNA cleavage. Subsequent DNA repair events juxtaposing IgH and c-myc are mediated by a break-induced replication pathway. Cycles of breakage-fusion-bridge result in amplification of IgH/c-myc while chromosome stabilization occurs through telomere capture. Thus, mice deficient in NHEJ provide excellent models to study the etiology of unbalanced translocations and amplification events during tumorigenesis.
Keywords: nonreciprocal translocations, gene amplification, bridge-fusion breakage, nonhomologous end-joining, tumorigenesis
Introduction
Genomic instability is a hallmark of cancer. Human hematopoietic malignancies are characterized by reciprocal translocations that juxtapose oncogenes and antigen receptor loci or create oncogenic fusion proteins. By contrast, solid tumors commonly contain a much more complex karyotype, consisting of nonreciprocal translocations, which typically result in chromosomal gains, losses and gene amplifications. Such chromosomal imbalances promote tumorigenesis by the underexpression of tumor suppressor genes as well as disregulated expression of oncogenes. Although studies in vitro have shown that site-specific breaks may be the initiating lesion that triggers these events (1, 2), the mechanisms leading to gross chromosomal rearrangements remain to be characterized.
Nonhomologous end joining (NHEJ)* components, including Ku70, Ku80, DNA-PKcs, DNA ligase IV, Xrcc4, and Artemis, are essential for the repair of double-strand breaks (DSBs) during V(D)J recombination (3). Additionally, this DNA repair pathway has been implicated in maintaining genomic integrity via suppression of chromosomal rearrangements (4–6). Consistent with this idea, mice deficient in NHEJ components are characterized by increased sensitivity to DNA damaging agents, chromosomal instability, immunodeficiency, and predisposition to thymic lymphomas (3). Further loss of cell cycle checkpoints in NHEJ-deficient mice results in a shift from thymomas to pro-B cell lymphomas (4, 7–12) or to sarcomas (10, 13). Defects in NHEJ components have also been observed in patients with multiple myelomas (14), leukemia (15), and a disorder resembling Nijmegen breakage syndrome (NBS) (16). Thus, loss of the NHEJ repair pathway has implications in a broad spectrum of tumor types.
To understand the mechanisms by which aberrant chromosome rearrangements occur in the absence of NHEJ, we molecularly characterized pro-B cell lymphomas from mice deficient for Ku and p53. All of these tumors harbor coamplification of the IgH and c-myc loci on either derivative chromosomes 12 or 15 (4). Our data provide evidence that unresolved RAG-mediated breaks during V(D)J recombination trigger a nonreciprocal replication pathway (i.e., break-induced-replication), leading to the juxtaposition of IgH and c-myc loci. Subsequent attempts to stabilize the end of the derivative chromosome result in amplification of these genes by cycles of breakage-fusion-bridge and capture of telomere sequences from other chromosomes.
Materials and Methods
Generation and Screening of Mice.
Ku70 mutant mice were generously provided by Dr. Fred Alt, The Children's Hospital and The Center for Blood Research, Boston, MA (17). p53−/− mice were obtained from Taconic Laboratories, and p53 genotyping was performed as described (18, 19). Mice heterozygous for Ku70 and p53 were crossed to obtain progeny homozygous for both Ku70 and p53 null alleles. Ku80−/−p53−/− mice were described previously (4) while the Ku80−/− mice were generated by intercrossing Ku80+/−ATM+/− mice and were either heterozygous or homozygous wild-type at the ATM locus. Mice heterozygous for Ku80 and p53 (20) were bred with Rad54−/− mice (21) to obtain F1 Rad54+/−Ku80+/−p53+/− offspring. The Rad54 mutation was fixed by crossbreeding F1 animals. Rag2−/− mice were purchased from Taconic Laboratories.
Spectral Karyotyping and FISH.
Suspensions of tumor cells were grown in RPMI plus 10% FBS plus 50 U/ml of IL-7 (R&D Systems) and 20 U/ml of IL-2 (Boehringer). Tumor cells were arrested at mitosis with Colcemid (GIBCO BRL) treatment at 0.1 μg/ml for 0.5–3 h. Mitotic chromosome spreads were prepared following standard procedures, and Spectral Karyotype (SKY) analysis was performed as described (22). For FISH analysis, metaphases were hybridized with BAC probes for the following regions: IgH Cα (from Cγ1 to 3′ of Cα) and IgH Cμ were obtained by PCR screening of a BAC library with gene-specific primers (22), IgH variable region representing the distal end of the V cluster (VJ588) (provided by Dr. Riblet, Torrey Pines Institute for Molecular Studies, San Diego, CA), c-myc, 15 subtelomeric region, and two BAC clones containing the breakpoint of chromosome 15 observed in PKT2 (see below). Flow sorted single chromosomes were used for painting probes. A telomere repeat-specific peptide nucleic acid (PNA) probe labeled with Cy3 (Applied Biosystems) was hybridized with PCR-generated chromosome-specific painting probes in 50% Formamide/20% dextran sulfate/2× SSC at 37°C overnight. Slides were then washed three times in 50% Formamide/2× SSC at 37°C, 2 times in 0.1× SSC at 60°C, and finally in 4× SSC/Tween 20 at 37°C. Slides were then processed following standard FISH detection of the painting probes.
Sequencing of Breakpoints and Identification of BAC Clones.
The breakpoint fusion from tumors PKT2, PKT7, and PKT13 were obtained by digestion circularization PCR (4, 23). A search of the mouse database with the breakpoint sequences enabled us to determine which bases were derived from chromosomes 12 and 15 and to determine the proximity and orientation of these sequences with regard to the c-myc gene. BAC clones for mapping of the breakpoints were obtained by screening the RPCI-23 mouse BAC library with a 23-mer oligonucleotide derived from the 174-bp chromosome 15-specific sequence at the PKT2 breakpoint. Fingerprint analysis enabled the selection of clones with a minimum overlap as well as clones in the contig that provided the largest coverage.
Results
Ku80−/− Thymic Lymphomas.
Several mouse strains (Ku70−/−, Ku80−/−, SCID, ATM−/−, PARP−/−) deficient in V(D)J recombination and/or DNA damage responses are susceptible to thymic lymphomas (4, 10, 17, 24–27). Cytogenetic analysis has only been described for ATM−/− lymphomas, where failure to correctly resolve V(D)J-induced double strand breaks at the TCRα/δ locus in developing thymocytes results in the translocation of chromosome 14 to other chromosomes (22, 24). Ku80-deficient mice succumb at a low frequency to late onset development of thymic lymphomas. FACS® analysis of cell surface markers indicates that Ku80−/− tumors arose during the double positive CD4+CD8+stage of T cell (Thy1.2+) development (not shown), similar to the phenotype observed in ATM−/− mice. Analysis of six Ku80−/− tumors revealed that although chromosome 14 was sometimes rearranged to other chromosomes (L796 and L2283), none of these translocation events involved either the TCRβ or TCRα loci (Table I). These results differ from those in ATM−/− thymomas (22) despite heterozygosity for the ATM gene in three of the mice. Thus, the absence of Ku80 at RAG-induced DSB during V(D)J recombination in developing T cells does not result in the aberrant ligation of TCR loci to another chromosome, as in ATM-deficient mice (22). A full description of the karyotype can be found at http://www.ncbi.nlm.nih.gov/sky/skyweb.cgi.
Table I.
Karyotypic Analysis of Pro-B and T Cell Lymphomas
| Locus involvement |
Amplification
|
|||||||
|---|---|---|---|---|---|---|---|---|
| Tumor no. |
Genotype | Immunophenotype | Composite karyotype | TCR | IgH | Age of onset |
IgH | c-myc |
| L796 | Ku80−/−ATM+/+ | Thymic lymphoma | 39,X,-X, or Y, T(2:13)(B;C1),Rb(7.15),T(13;2)(B;B),+15 | No | n.d. | 7 mo | No | No |
| L2282 | Ku80−/−ATM+/+ | Thymic lymphoma | 40,XX.T(3;14)(H4;E3) | No | n.d. | 6 mo | No | No |
| L2283 | Ku80−/−ATM+/+ | Thymic lymphoma | 40,XX,T(7;14)(B;D),Rb(13.17),T(14;7)(C;C),+15 | No | n.d. | 13 mo | No | No |
| L387 | Ku80−/−ATM+/− | Thymic lymphoma | 42,XY,del(12)(F),+Rb(14.14),-14[2],+15 | No | n.d. | 7 mo | No | No |
| L2240 | Ku80−/−ATM+/− | Thymic lymphoma | 40,XX,Rb(5,18)[1],Rb(13.14)[1],Rb(15.15),+15 | No | n.d. | 8 mo | No | No |
| L3835 | Ku80−/−ATM+/− | Thymic lymphoma | 42,X,T(X;4)(F;C),del(13)(A2→A5),+15,+T(15;1)(C;D) | No | n.d. | 7 mo | No | No |
| PKT1 | Ku80−/−p53−/− | Pro-B cell lymphoma | 39–49,XY,+1,+5,T(12;15;3;[1]) OR T(12;15;3)x2,+14 | n.d. | Yes | 2 mo | Yes | Yes |
| PKT2 | Ku80−/−p53−/− | Pro-B cell lymphoma | 39–41,XX,+3,T(12;15),dup(15) | n.d. | Yes | 2 mo | Yes | Yes |
| PKT3 | Ku80−/−p53−/− | Pro-B cell lymphoma | 38–42,XY,T(12;15),T(15;12) OR T(15;12;16) | n.d. | Yes | 2 mo | Yes | Yes |
| PKT4 | Ku80−/−p53−/− | Pro-B cell lymphoma | 39–40,XY,T(12;15),T(15;12),dup(16) | n.d. | Yes | 2 mo | Yes | Yes |
| PKT5–13 | Ku80−/−p53−/− | Pro-B cell lymphoma | n.d. | n.d. | Yes | 2 mo | Yes | Yes |
| PK7T1 | Ku70−/−p53−/− | Pro-B cell lymphoma | T(12;15),T(15;12;?) | n.d. | Yes | 2 mo | Yes | Yes |
| PK7T2 | Ku70−/−p53−/− | Pro-B cell lymphoma | T(12;15) | n.d. | Yes | 3 mo | Yes | Yes |
| PK7T3 | Ku70−/−p53−/− | Pro-B cell lymphoma | T(12;15),T(15;12;?) | n.d. | Yes | 3 mo | Yes | Yes |
| PK7T4 | Ku70−/−p53−/− | Pro-B cell lymphoma | n.d. | n.d. | Yes | 2 mo | Yes | Yes |
| N10527 | Ku80−/−p53−/−RAD54−/− | Pro-B cell lymphoma | T(12;15),T(15;12;3) | n.d. | Yes | 1.5 mo | Yes | Yes |
| N9984-B | Ku80−/−p53−/−RAD54−/− | Pro-B cell lymphoma | T(12;15) | n.d. | Yes | 3 mo | No | No |
| N9984-T | Ku80−/−p53−/−RAD54−/− | Thymic lymphoma | T(12;15),T(14;16),Rb(7.18) | No | n.d. | 3 mo | No | No |
| N10238 | Ku80−/−p53+/−RAD54−/− | Thymic lymphoma | T(4;14),T(14;4),T(12;13) | No | n.d. | 3 mo | No | No |
| N10449 | Ku80−/−p53+/−RAD54−/− | Thymic lymphoma | n.d. | n.d. | n.d. | 4 mo | No | No |
| N9454 | Ku80−/−p53−/−RAG−/− | Thymic lymphoma | Polyploid, aneuploid | No | n.d. | 3 mo | No | No |
| N8839 | Ku80−/−p53−/−RAG−/− | Thymic lymphoma | n.d. | n.d. | n.d. | 3 mo | No | No |
| N10611 | Ku80−/−p53−/−RAG−/− | Thymic lymphoma | Polyploid, aneuploid | No | n.d. | 3 mo | No | No |
| N10184 | Ku80−/−p53−/−RAG−/− a | Thymic lymphoma | Polyploid, aneuploid | No | n.d. | 3 mo | No | No |
| N9700 | Ku80+/−p53−/−RAG−/− a | Thymic lymphoma | Polyploid, aneuploid | No | n.d. | 4 mo | No | No |
| N10214 | Ku80+/−p53−/−RAG−/− a | Thymic lymphoma | Polyploid, aneuploid | No | n.d. | 5 mo | No | No |
The immunophenotype as determined by FACS® analysis is indicated for each of the tumors. Due to the instability of some of the tumors, composite karyotypes represent the most commonly observed clonal aberrations by SKY. Due to the high chromosome instability and polyploid nature of the Ku/p53/RAG tumors, it was not possible to derive a representative composite karyotype. Ku70−/−p53−/− tumors were analyzed with painting probes specific for chromosomes 12 and 15. Columns labeled TCR and IgH refer to the involvement of these loci in chromosome translocations as determined by SKY and/or gene specific (Tcrα, Tcrβ, Ig VH, and Ig Cα) BAC probes. n.d., not determined.
Mice additionally expressing a rearranged T cell receptor transgene (AND). The AND transgene did not change the age of onset, the type of tumor or karyotype.
IgH/c-myc Rearrangements Are Independent of Rad54 but Require Rag-mediated Cleavage.
On a p53-null background, loss of Ku70 promotes the rapid onset of pro-B cell lymphomas associated with rearrangement and amplification of IgH and c-myc loci (Table I). Alternative DNA repair pathways, such as homologous recombination (HR) could facilitate promiscuous chromosomal fusions in NHEJ/p53-deficient mice. To assess the role of Rad54-mediated HR in lymphoma development, we generated Rad54−/−Ku80−/−p53+/− and Rad54−/−Ku80−/−p53−/− mice. 20% of Rad54−/−Ku80−/−p53+/− mice died between the ages of 3–5 mo of thymic lymphoma (Table I). Similar to Ku80−/− thymic lymphomas, Rad54−/−Ku80−/− p53+/− tumors did not contain translocations involving TCRα or TCRβ loci (Table I). A shift to the rapid development of pro-B cell lymphomas, however, occurred in RAD54−/−Ku80−/−p53−/− mice. These tumors were similar to the Ku−/−p53−/− pro-B cell lymphomas in that they contained translocations involving chromosomes 12 and 15. Probes complementary to the IgH Cα and c-myc genes further demonstrated the juxtaposition and amplification of these genes in each tumor (Table I; see below). These results indicate that certain aspects of HR, such as Rad54-dependent sister chromatid exchanges (28), are not required for the chromosomal rearrangements characteristic of NHEJ/p53-deficient B-lineage tumors.
The IgH locus in the mouse is oriented such that transcription initiates at the telomeric V regions and proceeds downstream toward the more centromeric constant regions (see Fig. 4) . In Ku80−/−p53−/− lymphomas, chromosome 15 was translocated to chromosome 12 upstream of the JH segments while the more telomeric variable (VH) region was lost, suggesting that these events were initiated by Rag-mediated cleavage (4). To directly test the requirement for V(D)J recombination in the generation of pro-B cell lymphomas, we generated Ku80−/−p53−/−RAG2−/− mice. These mice did not succumb to pro-B cell lymphomas, but unexpectedly, developed thymic lymphomas at approximately the same time (3 mo) that pro-B cell lymphomas developed in Ku80−/−p53−/− mice (Table I). Chromosome aberrations in Ku80−/−p53−/−RAG2−/− thymomas were similar to those reported for p53−/− thymomas in that they exhibited polyploidy and aneuploidy, with very few translocations detectable by SKY analysis (not shown). We conclude that Rag-mediated cleavage is essential for the development of pro-B cell lymphomas in Ku80−/−p53−/− mice, but it is dispensable for the development of thymomas in Ku80−/−p53−/−Rag2−/− mice. Consistent with these results, no B-lineage tumors were observed in SCIDp53−/−Rag2−/− mice of which 43% developed thymic lymphomas (12).
Figure 4.
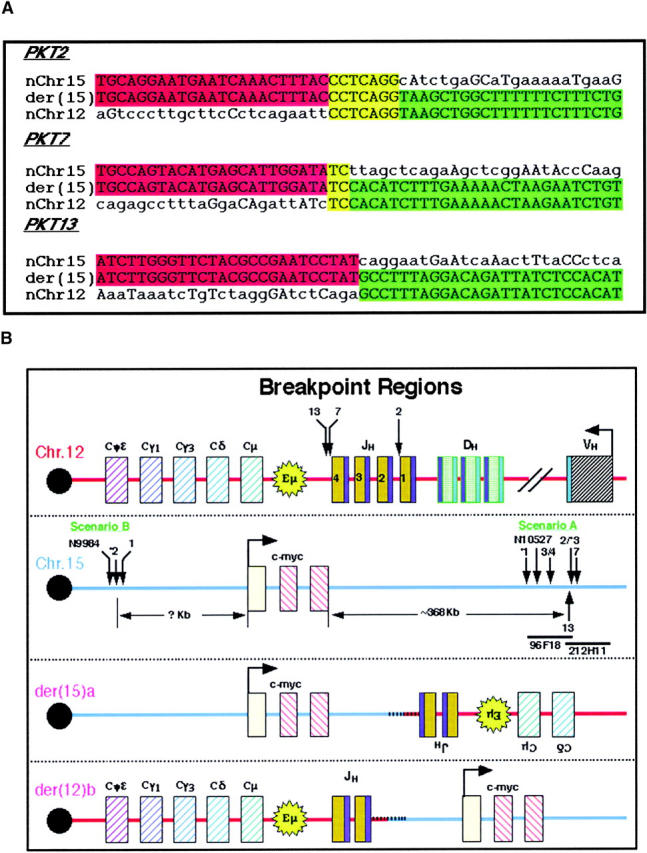
Sequence analysis and localization of fusion points and rearrangements in the IgH Ca and c-myc loci. (A) The fusion points of three Ku80−/−p53−/− pro-B cell lymphomas (PKT no. 2, 7, and 13) were cloned by digestion circularization PCR [der(15)a]. Sequence analysis of the three breakpoints from scenario A demonstrate microhomology (yellow) at the junction of chromosomes 12 (red) and 15 (green) in two cases (PKT2 and PKT7), but none in tumor PKT13. Homologous bases outside of this region are also indicated in uppercase letters. (B) Sequence analysis implied that the breakpoint was the V(D)J recombination signal sequence (RSS; blue) flanking IgH J1 in PKT2 and IgH J4 in PKT7 and PKT13 (top panel); however, exonuclease digestion of the cleaved ends resulted in loss of 45, 208, and 189 bp, respectively, from the original site of the RAG-mediated DSB to the fusion point (arrows). Approximate localization of the breakpoints on chromosome 15 (second panel) was determined by FISH hybridization with two BAC clones in this region (96F18 and 212H11) as illustrated in Fig. 5. Tumors 1–4, 7, and 13 are from Ku80−/−p53−/− mice, tumors *1–*3 are from Ku70−/−p53−/− mice and tumors N9984 and N10527 are from Ku80−/−p53−/−RAD54−/− mice. The resulting der(15)a and der(12)b chromosomes are illustrated in the bottom panels. In scenario A a portion of the IgH locus is copied to chromosome 15 downstream of c-myc and in the reverse orientation. In scenario B it appears that the copying of chromosome 15 to the der(12)b initiates centromeric of the c-myc locus and continues along toward the telomere.
Recurrent Patterns of Rearrangements, Amplifications and Telomere Capture in pro-B Cell Lymphomas.
Using a combination of whole-chromosome painting probes, a chromosome 15-specific subtelomeric probe, and probes for c-myc, IgH Cα, and IgH Cμ, we found two distinct types of configurations that juxtaposed IgH and c-myc in the pro-B cell lymphomas. Although we describe results for IgH Cα, identical results were obtained using BAC clones containing the most JH-proximal constant region (IgH Cμ). In the first configuration there is an exchange of material between chromosomes 12 and 15 resulting in the formation of two derivative chromosomes, der(12)a and der(15)a (Fig. 1 ; scenario A). IgH Cα was found near c-myc on the der(15)a and both genes were coamplified as determined by FISH and Southern blot analysis (reference 4, Table I, and data not shown). A single copy of the IgH Cα region, however, remained on der (12)a, indicating that a portion of chromosome 12 had been copied (not simply translocated) to der(15)a. Additionally, the region of chromosome 15 telomeric of c-myc was translocated to the der(12)a and was therefore absent from der(15)a. In fact, the subtelomeric region of chromosome 15, as indicated by hybridization with a region-specific BAC clone (15T), was found on the normal 15 [n15] as well as on the der(12)a (Fig. 1; scenario A). It should be noted that 15T is absent from the der(15)a. Scenario A–type rearrangements were found in 66% of the tumors analyzed. The alternative rearrangement pathway (Fig. 1; scenario B), was used by the remainder of the tumors. In these tumors c-myc and IgH Cα were coamplified on the derivative 12 [der(12)b] and no IgH Cα signal was detectable on chromosome 15. In fact, both copies of chromosome 15 appeared normal and hybridized with the c-myc and 15T BAC clones, while der(12)b did not contain any 15T sequences. In summary, in one class of tumors, IgH Cα was copied and amplified together with c-myc on the der(15)a (scenario A), whereas in the other class (scenario B) the c-myc oncogene was copied and amplified together with IgH Cα on der(12)b. These unbalanced rearrangements and amplifications are much more complex than the reciprocal translocations that juxtapose c-myc and IgH in human Burkitt's lymphoma and mouse plasmacytomas (29) but resemble the gene amplifications typically seen in solid tumors (30) and multiple myelomas (31).
Figure 1.

Two types of rearrangement scenarios observed in pro-B cell lymphomas from Ku−/−p53−/− and Ku−/−p53−/−Rad54−/− mice. In scenarios A and B, the IgH locus on mouse chromosome 12 and the c-myc locus on mouse chromosome 15 are brought into close proximity with each other. The top panel contains chromosomes hybridized with chromosome 12 (red) and 15 (green) specific painting probes. Chromosomes 12 (n12) and 15 (n15) illustrate the normal locations of the probes used in the FISH analysis. In scenario A, der(12)a results from translocation of chromosome 15 (distal to c-myc [pink]), including the subtelomeric region (15T - red), to chromosome 12. Copying of the IgH Cα locus (IgH - green) from chromosome 12 to chromosome 15 results in der(15)a. In scenario B, both homologues of chromosome 15 usually appear to be intact, whereas copying of the c-myc oncogene to chromosome 12 results in der(12)b. The extrachromosomal material at the end of the der(12)b is explained in Fig. 5.
Hybridization with the 15T BAC implied that the telomeric region of chromosome 15 was capping not only the normal chromosome 15, as expected, but also the der(12)a. Additionally, while both chromosomes 15 in scenario B were presumably protected from degradation by chromosome 15 telomeric sequences, der(12)b was not (Fig. 1). To determine if der(15)a and der(12)b were indeed protected from degradation by telomere sequences and from which chromosomes these might be derived, we used FISH and SKY analysis, respectively. In tumors in which IgH Cα was copied to chromosome 15 (scenario A), additional material at the ends of der(15)a was often found to be derived from different chromosomes (Fig. 2 A). Moreover, there were frequently variations in the length and composition at the end of der(15)a in different metaphases from the same tumor (Fig. 2 B). In some metaphases, the end of der(15)a consisted of amplified copies of IgH Cα and c-myc (Fig. 1, and Fig. 2 B, left panel); in others there appeared to be a single chromosome that capped der (15)a (Fig. 2 A, and Fig. 2 B, middle and right panels); while in still others we found tandem amplification of material from chromosomes 12 and 16 (Fig. 2 A). Additionally, in tumor N10527 we observed cells containing a dicentric chromosome with amplification of the IgH and c-myc loci (Fig. 2 C). Despite these differences in chromatin content at the end of der(15)a, all of the metaphases retained the clonal der(12)a containing 15T sequences (Fig. 2 B). The three tumors in scenario B also contained chromatin from chromosomes 1, 3 and/or 16 distal to the IgH Cα and c-myc genes (Fig. 2 A).
Figure 2.

Stabilization of broken chromosomes via chromatid fusion or chromosome capping. (A) Sequences from chromosomes 1, 3, and 16 are commonly used to cap the derivative chromosome. The presence of DNA from chromosomes 12 or 15 was confirmed by FISH when the region was too small to be visualized with painting probes. (B) Left panel: copying of the IgH locus onto chromosome 15 leaves der(15)a without a telomere (red) and results in amplification of both IgH Cα (green) and c-myc (pink). Stabilization of this chromosome occurs by the copying of telomeric sequences from other chromosomes (middle panel: unidentified with chromosome 3 paint; right panel: chromosome 3). (C) Dicentric chromosome stained with DAPI (blue) and hybridized with probes containing the IgH Cα (red) and IgH Cμ (green) loci.
A combination of painting probes for chromosomes 3 (green) and 15 (red) and a telomere repeat-specific PNA probe (white) was used to determine if the additional material capping der(15)a contained telomere sequences (Fig. 3) . We observed that in those instances when material from another chromosome (i.e., chromosome 3) capped der(15)a, telomere sequences were indeed present (Fig. 3 A). However, when der(15)a was not capped (as in Fig. 2 B, left panel where the end of der(15)a consisted of amplified copies of IgH Cα and c-myc) there was a distinct absence of telomere sequences (Fig. 3 B). Telomere sequences were also observed on the dicentric chromosomes, but as anticipated only at the two centromeric ends, not at the internal fusion point (Fig. 3 C). Thus, the derivative chromosome in which c-myc and IgH are coamplified is eventually stabilized by the capture of telomeric sequences from other chromosomes.
Figure 3.

Telomere healing via chromosome capture. Metaphase spreads were hybridized with painting probes specific for chromosomes 3 (green) and 15 (red) as well as a telomere sequence specific PNA probe (white). (A) The telomere for der(12)a is provided by chromosome 15, however, der(15)a requires the capture of a different chromosome, in this case chromosome 3, in order to obtain telomere repeat sequences. (B) Derivative chromosome 15 [der(15)a] with persistent broken chromatids is not protected from degradation by telomere repeats. (C) Telomere-free end fusion can then result in the formation of a dicentric chromosome.
Breakpoint Analysis.
Because tumors from NHEJ p53 double-deficient mice harbored a recurrent pattern of rearrangements and amplifications, we wanted to determine the precise nature of the breakpoints in the tumors. Using digestion circularization PCR (DC-PCR; references 4 and 23), we obtained breakpoints from tumors PKT2, PKT7, and PKT13 (Fig. 4 A). Consistent with the cytogenetic analysis, DNA sequencing revealed that the cloned rearrangements contained sequences from chromosome 12 and 15. Because the break in the IgH gene occurs during V(D)J recombination, the region of chromosome 12 to which chromosome 15 sequences were fused appeared near the J segments of the IgH locus (JH4 for PKT2, JH1 for PKT7 and PKT13; Fig. 4 B). However there was a loss of 45, 208, and 189 bp, respectively, for PKT2, PKT7, and PKT13 from the original site of the RAG-mediated double strand breakage. The precise position of the breakpoint on chromosome 15 was obtained by comparing the sequence of the cloned JH fusion partner with the mouse genome database (see Materials and Methods). The position of the breakpoint relative to c-myc was thus readily obtained, and was 368, 365, and 368 kb telomeric of c-myc for tumors PKT2, PKT7, and PKT13, respectively. The breakpoints on chromosome 15 for PKT2 and PKT13 were separated by only 23 bp (Fig. 4 B). Although we did not observe cryptic recombination signal sequences near the fusion points on chromosome 15, there was microhomology at the junction of chromosomes 12 and 15 in two cases (PKT2 and PKT7; Fig. 4 A). Taken together, these results are consistent with the finding that DNA repair in the absence of NHEJ is imprecise, subject to exonuclease digestion and frequently generated by microhomology-mediated joining (32–34).
As the breakpoints on chromosome 15 for each of the three tumors clustered within ∼3 kb of each other, it was possible that this region contained “fragile” sites for DNA breakage. To map the breakpoints in other tumors, we used the chromosome 15-specific sequence from the PKT2 DC-PCR product to design a 23-bp probe for the screening of a mouse BAC library (see Materials and Methods). A number of positive clones were identified and mapped to a contig in the existing mouse database. In particular, clones 96F18 and 212H11 contained the minimum amount of sequence overlap. FISH analysis with these two BACs as well as BAC clones for IgH Cα (17303), c-myc (17312), and the subtelomeric region of chromosome 15 (15T) revealed a number of different breakpoints and rearrangement products (Fig. 5) . For the tumors classified in scenario A, some of the breakpoints were mapped within the overlap region of the two BAC clones (PKT2, KP7T3), others occurred somewhere within BAC 96F18 (PKT3, PKT4, N10527), and another occurred either near the centromeric region of this BAC or in the intervening ∼170 kb between it and the c-myc gene (KP7T1; Fig. 5). As both copies of chromosome 15 appear normal for the tumors classified in scenario B (i.e., hybridized with the c-myc, 96F18, 22H11, and 15T BACs), sequences from this chromosome must have been copied to the der(12)b. However, it appears that the amount of chromatin copied varied among the different tumors, either stopping centromeric of 96F18 (PKT1) or continuing past BAC 22H11, but not including the subtelomeric region homologous to BAC 15T (KP7T2; Fig. 5). In summary, our analysis of the breakpoints in six different pro-B cell tumors (scenario A) revealed that there is not a single consistent rearrangement site on chromosome 15, but that multiple breaks occur within the BAC 96F18 chromosomal region which is ∼170–370 kb from the 3′ end of c-myc. Results from the remaining three tumors (scenario B) indicated that various amounts of chromosome 15 were copied onto the unresolved V(D)J-induced broken end of chromosome 12 and that the site of initiation of DNA synthesis was centromeric to c-myc.
Figure 5.
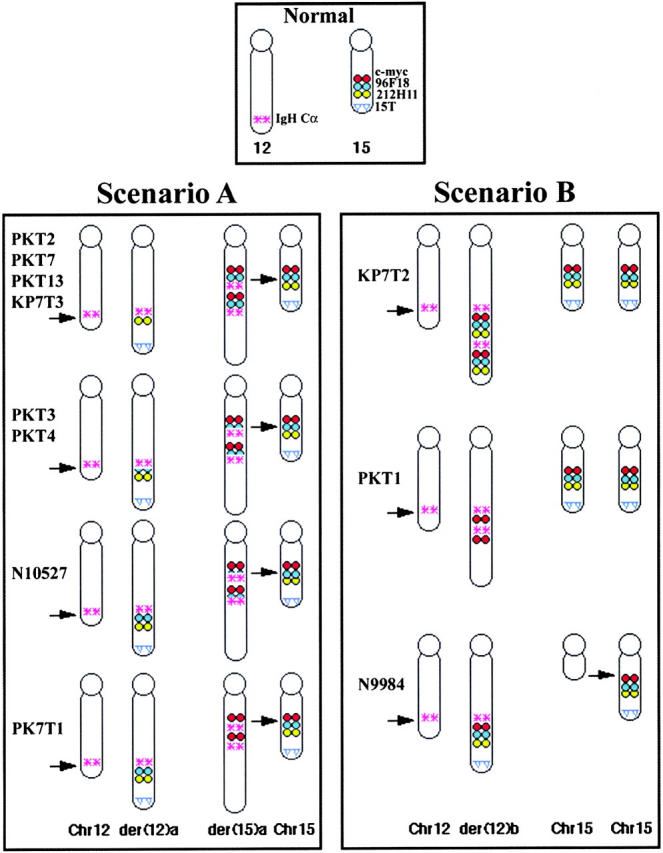
FISH mapping of the chromosome 12 and 15 breakpoints in 11 pro-B cell lymphomas. Top central panel illustrates the locations of the IgH Cα and c-myc loci, two overlapping BAC clones spanning the breakpoint in tumor PKT2 (96F18 and 212H11), and the hybridization site of a chromosome 15 subtelomeric probe (15T) on normal chromosomes. In the bottom panels, the breakpoints and rearrangements in the pro-B cell lymphomas are shown. The normal chromosomes appear to the far left (Chr. 12) and right (Chr. 15) of each box while the derivative chromosomes are illustrated between them. Arrows on the normal chromosomes indicate the location of the rearrangements on the derivative chromosomes.
Discussion
Our analysis of lymphomas from knockout mice deficient in NHEJ, HR, V(D)J recombination, and/or p53 has provided insights into chromosomal rearrangements that are associated with tumor formation during lymphocyte development. Although we previously demonstrated karyotype instability and gross chromosomal aberrations in fibroblasts from Ku80-deficient mouse embryos, the rate of tumorigenesis in these mice is surprisingly low (∼5–10%). Those thymomas that did arise were distinct from ATM−/− lymphomas (22), but similar to ATM−/−RAG2−/− thymomas (35), in that chromosome rearrangements involving the TCR loci were not observed. Similarly, our analysis of Ku80−/−p53−/−RAG2−/− and Ku80+/−p53−/−RAG2−/− thymic lymphomas revealed the absence of chromosome rearrangements involving the TCRα or TCRβ gene loci. The polyploidy and aneuploidy of these tumors was very similar to those observed in p53−/− thymic lymphomas (36), although Ku80−/−p53−/−RAG2−/− thymomas arose at an earlier age (3 mo) than observed in p53−/− mice or Ku80−/− mice (5–8 mo). Thus, although aberrant rearrangements of the TCR locus do not play a causal role in the development of thymomas in Ku80−/− mice, genomic instability promoted by absence of Ku80 synergizes with p53-deficiency to promote early thymomagenesis.
V(D)J recombination does, however, play a direct role in the development of Ku80−/−p53−/− pro-B cell lymphomas as demonstrated by the proximity of the translocations on chromosome 12 to the JH loci and the absence of these tumors in Ku80−/−p53−/−RAG2−/− mice. We thus demonstrate that RAG-induced DSBs at the IgH gene on chromosome 12 is the event initiating pro-B cell lymphomagenesis in Ku80−/−p53−/− mice (Fig. 6 , step 1). In the majority of tumors (scenario A), a DSB is also generated telomeric to c-myc (Fig. 6, step 2a), as evidenced by the translocation of the distal portion of chromosome 15 to the der(12)a (Fig. 6, step 3a). Thus, at least two DSBs are generated in these tumors, an event which is predicted to increase the frequency of translocation by several orders of magnitude relative to a single DSB (1). Our analysis of the regions involved in the breakpoint on chromosome 15 reveals that they cluster ∼170–370 kb from the 3′ end of c-myc in a region that appears devoid of recombination signal sequences. It is possible that the clustering reflects the presence of fragile sites in this region; alternatively, DNA cleavage on the partner chromosome may be targeted by RAG-mediated transposition (37, 38), an event that is stimulated by distorted DNA structures (39). Consistent with the latter possibility, the fusion points contained a combination of small inverted repeats and palindromic sequences (Fig. 4 A).
Figure 6.

Proposed model for rearrangement events in the Ku−/−p53−/− and Ku−/−p53−/− Rad54−/− pro-B cell lymphomas. RAG proteins initiate DNA DSB at the IgH D and J gene segments (step 1). Due to the absence of the Ku holoenzyme, the DSB cannot be efficiently and correctly repaired. The IgH DSB attacks sequences on chromosome 15. In scenario A, this results in a DSB in which the telomeric fragment of chromosome 15 is translocated to chromosome 12 while repair of the lesion on chromosome 15 occurs through a break-induced replication pathway such that sequences from chromosome 12 are copied to the der(15)a (steps 2a and 3a). In scenario B, c-myc is copied to der(12)b without disruption of the normal chromosome 15 (steps 2b and 3b). In both cases, however, the derivative chromosome is left with a persistent DSB. The broken chromatid is replicated during S-phase (step 4) and the free chromatid ends are ligated together to form a bridge (step 5). During mitosis when the chromatids are separated, a tug-of-war ensues resulting in the breaking of the derivative chromosome (step 6). Depending on where the break occurs, one cell may contain a chromosome with amplification of the genes as an inverted repeat (step 7). Because this chromosome still does not have a telomere sequence, the cycle continues until a telomere is captured (step 8).
Loss of p53 suppresses apoptosis in pro-B cells as evidenced by expansion of the pro-B cell compartment in p53−/− mice (40) and the enhanced survival of pro-B (but not pro-T) cells in Ku80−/−p53−/− mice (4). Furthermore, the absence of a p53 replication checkpoint would permit the broken IgH locus to persist into S-phase, allowing for a replicative repair pathway. The most striking evidence in support of replicative repair comes from observations of the breakpoints by FISH with BAC probes which revealed the presence of the IgH Cμ and Cα regions on the unrearranged chromosome 12 as well as on both derivative chromosomes 12 [der(12)a] and 15 [der(15)a]. Thus, this region of the genome was present on three different chromosomes (Figs. 1 and 2). Furthermore, the relative orientation of c-myc and IgH loci on der(15)a (Fig. 4 B) suggests that chromosome 12 was copied from the JH breakpoint toward the centromere onto der(15)a (Fig. 6, steps 2a and 3a). The same was true of the c-myc locus in scenario B, being present on both normal copies of chromosome 15 as well as on the derivative chromosome 12 [der(12)b] (Fig. 6, steps 2b and 3b). Importantly, at least one rearrangement in each tumor, therefore, cannot be explained by a simple translocation event involving the swapping of material between two broken chromosomes.
Sequences near a site-specific DSB can invade and copy sequences from another chromosome, leading to nonreciprocal translocations (41). Replication of the invading DNA can extend as far as several hundred kilobases to a chromosome end (42). These models are consistent with the varying amount of chromosome 15 copied onto der(12)b in tumors PKT1 and KP7T2. Signon et al. (43) have recently demonstrated that such a break-induced replication mechanism in yeast occurs in a Rad51- and Rad54-independent manner, probably mediated by the Rad50/Nbs1/Mre11 repair pathway which utilizes shorter stretches of homology. This also is consistent with our findings that pro-B cell lymphomas from both Ku−/−p53−/− and Ku80−/−p53−/− Rad54−/− mice had similar chromosome rearrangements. Despite the absence of significant regions of homology near the fusion points, we postulate that a break-induced replication pathway, possibly involving the Rad50/Nbs1/Mre11 complex, replicates either IgH sequences from chromosome 12 onto chromosome 15 telomeric of the c-myc locus (Fig. 6, step 2a) or c-myc sequences onto chromosome 12 near the site of V(D)J-induced DSB (Fig. 6, step 2b).
Gene amplification through breakage-fusion-bridge cycles is well documented in tissue culture cells (44, 45), and has been proposed to contribute to tumorigenesis (46). Loss of DNA damage responsive cell cycle checkpoints (either by inactivation of p53 or deregulation of c-myc) allows cells to be permissive to gene amplification in vitro (47–49). We propose that after completion of break-induced replication at least one of the chromosomes, namely the one containing IgH and c-myc, is still free of telomeric sequences (Figs. 3 B and 6, step 3). Replication of this “damaged” DNA could be permitted because cells lack the p53-dependent G1 checkpoint, and results in a chromosome containing two sister chromatids, each without a telomere cap (Fig. 6, step 4). The loss of a telomere cap leads to ligation of the free ends and the covalent fusion of the chromatids (50; Fig. 6, step 5). During mitosis when normal sister chromatids are separated, this particular chromosome encounters difficulties because the chromatids are actually one continuous DNA molecule. The forces exerted by the mitotic spindles at the kinetichore usually cause this molecule to break (Fig. 6, step 6). Depending on where the break occurs, one of the chromatids may contain two copies of IgH and c-myc oriented as an inverted repeat (Fig. 2 B) whereas the other chromatid contains neither of these genes (Fig. 6, step 7). Observation of dicentric chromosomes with multiple copies of the IgH and c-myc genes between the centromeres (Figs. 2 C and 3 C) further supports the mechanism of bridge-fusion-breakage in these gene amplification events.
During the subsequent cell cycle (Fig. 6, dashed lines) the broken end would be available to enter another round of break-induced replication, breakage-fusion-bridge, or both. Evidence for the hypothesis that the missing telomeric region is promoting amplification comes from the fact that the only tumor without amplification, N9984, resulted in a “simple” translocation event and contains the telomere of chromosome 15 (Fig. 5). Furthermore, while all metaphase spreads from the same tumor showed the clonal T(12,15), individual metaphases exhibited varying degrees of amplification, and some still did not carry a “telomere cap” on der(15)a. In these metaphases, repeated cycles of breakage-fusion-bridge would explain the observed amplification of IgH/c-myc loci extending to the end of the chromosome.
Resolution of the breakage-fusion-bridge cycle can occur when the derivative chromosome is capped by telomeric sequences (51). Such capping may also occur by break-induced replication (Fig. 6, step 8). Why chromosomes 1, 3 and 16 (Fig. 2) are consistently used for the purpose of telomere capture remains to be determined; however, one can postulate that they either harbor sequences which are homologous to chromosomes 12 or 15, are in close proximity within the nucleus or a combination of these two possibilities.
A similar question is often raised as to why c-myc is the oncogene so frequently observed in juxtaposition with IgH in tumors of B cell origin. Given the fact that there are around 1.4 × 106 B220+μ− pro-B cells in the bone marrow (BM) of p53-deficient mice (40), a one in a million event would be sufficient to give rise to a tumor precursor cell in which the IgH enhancer drives expression of the c-myc oncogene. Yu and Thomas-Tikhonenko injected BM cells from p53−/− mice into syngeneic recipients after infecting them with a c-myc expressing retrovirus (52). Given their 60% infection rate, the ∼600,000 c-myc expressing BM cells gave rise to tumors within 5 weeks post-injection. Only 19 cell divisions would be required to get from a one in a million precursor cell to the 600,000 cells injected into these mice. At one cell division per day, a precursor cell in a newborn mouse could give rise to a pro-B cell lymphoma within 8 wk, a time consistent with our observations in Ku/p53-deficient mice. Thus, one need not postulate that the region near c-myc is particularly prone to rearrangement with the IgH locus, but that this extremely random event juxtaposing c-myc and IgH is sufficient to allow cell survival. Subsequent amplification of this rearranged locus and deregulation of c-myc expression would then allow for the rapid expansion of this cell into a tumor population.
In summary, the Ku−/−p53−/− mouse provides a genetic system in which the etiology of chromosomal aberrations in the absence of NHEJ can be studied. We demonstrate that the formation of oncogenic nonreciprocal translocations and amplifications in NHEJ-deficient cells are triggered by a Rad54-independent break-induced replication mechanism that involves invasion of a heterologous chromosome using DSBs created through RAG-mediated cleavage. The subsequent amplification of c-myc/IgH results from cycles of bridge-fusion-breakage. Resolution and protection from exonucleolytic degradation is eventually achieved when the recombinogenic chromatid ends invade and copy sequences from an unbroken chromosome containing a telomere. The resultant nonreciprocal translocation and gene amplifications resemble the complex cytogenetic profiles frequently observed in human cancers. Ku−/− p53−/− mice will therefore serve as useful model systems for further studies aimed at elucidating the genetic requirements of replication-based recombination mechanisms leading to malignancy.
Acknowledgments
We thank F. Alt for kindly providing the Ku70−/− mice; J. Thomas for help in screening the BAC library; K. Hathcock for providing the PNA telomere probe; E. Max for helpful discussions; and M.C. Nussenzweig, A. Singer, and R. Hodes for critical reading of the manuscript.
Note added in proof: After acceptance of this manuscript, similar findings were published by Fred Alt's group in pro-B cell lymphomas from Xrcc4−/−p53−/− and DNA LigIV−/−p53−/− mice.
Footnotes
Abbreviations used in this paper: BM, bone marrow; DSB, double-strand break; HR, homologous recombination; NHEJ, nonhomologous end joining; PNA, peptide nucleic acid; SKY, Spectral Karyotype.
References
- 1.Richardson, C., and M. Jasin. 2000. Frequent chromosomal translocations induced by DNA double-strand breaks. Nature. 405:697–700. [DOI] [PubMed] [Google Scholar]
- 2.Pipiras, E., A. Coquelle, A. Bieth, and M. Debatisse. 1998. Interstitial deletions and intrachromosomal amplification initiated from a double-strand break targeted to a mammalian chromosome. EMBO J. 17:325–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bassing, C.H., W. Swat, and F.W. Alt. 2002. The mechanism and regulation of chromosomal V(D)J recombination. Cell. 109(Suppl.):S45–S55. [DOI] [PubMed] [Google Scholar]
- 4.Difilippantonio, M.J., J. Zhu, H.T. Chen, E. Meffre, M.C. Nussenzweig, E.E. Max, T. Ried, and A. Nussenzweig. 2000. DNA repair protein Ku80 suppresses chromosomal aberrations and malignant transformation. Nature. 404:510–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karanjawala, Z.E., U. Grawunder, C.L. Hsieh, and M.R. Lieber. 1999. The nonhomologous DNA end joining pathway is important for chromosome stability in primary fibroblasts. Curr. Biol. 9:1501–1504. [DOI] [PubMed] [Google Scholar]
- 6.Ferguson, D.O., J.M. Sekiguchi, S. Chang, K.M. Frank, Y. Gao, R.A. DePinho, and F.W. Alt. 2000. The nonhomologous end-joining pathway of DNA repair is required for genomic stability and the suppression of translocations. Proc. Natl. Acad. Sci. USA. 97:6630–6633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frank, K.M., N.E. Sharpless, Y. Gao, J.M. Sekiguchi, D.O. Ferguson, C. Zhu, J.P. Manis, J. Horner, R.A. DePinho, and F.W. Alt. 2000. DNA ligase IV deficiency in mice leads to defective neurogenesis and embryonic lethality via the p53 pathway. Mol. Cell. 5:993–1002. [DOI] [PubMed] [Google Scholar]
- 8.Gao, Y., D.O. Ferguson, W. Xie, J.P. Manis, J. Sekiguchi, K.M. Frank, J. Chaudhuri, J. Horner, R.A. DePinho, and F.W. Alt. 2000. Interplay of p53 and DNA-repair protein XRCC4 in tumorigenesis, genomic stability and development. Nature. 404:897–900. [DOI] [PubMed] [Google Scholar]
- 9.Guidos, C.J., C.J. Williams, I. Grandal, G. Knowles, M.T. Huang, and J.S. Danska. 1996. V(D)J recombination activates a p53-dependent DNA damage checkpoint in scid lymphocyte precursors. Genes Dev. 10:2038–2054. [DOI] [PubMed] [Google Scholar]
- 10.Lim, D.S., H. Vogel, D.M. Willerford, A.T. Sands, K.A. Platt, and P. Hasty. 2000. Analysis of ku80-mutant mice and cells with deficient levels of p53. Mol. Cell. Biol. 20:3772–3780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nacht, M., A. Strasser, Y.R. Chan, A.W. Harris, M. Schlissel, R.T. Bronson, and T. Jacks. 1996. Mutations in the p53 and SCID genes cooperate in tumorigenesis. Genes Dev. 10:2055–2066. [DOI] [PubMed] [Google Scholar]
- 12.Vanasse, G.J., J. Halbrook, S. Thomas, A. Burgess, M.F. Hoekstra, C.M. Disteche, and D.M. Willerford. 1999. Genetic pathway to recurrent chromosome translocations in murine lymphoma involves V(D)J recombinase. J. Clin. Invest. 103:1669–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sharpless, N.E., D.O. Ferguson, R.C. O'Hagan, D.H. Castrillon, C. Lee, P.A. Farazi, S. Alson, J. Fleming, C.C. Morton, K. Frank, et al. 2001. Impaired nonhomologous end-joining provokes soft tissue sarcomas harboring chromosomal translocations, amplifications, and deletions. Mol. Cell. 8:1187–1196. [DOI] [PubMed] [Google Scholar]
- 14.Tai, Y.T., G. Teoh, B. Lin, F.E. Davies, D. Chauhan, S.P. Treon, N. Raje, T. Hideshima, Y. Shima, K. Podar, and K.C. Anderson. 2000. Ku86 variant expression and function in multiple myeloma cells is associated with increased sensitivity to DNA damage. J. Immunol. 165:6347–6355. [DOI] [PubMed] [Google Scholar]
- 15.Riballo, E., S.E. Critchlow, S.H. Teo, A.J. Doherty, A. Priestley, B. Broughton, B. Kysela, H. Beamish, N. Plowman, C.F. Arlett, et al. 1999. Identification of a defect in DNA ligase IV in a radiosensitive leukaemia patient. Curr. Biol. 9:699–702. [DOI] [PubMed] [Google Scholar]
- 16.O'Driscoll, M., K.M. Cerosaletti, P.M. Girard, Y. Dai, M. Stumm, B. Kysela, B. Hirsch, A. Gennery, S.E. Palmer, J. Seidel, et al. 2001. DNA ligase IV mutations identified in patients exhibiting developmental delay and immunodeficiency. Mol. Cell. 8:1175–1185. [DOI] [PubMed] [Google Scholar]
- 17.Gu, Y., K.J. Seidl, G.A. Rathbun, C. Zhu, and J.P. Manis. N.v.D. Stoep, L. Davidson, H.-L. Cheng, J.M. Sekiguchi, K. Frank, P. Stanhope-Baker, M.S. Schlissel, D.B. Roth, and F.W. Alt. 1997. Growth retardation and leaky SCID phenotype of Ku70-deficient mice. Immunity. 7:653–665. [DOI] [PubMed] [Google Scholar]
- 18.Timme, T.L., and T.C. Thompson. 1994. Rapid allelotype analysis of p53 knockout mice. Biotechniques. 17:460, 462–463. [PubMed]
- 19.Donehower, L.A., M. Harvey, B.L. Slagle, M.J. McArthur, C.A. Montgomery, Jr., J.S. Butel, and A. Bradley. 1992. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 356:215–221. [DOI] [PubMed] [Google Scholar]
- 20.Lowe, S.W., E.M. Schmitt, S.W. Smith, B.A. Osborne, and T. Jacks. 1993. p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature. 362:847–849. [DOI] [PubMed] [Google Scholar]
- 21.Essers, J., R.W. Hendriks, S.M. Swagemakers, C. Troelstra, J. de Wit, D. Bootsma, J.H. Hoeijmakers, and R. Kanaar. 1997. Disruption of mouse RAD54 reduces ionizing radiation resistance and homologous recombination. Cell. 89:195–204. [DOI] [PubMed] [Google Scholar]
- 22.Liyanage, M., Z. Weaver, C. Barlow, A. Coleman, D.G. Pankratz, S. Anderson, A. Wynshaw-Boris, and T. Ried. 2000. Abnormal rearrangement within the alpha/delta T-cell receptor locus in lymphomas from Atm-deficient mice. Blood. 96:1940–1946. [PubMed] [Google Scholar]
- 23.Chu, C.C., W.E. Paul, and E.E. Max. 1992. Quantitation of immunoglobulin m-g1 heavy chain switch region recombination by a digestion-circularization polymerase chain reaction method. Proc. Natl. Acad. Sci. USA. 89:6978–6982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barlow, C., S. Hirotsune, R. Paylor, M. Liyanage, M. Eckhaus, F. Collins, Y. Shiloh, J.N. Crawley, T. Ried, D. Tagle, and A. Wynshaw-Boris. 1996. Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell. 86:159–171. [DOI] [PubMed] [Google Scholar]
- 25.Jhappan, C., H.C. Morse III, R.D. Fleischmann, M.M. Gottesman, and G. Merlino. 1997. DNA-PKcs: a T-cell tumour suppressor encoded at the mouse scid locus. Nat. Genet. 17:483–486. [DOI] [PubMed] [Google Scholar]
- 26.Morrison, C., G.C. Smith, L. Stingl, S.P. Jackson, E.F. Wagner, and Z.Q. Wang. 1997. Genetic interaction between PARP and DNA-PK in V(D)J recombination and tumorigenesis. Nat. Genet. 17:479–482. [DOI] [PubMed] [Google Scholar]
- 27.Ouyang, H., A. Nussenzweig, A. Kurimasa, V.C. Soares, X. Li, C. Cordon-Cardo, W. Li, N. Cheong, M. Nussenzweig, G. Iliakis, et al. 1997. Ku70 is required for DNA repair but not for T cell antigen receptor gene recombination in vivo. J. Exp. Med. 186:921–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dronkert, M.L., H.B. Beverloo, R.D. Johnson, J.H. Hoeijmakers, M. Jasin, and R. Kanaar. 2000. Mouse RAD54 affects DNA double-strand break repair and sister chromatid exchange. Mol. Cell. Biol. 20:3147–3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taub, R., I. Kirsch, C. Morton, G. Lenoir, D. Swan, S. Tronick, S. Aaronson, and P. Leder. 1982. Translocation of the c-myc gene into the immunoglobulin heavy chain locus in human Burkitt lymphoma and murine plasmacytoma cells. Proc. Natl. Acad. Sci. USA. 79:7837–7841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Savelyeva, L., and M. Schwab. 2001. Amplification of oncogenes revisited: from expression profiling to clinical application. Cancer Lett. 167:115–123. [DOI] [PubMed] [Google Scholar]
- 31.Bergsagel, P.L., and W.M. Kuehl. 2001. Chromosome translocations in multiple myeloma. Oncogene. 20:5611–5622. [DOI] [PubMed] [Google Scholar]
- 32.Liang, F., P.J. Romanienko, D.T. Weaver, P.A. Jeggo, and M. Jasin. 1996. Chromosomal double-strand break repair in Ku80-deficient cells. Proc. Natl. Acad. Sci. USA. 93:8929–8933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taccioli, G.E., G. Rathbun, E. Oltz, T. Stamato, P.A. Jeggo, and F.W. Alt. 1993. Impairment of V(D)J recombination in double-strand break repair mutants. Science. 260:207–210. [DOI] [PubMed] [Google Scholar]
- 34.Kabotyanski, E.B., L. Gomelsky, J.O. Han, T.D. Stamato, and D.B. Roth. 1998. Double-strand break repair in Ku86- and XRCC4-deficient cells. Nucleic Acids Res. 26:5333–5342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Petiniot, L.K., Z. Weaver, C. Barlow, R. Shen, M. Eckhaus, S.M. Steinberg, T. Ried, A. Wynshaw-Boris, and R.J. Hodes. 2000. Recombinase-activating gene (RAG) 2-mediated V(D)J recombination is not essential for tumorigenesis in Atm-deficient mice. Proc. Natl. Acad. Sci. USA. 97:6664–6669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liao, M.J., X.X. Zhang, R. Hill, J. Gao, M.B. Qumsiyeh, W. Nichols, and T. Van Dyke. 1998. No requirement for V(D)J recombination in p53-deficient thymic lymphoma. Mol. Cell. Biol. 18:3495–3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Agrawal, A., Q.M. Eastman, and D.G. Schatz. 1998. Transposition mediated by RAG1 and RAG2 and its implications for the evolution of the immune system. Nature. 394:744–751 (see comments). [DOI] [PubMed] [Google Scholar]
- 38.Hiom, K., M. Melek, and M. Gellert. 1998. DNA transposition by the Rag1 and Rag2 proteins: A possible source of oncogenic translocations. Cell. 94:463–470. [DOI] [PubMed] [Google Scholar]
- 39.Lee, G.S., M.B. Neiditch, R.R. Sinden, and D.B. Roth. 2002. Targeted transposition by the V(D)J recombinase. Mol. Cell. Biol. 22:2068–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu, L., D. Lejtenyi, and D.G. Osmond. 1999. Regulation of cell survival during B lymphopoiesis: suppressed apoptosis of pro-B cells in P53-deficient mouse bone marrow. Eur. J. Immunol. 29:2484–2490. [DOI] [PubMed] [Google Scholar]
- 41.Bosco, G., and J.E. Haber. 1998. Chromosome break-induced DNA replication leads to nonreciprocal translocations and telomere capture. Genetics. 150:1037–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morrow, D.M., C. Connelly, and P. Hieter. 1997. “Break copy” duplication: a model for chromosome fragment formation in Saccharomyces cerevisiae. Genetics. 147:371–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Signon, L., A. Malkova, M.L. Naylor, H. Klein, and J.E. Haber. 2001. Genetic requirements for RAD51- and RAD54-independent break-induced replication repair of a chromosomal double-strand break. Mol. Cell. Biol. 21:2048–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith, K.A., M.L. Agarwal, M.V. Chernov, O.B. Chernova, Y. Deguchi, Y. Ishizaka, T.E. Patterson, M.F. Poupon, and G.R. Stark. 1995. Regulation and mechanisms of gene amplification. Philos. Trans. R. Soc. Lond. B Biol. Sci. 347:49–56. [DOI] [PubMed] [Google Scholar]
- 45.Coquelle, A., E. Pipiras, F. Toledo, G. Buttin, and M. Debatisse. 1997. Expression of fragile sites triggers intrachromosomal mammalian gene amplification and sets boundaries to early amplicons. Cell. 89:215–225. [DOI] [PubMed] [Google Scholar]
- 46.Schimke, R.T. 1982. Gene Amplification. Cold Spring Harbor Lab. Press, Plainview, NY. 339 pp.
- 47.Livingstone, L.R., A. White, J. Sprouse, E. Livanos, T. Jacks, and T.D. Tlsty. 1992. Altered cell cycle arrest and gene amplification potential accompany loss of wild-type p53. Cell. 70:923–935. [DOI] [PubMed] [Google Scholar]
- 48.Yin, Y., M.A. Tainsky, F.Z. Bischoff, L.C. Strong, and G.M. Wahl. 1992. Wild-type p53 restores cell cycle control and inhibits gene amplification in cells with mutant p53 alleles. Cell. 70:937–948. [DOI] [PubMed] [Google Scholar]
- 49.Denis, N., A. Kitzis, J. Kruh, F. Dautry, and D. Corcos. 1991. Stimulation of methotrexate resistance and dihydrofolate reductase gene amplification by c-myc. Oncogene. 6:1453–1457. [PubMed] [Google Scholar]
- 50.McClintock, B. 1951. Chromosome organization and genetic expression. Cold Spring Harb. Symp. Quant. Biol. 16:13–47. [DOI] [PubMed] [Google Scholar]
- 51.Meltzer, P.S., X.Y. Guan, and J.M. Trent. 1993. Telomere capture stabilizes chromosome breakage. Nat. Genet. 4:252–255. [DOI] [PubMed] [Google Scholar]
- 52.Yu, D., and A. Thomas-Tikhonenko. 2002. A non-transgenic mouse model for B-cell lymphoma: in vivo infection of p53-null bone marrow progenitors by a Myc retrovirus is sufficient for tumorigenesis. Oncogene. 21:1922–1927. [DOI] [PubMed] [Google Scholar]