

Abstract
Glutamic acid decarboxylase (GAD)65 is an early and important antigen in both human diabetes mellitus and the nonobese diabetic (NOD) mouse. However, the exact role of GAD65-specific T cells in diabetes pathogenesis is unclear. T cell responses to GAD65 occur early in diabetes pathogenesis, yet only one GAD65-specific T cell clone of many identified can transfer diabetes. We have generated transgenic mice on the NOD background expressing a T cell receptor (TCR)-specific for peptide epitope 286–300 (p286) of GAD65. These mice have GAD65-specific CD4+ T cells, as shown by staining with an I-Ag7(p286) tetramer reagent. Lymphocytes from these TCR transgenic mice proliferate and make interferon γ, interleukin (IL)-2, tumor necrosis factor (TNF)-α, and IL-10 when stimulated in vitro with GAD65 peptide 286–300, yet these TCR transgenic animals do not spontaneously develop diabetes, and insulitis is virtually undetectable. Furthermore, in vitro activated CD4 T cells from GAD 286 TCR transgenic mice express higher levels of CTL-associated antigen (CTLA)-4 than nontransgenic littermates. CD4+ T cells, or p286-tetramer+CD4+ Tcells, from GAD65 286–300-specific TCR transgenic mice delay diabetes induced in NOD.scid mice by diabetic NOD spleen cells. This data suggests that GAD65 peptide 286–300-specific T cells have disease protective capacity and are not pathogenic.
Keywords: T lymphocytes, regulatory; autoimmunity; mouse, NOD; IL-10; CD152
Introduction
Type I diabetes mellitus is an autoimmune disease in which lymphocytes infiltrate the pancreatic islets and destroy the insulin-producing β cells (1). Nonobese diabetic (NOD)* mice develop a similar destructive pancreatic infiltrate which results in diabetes (2). NOD mice also develop lymphocytic infiltrates in other organs such as salivary glands and thyroid, which parallel other autoimmune syndromes that develop in some humans with diabetes. T cells are central to diabetes pathogenesis, as evidenced by the ability of purified CD4 and CD8 T cells to cause diabetes upon adoptive transfer (3, 4).
In NOD mice, insulitis is first observed at 3–8 wk of age, but overt diabetes does not occur until 12–25 wk of age (5). Humans with type I diabetes also have a lag between onset of autoimmunity and overt diabetes; autoreactive T cell and antibody responses can precede clinical diabetes, sometimes by years (6). A regulatory cell type present early, but which disappears or becomes ineffective as the disease progresses, may be responsible for the delay of diabetes initiation. Transfer studies show that CD4+ T cells from young prediabetic mice can delay the transfer of diabetes to irradiated adult NOD mice by spleen cells from recently diabetic mice (7). Many different cell types may regulate diabetes pathogenesis. For example, NOD mice have lower levels of CD4+CD25+ cells, and increasing the number of these cells delays diabetes onset (8, 9) (unpublished data). A shift from production of Th1 cytokines to Th2 cytokines is also protective in diabetes models (10). Another CD4+ subset identified in mice and humans, named Tr1, makes IFN-γ and IL-10, but not IL-4, and can downregulate immune responses (11). In some situations, even IFN-γ–producing cells may have a regulatory role in diabetes development. This is suggested by the ability of IFN-γ deficient (but not IL-10 or IL-4 deficient) NOD mice to develop diabetes after treatment with bacillus Calmette-Guerin (BCG), an immune stimulant which normally prevents diabetes development (12). The relative importance of the different regulatory populations in maintaining tolerance to islet antigens has not been determined.
Studies using mice transgenic for different TCRs show that even a restricted TCR repertoire can lead to development of diabetes. For example, expression of nondisease-related TCR-α and -β chains in NOD mice does not reduce diabetes incidence (13). Similarly, NOD mice expressing a MHC class I–restricted, LCMV-specific TCR develop diabetes as long as endogenous TCRs can still rearrange (14). This suggests that the limited diverse repertoire, most likely the result of endogenous TCR-α chains pairing with the transgenic β chain, generates sufficient self-reactive, pathogenic T cell responses to result in diabetes. Mice expressing TCR transgenes specific for unidentified β cell antigens show that even a single specificity can generate a T cell response adequate for disease progression. Mice expressing the BDC2.5 TCR, derived from a diabetogenic CD4+ clone isolated from NOD spleen, develop insulitis and diabetes (15, 16). Likewise, mice expressing the 4.1 TCR, which uses a TCR isolated from a CD4+ T cell clone found in the islets of a diabetic NOD mouse, rapidly develops diabetes in 75% of female mice (17). Studies with these two TCR transgenics have been instrumental in defining many aspects of diabetes pathogenesis (5). However, these studies are limited by the unknown antigen specificity of these transgenic TCRs.
Two of the most important autoantigens in diabetes pathogenesis, in both mice and humans, are insulin and glutamic acid decarboxylase (GAD)65 (18, 19). The presence of antibodies specific for these two antigens is predictive of future diabetes onset in individuals with a permissive HLA type and a family history of diabetes (20). Insulin is the major secreted protein of the β cell and is also expressed in the thymus. Both CD4+ T cell clones specific for the insulin immunodominant epitope B(9–23), and CD8+ cells specific for an overlapping epitope, B (15–23), can induce diabetes upon adoptive transfer (21, 22). GAD65, the enzyme which makes the inhibitory neurotransmitter GABA from glutamic acid, is expressed only in islet cells and the brain (23). Both antibody and T cell responses to GAD65 occur at the earliest detectable stages of diabetes pathogenesis in mice and humans (24–26), leading to speculation that GAD65 may be the first antigen for which tolerance is broken. As with insulin, inducing tolerance to GAD65 by injecting GAD65 or its peptide epitopes can prevent diabetes (27, 28). Furthermore, diabetes did not develop in mice in which GAD65 expression in the islets was ablated by expression of GAD anti-sense RNA under the insulin promoter. (29). Despite this evidence, only one diabetogenic GAD-specific clone has been identified (30). Clearly much is still unknown about the role of GAD65-specific T cells in diabetes pathogenesis.
The goal of this study is to determine the role of T cells specific for one GAD65 epitope, peptide 286–300 (p286), in the pathogenesis of diabetes in NOD mice. p286 is one of three immunodominant epitopes of GAD65 previously identified by this laboratory by screening T cell hybridomas derived from 10–12-wk-old NOD mice immunized with GAD65 protein emulsified in IFA (31, 32). T cells specific for this region of GAD65 (p286 in one case, peptide 290–309 in another) were also isolated in two separate systems from the spleens of unimmunized NOD mice, confirming the presence of these T cells in unmanipulated mice (unpublished data) (10). We have developed and characterized NOD mice expressing a p286-specific TCR transgene, and found that these mice do not develop diabetes. Our results suggest instead, that GAD65-p286-specific T cells may play a protective role in the development of diabetes in NOD mice.
Materials and Methods
Cloning of 286-specific TCRs into TCR-α and -β Expression Vectors.
RT-PCR using primer pairs specific for each Vα and Vβ was used to determine that B16.3, a p286-specific hybridoma, expresses Vα4.5-Jα6 and Vβ1-Jβ2.7 (unpublished data). To clone this TCR, PCR was performed on genomic DNA preparations from B16.3. Primers were designed at the beginning of the leader segment of V, and the second primer located in the intron ∼200-bp downstream of the J segment for both α and β chains. Primer sequences were designed to engineer restriction sites (underlined) for subcloning as follows: β chain forward, CCGTCTGGAGTC GACTTCCACCATGAGC; β chain reverse, CTGGTCTACCCGCGGCTACTCCAGGGACCCAGGAATTTG; α chain forward, TCTCACTGCCTAGCCATGAACACTTCTCCAGCTTTA; α chain reverse, AACCTCGGTGCGGCCGCTTTGGATATTTGTCCTTTG. The α chain PCR product was subjected to an additional round of PCR with a second forward primer, TGCAGAACCCGGGCTTCTCACTGCCTAGCCATG, coupled with the α chain reverse primer to introduce the XmaI site. Once the V(D)J sequence was confirmed, the insert was digested with XmaI and NotI (α) or XhoI and SacII (β) and then subcloned into previously described expression vectors containing TCR regulatory and constant regions (pTαcass, pTβcass; reference 33).
Generation of Mice Expressing p286-specific TCR-α and -β Transgenic Mice.
The TCR-α and -β chain vectors were cut with SalI and KpnI respectively to remove prokaryotic sequences. The purified 18-kb (TCR-α) and 17-kb (TCR-β) linearized vectors were then injected into NOD embryos. The DNA was injected into NOD embryos (instead of NOD×FVB F1 embryos) to avoid potentially introducing diabetes protective loci from the FVB strain linked to the site of transgene integration.
Potential founders were screened for integration of the transgenes by PCR using the same primers described above. Expression of αβ pairs was confirmed by staining with I-Ag7/p286 tetramers. Two lines were established (A and B). Unless otherwise indicated, all data shown here were obtained using A line mice. These TCR transgenic mice expressing p286-specific TCR-α and -β transgenes will be subsequently referred to as G286 (GAD65–286–300). All animal studies have been approved by Stanford University's Administrative Panel for Laboratory Animal Care (A-PLAC).
Cell Isolation.
Where indicated, pooled lymph nodes, specific lymph nodes, or spleen cells were macerated and washed in RPMI 1640 media containing 200 U/ml penicillin, 200 μg/ml streptomycin, 10 mM Hepes, 0.06 μg/ml l-glutamine, 10−5 mM 2-ME, and 10% FCS. For spleen cells, red blood cells were lysed by incubation in ACK lysing buffer (0.15 M NH4Cl, 1 mMKHCO3, 0.1 mM Na2EDTA; reference 34), 4 ml/spleen for 4 min, then washed in media. Where indicated, CD4+ T cells were purified by negative selection using magnetic beads conjugated with CD8 and B220 antibodies (Dynal).
Flow Cytometry.
The following antibodies were used for FACS® analysis: anti-CD4 (RM4–5) and anti-CD8 (5H-10); FITC and PE conjugated, were purchased from Caltag Laboratories. Antibodies against CD4 (GK1.5) and CD8 (53–6.7) conjugated to Texas Red and APC were obtained from the laboratory of Irving Weissman. Antibodies against the following Vβ8 (MR5–2), CD3 (145–2C11), CD25 (7D4), CD44 (IM7), CD62L (MEL-14), CD69 (H1.2F3), all FITC conjugated; Vα2-PE (B20.1) CTL-associated antigen (CTLA)-4-PE (UC10–4F10–11) were all purchased from BD PharMingen. All washes and staining were performed in PBS with 1% FBS. All staining was done at 4° for 30 min following standard protocols (BD PharMingen).
To stain p286-specific T cells, a tetramer was assembled by associating streptavidin-PE or streptavidin-APC with soluble biotinylated I-Ag7 monomers to which GAD peptide 286–300 was covalently linked. The soluble monomers were produced by Luc Teyton (Scripps Research Institute, La Jolla, CA) and synthesized as indicated previously (35). Any staining which included p286-specific tetramer was done at room temperature for 45 min.
For assessment of CTLA-4 expression, pooled lymph node cells from G286 mice and littermate transgene negative mice were activated in vitro with plate-bound anti-CD3 (2 μg/ml) and anti-CD28 (4 μg/ml). Cells were harvested after 48 h and stained with CD4-FITC and p286-tetramer. Cells were then fixed for 20 min in 2% formaldehyde and permeabilized with 0.5% saponin. Anti–CTLA-4 was added for 30 min at room temperature.
Histological Analysis.
Pancreas or salivary gland tissue was fixed in 10% formalin and paraffin-embedded sections were stained with H&E. Tissue cuts were made 100 microns apart to avoid counting any islets twice. Insulitis was assessed for each islet, and scored as: no insulitis, periinsulitis, and intrainsulitis.
Proliferation and Cytokine Detection.
To activate cells in vitro, 2.5 × 105 pooled lymph node or spleen cells from G286 and nontransgenic mice were activated in triplicate with the indicated dose of GAD65 p286 and 2.5 × 105 irradiated (3,000 rads) NOD spleen cells (alterations to this protocol are indicated). Proliferation was assessed by 3[H]thymidine incorporation. 0.5 μCi/well 3[H]thymidine was added at 72 h, and plates were harvested 12 h later unless otherwise indicated. To determine expression of IL-2, IL-4, IL-5, TNF-α, and IFN-γ, antibody pairs from BD PharMingen were used, and a sandwich immunoassay was performed following standard protocols with supernatants from in vitro activated cultures. Streptavidin-Europium was used to quantitate the amount of bound biotinylated secondary antibody. IL-10 ELISAs were performed with precoated plates and reagents from Endogen. The cultures used to determine IL-10 expression contained 6 × 105 lymph node cells plus 6 × 105 irradiated NOD spleen cells.
Cell Transfer Studies and Monitoring of Diabetes.
2–10 × 106 unfractionated spleen cells from newly diabetic NOD mice were injected intravenously (tail vein) into 5–10-wk-old female NOD scid mice. As indicated for each experiment, various lymphocyte populations from G286 mice were mixed with these spleen cells. All G286 mice used as donors were female. When indicated, spleen or lymph node cells from G286 mice were activated with anti-CD3 (2 μg/ml) and anti-CD28 (4 μg/ml) or with 10 μg/ml p286 for 48 h. The final volume of all injections was 100 μl per mouse in PBS/0.1% BSA. Urine glucose levels were monitored using Chemstrips (Roche Laboratories) which have a glucose threshold of 100 mg/dL. Mice were considered diabetic on the first of three consecutive high glucose readings and were killed after the third high reading.
Statistical Analysis.
Statistical significance of delay in diabetes transfer was determined at each time point by Fisher's exact test and is indicated on the incidence plots. All comparisons are between the experimental condition indicated and the control condition consisting of spleen cells from diabetic NOD females transferred alone, except in Fig. 6 A where samples containing tetramer positive cells are compared with those containing tetramer-negative cells. P values < 0.05 are indicated by the symbol *, whereas P values <0.01 are indicated by the symbol #.
Figure 6.
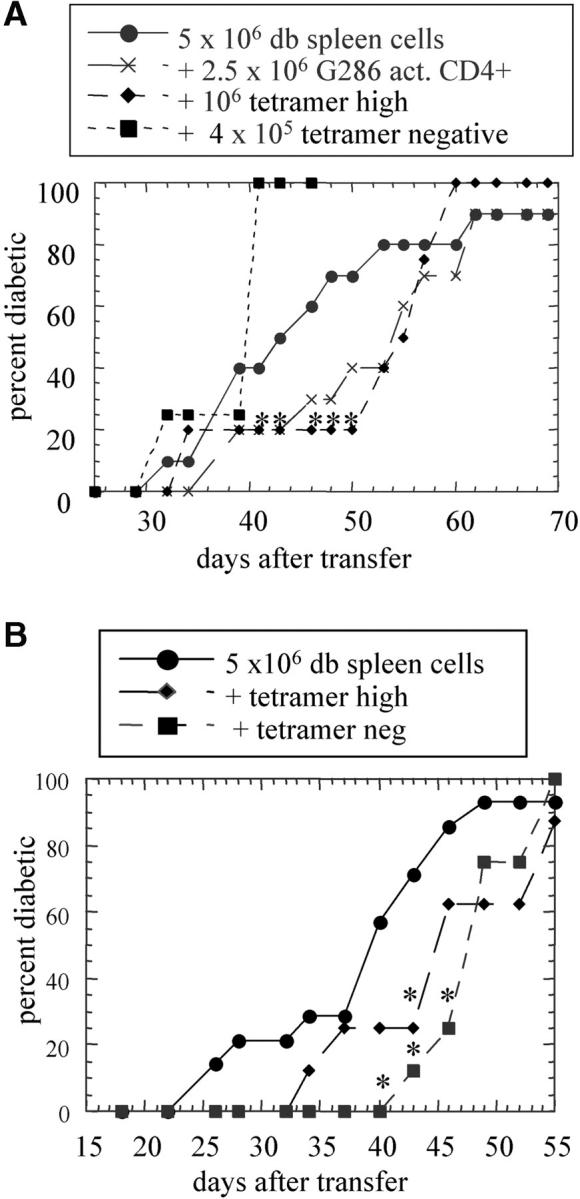

p286/I-Ag7 tetramer-positive CD4+ T cells from G286 mice delay diabetes transfer. Tetramer-positive and -negative cells were sorted from G286 lymph node cells which were activated with irradiated NOD spleen and p286 for 48 h. B220 and CD8 cells were depleted, and the remaining cells were stained with p286 tetramer and CD4 (C, left panel). Gates were set as indicated (C, middle panel), and cells transferred had purity of 98% for sorted populations. The indicated populations were transferred to NOD.scid mice in two separate experiments (A and B). Group sizes are as follows: In A, diabetic spleen cells (n = 10), G286 CD4+ (n = 10), tetramer-high (n = 4), tetramer-negative (n = 4). In B, diabetic spleen cells (n = 15), tetramer-high (n = 8), tetramer-negative (n = 8).
Results
Generation of TCR Transgenic Mice Specific for Peptide Epitope 286–300 of GAD65.
To study the function of GAD65-specific T cells in NOD mice, TCR transgenic mice were produced with TCR-α and -β chains cloned from a hybridoma (B16.3) expressing a TCR specific for p286. This transgenic line will subsequently be referred to as G286. B16.3 expresses one functional TCR-α (Vα4.5-Jα6) and TCR-β (Vβ1-Jβ2.7) rearrangement. The TCR-α and -β variable region sequences were cloned by genomic PCR, and the α and β chains were then ligated into separate TCR expression vectors containing the constant region and regulatory elements necessary for normal T cell–restricted TCR expression (33).
Incorporation and expression of the transgene was assessed by genomic PCR and flow cytometry, respectively. Transgenic TCR-α and -β specific primers amplify PCR product from genomic DNA isolated from transgene-positive but not transgene-negative littermates (Fig. 1 A). Because a Vβ1-specific antibody is not available, expression of the TCR-β chain transgene was determined by a lack of expression of other Vβ proteins due to allelic exclusion. In NOD mice, the normal expression of CD4+ Vβ8+ cells is 8–12% of peripheral blood lymphocytes, but in G286 mice, the percentage of CD4+Vβ8+ cells is <0.5% of PBLs (Fig. 1 B). Vβ5 staining gave similar results (unpublished data). This suggests that 90–95% of the T cells express the transgenic β chain. The α chain of this TCR is Vα4.5, for which there also is not a specific antibody. Because allelic exclusion is not as complete for TCR-α chain, one cannot assess expression of the TCR-α transcript using this same approach.
Figure 1.



Characterization of G286 mice. (A) Integration of TCR-Vα and -Vβ constructs is shown by genomic PCR. (Left) DNA amplified with TCR-α specific primers from transgene positive (lane 1) and transgene negative (lane 2) mice. (Right) DNA amplified with TCR-β specific primers from transgene positive (lanes 3 and 4) and transgene negative (lane 5) mice. (B) Staining of peripheral blood lymphocytes with antibodies against Vβ8 (x axis) and CD4 (y axis) for NOD littermate (left) and transgene positive (right). (C) Staining of spleen cells from NOD (top) or G286 mice (bottom) with I-Ag7-286 tetramer and CD4 or CD8 (right). As a control, cells were also stained with an I-Ag7 tetramer containing peptide 323–339 of OVA (left). (D) Thymocytes from NOD (left) or G286 (right) mice were stained with antibodies against CD4 and CD8. The percentage of cells in different boxed gates are shown.
To assess α chain expression and determine the expression level of p286-specific TCR in G286 mice, a tetramer reagent containing a soluble biotinylated I-Ag7 molecule with covalently linked p286 was employed (35). This soluble I-Ag7/p286 molecule was then conjugated to streptavidin-PE. T cells from G286 mice stain specifically with p286-tetramer, as compared with the OVA-specific tetramer control, and <0.5% of CD4+ T cells from nontransgenic NOD mice stain with the p286-specific tetramer (Fig. 1 C). 10–20% of CD4+ and 40% of CD8+ T cells are positive for p286-tetramer staining. The percentage of CD4+ cells that are tetramer positive increases when T cells from G286 mice are activated with p286 and NOD APCs (Fig. 6 C, left).
Lymphocyte Populations in G286 Mice Are Normal.
Lymphocyte populations in G286 mice and nontransgenic NOD littermates were compared. The total percentage of activated cells in peripheral lymphoid organs was similar in the transgenic and littermate controls, as determined by expression of CD69, CD25, and CD62L (unpublished data). In G286 mice however, a slightly higher percentage of CD4+tetramer+ cells displayed an activated phenotype (as indicated by expression of CD25 and CD44) when compared with CD4+tetramer−cells (unpublished data). In contrast to many MHC class II–restricted TCR transgenics, which have a skewing of CD4:CD8 ratios in the thymus and in the periphery (36), little or no skewing in the CD4:CD8 ratio was observed in lymph node, spleen, or thymus of G286 mice when compared with NOD littermates (Fig. 1 D and unpublished data).
Endogenous TCR-α Chain Expression Is Necessary for Selection of CD4 T Cells in G286 Mice.
G286 mice were crossed onto NOD TCR Cα−/− mice to determine the role of T cells expressing endogenous TCR-α chain in selection and disease. In MHC class II–restricted TCR transgenics crossed onto Cα−/−, one would expect CD4+ cells to increase and CD8+ cells to decrease due to a decrease in the endogenous repertoire. Surprisingly, fewer CD4+ T cells were found in the periphery, and the percentage of CD8+ cells was not diminished in G286 Cα−/− mice. The lack of Vα2+ cells confirms that these mice lack expression of endogenous α chains and are TCR-Cα−/−. Many (20%) of the CD4+ cells from G286 Cα−/− mice express Vβ8, indicating that allelic exclusion of Vβ is not complete (Fig. 2 A). The most likely explanation for this result is that endogenous TCR-α chain expression in G286 Cα+/+ NOD mice allows this autoreactive T cell to survive thymic negative selection; the few CD4+ cells in G286 Cα−/− NOD mice are compensating by rearranging endogenous Vβ genes. Coexpression of an endogenous TCR-α chain paired with a transgenic TCR-β chain may allow for sufficient downregulation of the transgenic autoreactive TCR to allow escape from thymic negative selection without failure to be positively selected. This phenomenon has been observed with other autoreactive TCRs (37).
Figure 2.


Evidence for expression of multiple TCRs in G286 mice. (A) Peripheral blood lymphocytes from G286 Cα−/− (left) G286 Cα+/− (middle) and NOD Cα−/− (right) mice were stained with antibodies for CD4 and Vβ8 (top) or CD8 and Vα2 (bottom). The percentage of cells in each quadrant are indicated in each corner of the plots. (B) Cells from lymph node of a G286 mouse or thymus of a G286 Cα+/− mouse were stained with antibodies for CD4, CD8, and CD3. Cells were gated on CD4+ or CD8+. Expression of CD3 versus I-Ag7-286 tetramer is shown. Cells in the diagonal gate represent cells for which the p286-specific TCR is the predominant TCR expressed. Percentage of cells in the gates drawn are indicated on the plots.
Expression of one specific TCR in comparison to total TCR can be assessed by comparing TCR-specific staining (with a tetramer or clonotypic antibody) with the total TCR expression level (represented by CD3 expression; reference 38). Cells for which the majority of the surface TCR is p286-specific will lie on the diagonal of CD3 and tetramer staining. For thymic and lymph node CD8+ cells from G286 mice, the majority of tetramer-staining cells are in this diagonal gate (Fig. 2 B). This unusual CD8+ population binds I-Ag7/p286 tetramer and therefore, is restricted by a MHC class II molecule. Without the help of CD4 binding, these CD8+ cells are likely to have a lower affinity TCR-MHC/peptide interaction, and may thus allow CD8+ T cells with a higher expression of p286-specific TCR to survive negative selection.
For CD4+ cells, the majority of lymph node tetramer-staining cells have lower tetramer staining without a reduction in CD3 staining, indicating that significant levels of other TCRs are expressed on the surface of most of the p286-specific CD4+ cells. However, a minority of CD4+ cells from G286 mice express high levels of p286-specific TCR without other TCRs, as indicated by cells in the diagonal gate. It is not clear how these cells survive selection, but a similar result has been seen with another self-specific TCR (39). It is interesting to note that the percentage of CD4+ cells, which only express a p286-specific TCR (in the diagonal gate), is lower in the lymph node as compared with the thymus; this may be a result of peripheral tolerance mechanisms. Because G286 Cα−/− mice have very few CD4+ T cells, all subsequent experiments were performed with Cα+/− or Cα+/+ mice.
NOD Mice Expressing the p286-specific TCR Transgenes Do Not Develop Diabetes.
G286 females and transgene negative littermates were followed for 30 wk for diabetes. 80% of the nontransgenic and 0% of the G286 mice developed diabetes (Fig. 3 A). At 30 wk, the six G286 mice and two remaining nondiabetic nontransgenic mice were killed and insulitis assessed. Only 10% of islets in transgenic mice displayed any insulitis, whereas both of the nontransgenics had severe insulitis in the few islets that remained. A similar pattern of insulitis was seen in 17-wk-old mice (Fig. 3 B). Subsequently, none of the dozens of G286 mice used for other experiments have developed diabetes, and the second G286 transgenic B line also does not develop diabetes. Because mice which express an unrelated transgenic TCR can still develop diabetes (13, 14), and because G286 mice do not develop diabetes, it is likely that the p286-specific TCR plays a protective role in diabetes pathogenesis. In addition, both NOD and G286 mice develop lymphocytic infiltration in the salivary glands, although sialtis in G286 mice is less severe (Fig. 3 C). This suggests that the lack of inflammatory infiltration in the pancreas of G286 mice may be due to specific suppression of islet-specific responses and not due to the absence of a pathogenic repertoire or to a general immune disregulation.
Figure 3.
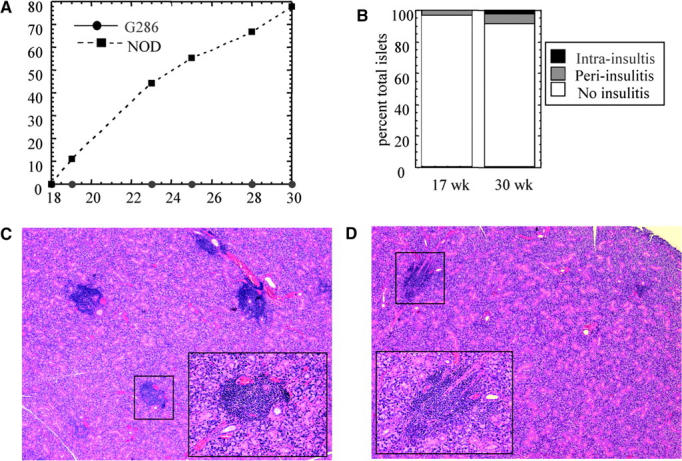
Incidence of diabetes and insulitis in G286 mice. (A) Diabetes incidence in female G286 (circles, n = 6) or NOD littermates (squares, n = 9). (B) Insulitis in 17-wk-old (left) or 30-wk-old (right) female G286 mice. Islets were scored as either no insulitis, periinsulitis, or intrainsulitis. A total of 42 and 160 islets were scored in the 17-wk-old and 30-wk-old mice, respectively. Salivary gland tissue sections from 22-wk-old NOD (C) and 23-wk-old G286 (D) mice. Original magnification is 40× for the large picture, and the inset (location in large picture indicated by the black box) shows one focus of infiltrating lymphocytes at an original magnification of 200×.
Cyclophosphamide (CY) treatment of NOD mice accelerates diabetes onset (40). G286 mice and littermate controls were treated with (CY) at 8 and 10 wk of age. Within 17 d after the second CY injection, 11/11 nontransgenic littermate mice and 0/10 of G286 mice were diabetic. The transgenic mice were followed for 40 d and never developed diabetes. At this time point, five of the transgenic mice were assayed for insulitis, and one mouse tested positive (unpublished data).
Transgenic T Cells Proliferate and Produce Cytokines In Response to p286 In Vitro.
To show that T cells from G286 mice were functional and not tolerant to peptide stimulation, lymph node and spleen cells were isolated and stimulated with p286. The peak stimulation index (SI), as determined by 3[H]thymidine incorporation, was 17 for lymph node cells, and occurred at the 10 μg/ml peptide dose (Fig. 4 A). G286 T cells also responded to GAD65 protein, stimulation index = 7 (unpublished data). When CD4+ T cells from G286 mice were sorted into tetramer-positive and tetramer-negative populations, only the tetramer-positive cells showed significant proliferation (Fig. 4 B). No IL-4 or IL-5 was detected in response to p286 in either the nontransgenic or G286 mice at any dose (unpublished data). Lymph node cells from G286 mice produced TNF-α, IL-2, IFN-γ, and IL-10 in response to p286 (Fig. 4 C and unpublished data). The levels of IFN-γ produced were high; in one experiment, an antigen dose of 15 μg/ml elicited 265 ng/ml of IFN-γ at 48 h. Lymphocytes from nontransgenic littermates produced no detectable IFN-γ, TNF-α, IL-2, or IL-10 in response to p286 (unpublished data). In conclusion, T cells from G286 mice are functional and respond in vitro to their cognate antigen with proliferation and cytokine production.
Figure 4.

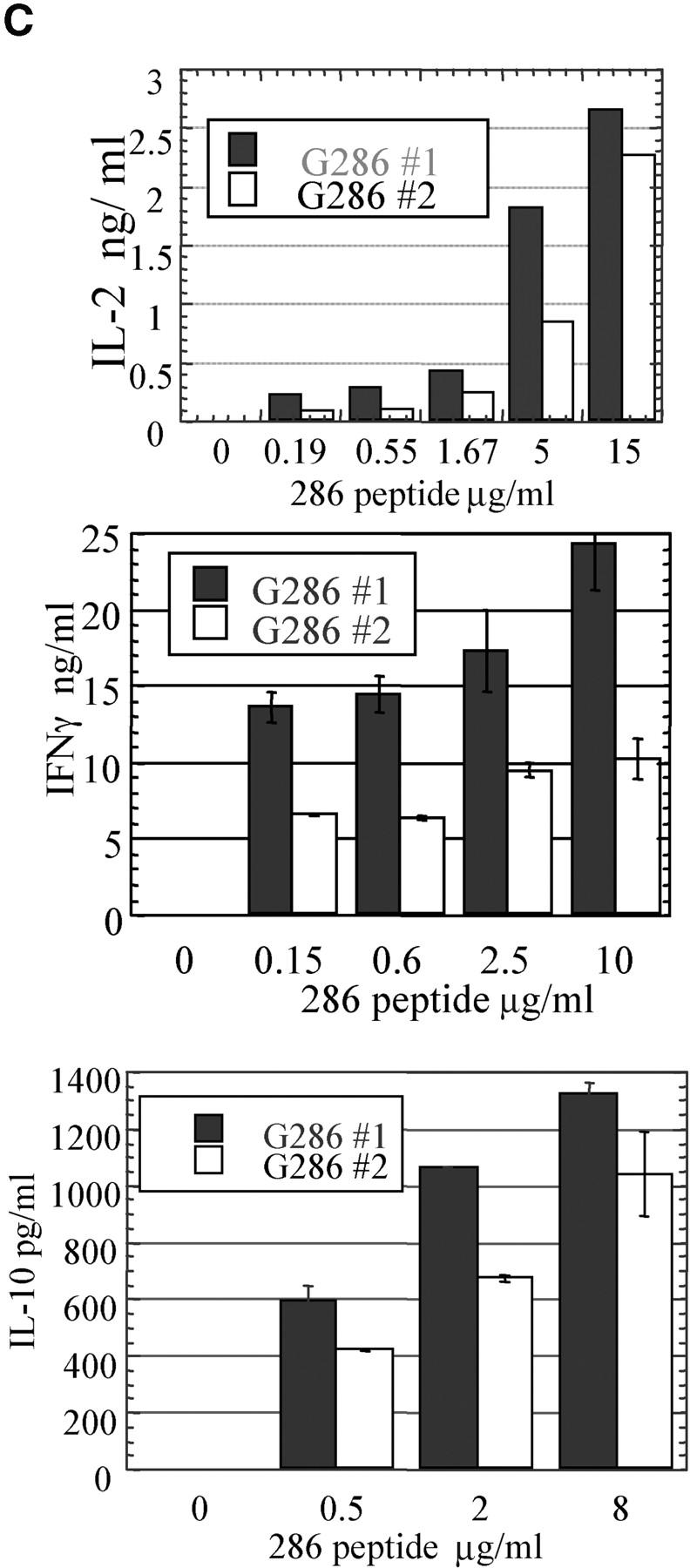

Activation and cytokine production by lymphocytes from G286 mice. Lymph node cells from G286 or NOD mice were activated in vitro with the indicated concentration of p286. Proliferation for total LN cells (A), or purified populations (B) was determined by 3[H]thymidine incorporation. In B, CD4+ p286 tetramer-positive and CD4+ p286 tetramer-negative cells were sorted and activated with 20 μg/ml p286 and irradiated NOD spleen cells; 3[H]thymidine was added for the last 16 of a 96 h incubation. Results are expressed as average stimulation index. The average background counts for the no antigen controls were 1,396 cpm (A) and 58 cpm (B). (C) Expression of IL-2 at 48 h (top), IFN-γ at 62 h (middle), and IL-10 at 48 h (bottom) was determined by ELISA as described in Methods. (D) T cells expressing p286-specific TCR are activated in vivo by p286 peptide. 5-wk-old female G286 mice or transgene negative littermates were immunized in the footpad with either CFA or CFA with 10 μg/ml p286. 60 h later, cells from the draining lymph node were harvested. CD4+ gated cells are shown with p286 tetramer-PE (y axis), and either CD44-FITC or CD62L-FITC (x axis).
In some systems, T cells that are capable of responding to their antigen in vitro cannot respond to the same antigen in vivo (41, 42). To determine if p286-specific T cells are capable of being activated in vivo, G286 mice were immunized with CFA or CFA and p286. Draining lymph node cells were harvested 60 h after immunization. CD4+ tetramer+ cells from mice immunized with p286 expressed higher levels of CD44 and lower levels of CD62L than those isloated from mice which received CFA alone (Fig. 4 D), suggesting that p286-specific cells are not tolerant in vivo. To determine if in vivo activation could induce G286 T cells to induce diabetes, 8–10-wk-old G286 mice were immunized with IFA and 100 μg p286. CFA was not used because it alone can prevent diabetes (43, 44). Of six G286 mice followed, none became diabetic.
T Cells from G286 Mice Do Not Transfer Diabetes but Protect Against Diabetes Transfer by Splenocytes from Diabetic NOD Mice.
To determine if this p286-specific CD4+ T cell response has a protective role in diabetes pathogenesis, G286 spleen cells were transferred together with spleen cells from diabetic NOD females. Splenocytes from diabetic NOD females transfer diabetes into 80–100% of NOD.scid recipients within 40 d of transfer (Fig. 5) . By contrast, transfer of splenocytes from G286 mice alone does not transfer diabetes. The addition of G286 splenocytes to diabetogenic cells results in a significant delay in diabetes transfer (Fig. 5 A). In most of the transfer experiments, even the mice which received lymphocytes from G286 mice eventually developed diabetes. This may be because the pathogenic cells from diabetic NOD mice divide faster or take longer to die than the protective G286 cells, allowing the pathogenic cells to overwhelm the G286 cells over time.
Figure 5.

Lymphocytes from G286 mice delay transfer of diabetes. Transfer of lymphocytes from recently diabetic NOD females and from G286 mice into NODscid females. The following populations were transferred: (A) 107 diabetic spleen cells (n = 8; circles); or 107 spleen cells from G286 mice (n = 8; squares); or both populations together (n = 8; diamonds). (B) 2 × 106 diabetic spleen cells (circles); 2 × 106 diabetic spleen cells plus 2 × 105 peptide activated CD4+ G286 cells (squares). C) 107 diabetic spleen cells n = 9 (circles); 107 diabetic spleen cells + 107 G286 spleen cells n = 11 (squares); 107 diabetic spleen cells plus 107 G286 spleen cells activated with anti-CD3 plus anti-CD28 n = 9 (diamonds); 107 anti-CD3 activated G286 spleen cells (n = 4; triangles). (D) 107 diabetic spleen cells (n = 11; circles); 107 diabetic spleen cells plus 107 CD3-activated G286 spleen cells (squares); 107 diabetic spleen cells plus 5 × 106 peptide activated G286 CD4+ spleen cells (n = 11; diamonds). (E) 107 diabetic spleen cells (n = 15; circles); 107 diabetic spleen cells plus 107 CD3-activated G286 spleen cells (n = 13; diamonds); 107 diabetic spleen cells plus 107 CD3-activated NOD spleen cells (n = 9; squares).
In one experiment, the dose of diabetogenic spleen cells was reduced to 2 × 106 and the dose of activated G286 cells was reduced to 2 × 105; only 2/6 of mice receiving both populations developed diabetes compared with 10/10 mice which received only the diabetogenic cells (Fig. 5 B). The ability of a lower ratio of G286 cells/diabetogenic cells to still protect indicates that the delays in disease progression observed are not due to a dilution of the diabetogenic cells by T cells with a neutral role in diabetes pathogenesis.
The delay in diabetes is increased when CD4+ cells from G286 mice are activated (Fig. 5, C and D). CD4+ populations activated using irradiated NOD spleen cells as APCs with p286 protected longer than those activated with a nonspecific stimulus such as anti-CD3. Whereas stimulation with p286 only stimulates p286-specific T cells, anti-CD3 stimulation should stimulate all T cells. The G286 transfer studies demonstrate that CD4+ T cells from G286 mice are not diabetogenic and are protective against diabetes transfer.
To show that the delays observed are specific to G286, spleen cells from 8-wk-old G286 and NOD mice were both activated with anti-CD3 (Fig. 5 E). G286 spleen cells displayed significantly better protection than NOD spleen cells. Previous studies show that NOD lymphocytes from young mice are capable of diabetes protection, but this capacity diminishes with age (7). This explains the slight delay in diabetes when NOD spleen cells from 8-wk-old mice are added. Most of the G286 mice used in these transfer experiments were older–an age at which NOD mice do not normally show protection.
Tetramer-Positive CD4+ Cells from G286 Mice Can Delay Diabetes Transfer.
CD4+ T cells from G286 mice contain p286-specific cells as well as T cells of other specificities (Fig. 2). Therefore, the observed ability of T cells from G286 mice to delay diabetes transfer could be due to either p286-specific T cells or T cells with other specificities. To study this, lymph node cells from G286 mice were activated with p286 and sorted into CD4+ tetramer-positive and CD4+ tetramer-negative fractions. These populations were added to spleen cells from diabetic NOD females and transferred to NOD scid mice. The tetramer positive fraction was able to delay diabetes transfer in all three experiments performed. The effect of tetramer negative cells was mixed, but in two of three experiments, tetramer negative cells did not delay diabetes transfer (Fig. 6 and unpublished data). Therefore, the delay in diabetes mediated by activated CD4+ T cells from 286 transgenic mice is due, at least in part to cells expressing GAD65 p286-specific TCR.
CTLA-4 Expression on Activated G286 CD4+ Cells Is Higher Than on Nontransgenic Cells.
Expression of the regulatory molecule CTLA-4 is one possible mechanism by which CD4+ T cells from G286 mice could mediate protection from diabetes transfer. For example, increased CTLA-4 expression could lead to downmodulation of other pathogenic cells by inducing TGF-β expression (45, 46). When T cells were activated with anti-CD3 and anti–CD28-coated plates, CD4+ lymph node cells from G286 mice expressed higher levels of CTLA-4 than nontransgenic littermates. In addition, when CD4+ T cells from transgenic mice were divided into tetramer positive and tetramer negative subsets, the tetramer positive subset expressed higher levels of CTLA-4 (Fig. 7 A). For the plots shown, the mean fluorescence intensity (MFI) of CTLA-4 expression was 4.3 for CD4+ cells from the transgene negative mice. For G286 mice, the MFI of CD4+ tetramer-negative cells was 5.8, and for CD4+ tetramer-positive cells was 9.0. Furthermore, cells expressing higher levels of tetramer staining had higher MFI for CTLA-4 expression; for the 1% of cells with the highest tetramer staining, the MFI was 12. The ranges of these increases in CTLA-4 expression are similar to those seen when comparing expression on CD4+CD25− versus CD4+CD25+ cells (47).
Figure 7.
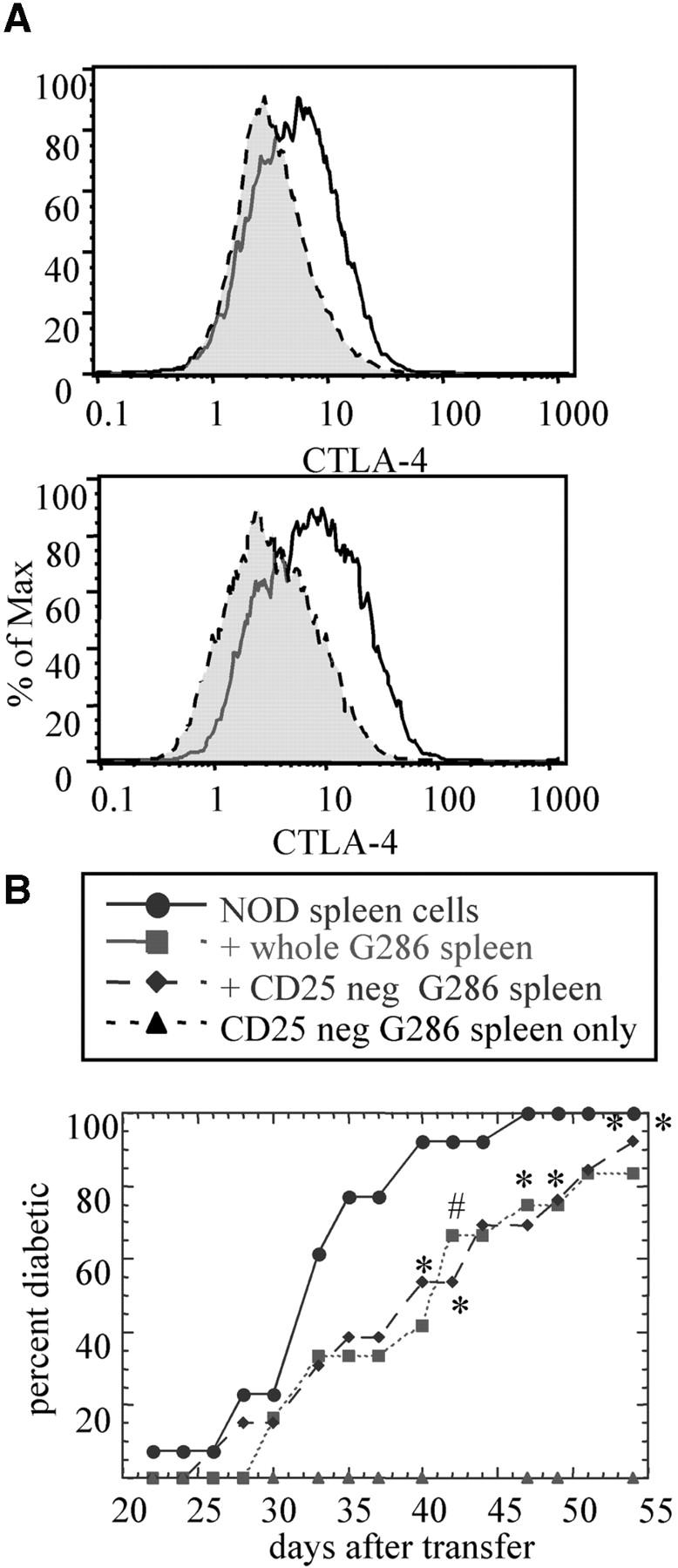
Potential mechanisms by which G286 T cells delay diabetes transfer. (A) CTLA-4 expression on CD4+ cells activated with anti-CD3 and anti-CD28. Results shown were gated on CD4+. The top panel shows CTLA-4 expression for nontransgenic (gray) and G286 (white). Similar results were found in three separate experiments. The bottom panel shows CD4+ cells from 286 mice gated on tetramer-negative (gray) or -positive (white). (B) The role of CD25+ cells in diabetes transfer. The G286 spleen cell population was incubated with biotinylated anti-CD25 and these cells were depleted using streptavidin magnetic beads (Dynal). Diabetic spleen (n = 13; circles); diabetic spleen positive 286 spleen (n = 12; squares); diabetic spleen plus CD25-negative 286 spleen (n = 13; diamonds); CD25-negative 286 spleen (n = 12; triangles).
Depletion of CD25+ Cells Does Not Alter the Protective Capacity of Cells from G286 Mice.
CD4+CD25+ cells have been shown to be protective against autoimmunity in many systems. As indicated previously, no difference was observed in the total percentage of CD4+CD25+ cells between G286 and nontransgenic NOD mice. To further evaluate the role of this population, G286 spleen cells depleted of CD25+ cells were compared with whole spleen cells in their ability to delay diabetes transfer. Depletion of CD25+ cells had no effect on the kinetics of diabetes transfer (Fig. 7 B). Thus, the protective population found within the CD4+ subset from G286 mice is not likely to be the CD4+CD25+ regulatory population, and probably represents a different type of CD4+ cell-mediated protection.
Discussion
Protection Mediated by 286-specific T Cells.
Our results demonstrate that T cells from G286 mice have specific diabetes protective capacity. If 286-specific T cells were neutral in their role in diabetes pathogenesis, one would expect these transgenic mice to develop diabetes mediated by the residual TCR repertoire, as has been observed with other NOD TCR transgenics (13, 14). This is especially true with G286, because the majority of CD4+ cells express TCRs of other specificities. In addition, one can see the pathogenic potential of the residual repertoire by the presence of sialitis in these mice; only the T cell infiltrate in the pancreas is absent. Most important, p286-activated tetramer-positive CD4+ T cells can delay diabetes transfer into NOD scid mice, indicating that p286-specific T cells are regulatory and not pathogenic.
The mechanism by which activated CD4+ T cells from G286 mice delay diabetes is unknown. The increased expression of CTLA-4 on p286-specific T cells (Fig. 6 A) may be one mechanism by which CD4+ T cells from G286 mice regulate diabetes pathogenesis. Since NOD mice normally have lower levels of CTLA-4 as compared with nonautoimmune strains (48), this increase in expression may restore the level of CTLA-4 necessary to regulate autoreactive T cell responses. The expression of IL-10 by G286 lymphocytes may also mediate downregulation of pathogenic cells, as has been shown for Tr1 cells in EAE and colitis (11, 49). Currently, we are crossing G286 mice to NOD mice deficient in either CTLA-4 or IL-10 to determine the role of these proteins in the protection mediated by p286-specific T cells.
Preliminary evidence suggests that lymphocytes from G286 mice produce TGF-β, which may also be important for regulation. It is also possible that IFN-γ expression may be important for this diabetes regulation, as was shown for BCG-mediated diabetes protection (12). Protection is not likely to be mediated by a shift of T cells to a Th2 phenotype, since these cells did not express IL-4 or IL-5. Because the total number of CD25+ cells are not increased in G286 mice, and depletion of CD25+ cells does not affect protection (Fig. 6 B), the protection observed here is not due to the CD4+CD25+ regulatory subset.
T Cell Responses To Insulin and GAD65 and Their Role in Diabetes Pathogenesis.
The ability to block diabetes progression by tolerance induction to both GAD65 and insulin has been given as evidence of the pathogenic importance of T cell responses to these antigens, such tolerance could be the result of a direct effect on autoreactive T cells through anergy or deletion of a naturally pathogenic response. Alternatively tolerance could arise through stimulation of an insulin or GAD-specific regulatory response. Such a regulatory response could represent a shifting of a pathogenic response to a protective response, perhaps by a shift in cytokine production; or treatment of mice with GAD65 or insulin in a tolerizing form could be enhancing preexisting regulatory responses specific for that autoantigen. It is interesting that some tolerizing protocols administer the antigen in IFA, an adjuvant often used to enhance immunity (27, 50).
Much is known about T cell responses to these two antigens which indicates their role in diabetes pathogenesis. First, T cell responses to insulin are more readily found in the islets and pancreatic lymph node than in the spleen (51), while most GAD65-specific clones have been isolated from spleen cells. It is probable that pathogenic responses would be found at higher frequency at the site of antigen than in other lymphoid organs. Second, many pathogenic and protective insulin-specific clones have been isolated (52), but only one GAD65-specific clone isolated to date is pathogenic and quite a few are protective (10, 30). Third, it has recently been shown that a DNA vaccine encoding many of the major GAD65 epitopes and IL-4 is protective (53). When similar DNA vaccines are used encoding insulin and IL-4, diabetes onset is accelerated (54). Finally, an attempt to induce tolerance to GAD65 by systemic expression of the protein failed. In fact, disease was exacerbated in the transgenic line which expressed the highest levels of GAD65 (55). Preliminary studies with a NOD TCR-transgenic line specific for another epitope of GAD65 show that T cell responses from these mice are not pathogenic (unpublished data). These results, along with the phenotype of the p286-specific TCR transgenic described here, raise the possibility that GAD65-specific T cell responses may display a more regulatory phenotype in NOD mice, whereas insulin-specific T cell responses may be more pathogenic. Pathogenic responses that develop later may overwhelm these early protective responses resulting in β cell destruction and diabetes. Of course, this protective phenotype may not be representative of all GAD65-specific T cell responses.
Autoimmunity and Expression of Multiple TCR-α Chains.
In G286 mice, the staining pattern of CD3 versus tetramer (Fig. 2 B) suggests that dual-specificity T cells are present at a high frequency. Allelic exclusion of the TCR-α chain locus is not nearly as complete as for TCR-β chain; it has been estimated that between 10–30% of peripheral T cells express more than one α chain on the cell surface (56, 57). Expression of two specificities has been proposed as a possible explanation of how nontolerant autoreactive T cells survive selection to populate the periphery (58, 59), and expression of a second TCR has been shown to rescue a self-specific TCR from negative selection (37). This is consistent with results obtained comparing selection of p286-specific T cells in NOD and NOD Cα−/− mice, which show that very few T cells expressing only the p286-specific TCR survive thymic selection (Fig. 2 A). In other systems, similar cells with more than one TCR expressed are hyporesponsive (39). The lower expression of the autoreactive TCR that allows the T cell to survive selection may also prevent peripheral pathogenicity. It has been proposed that T cells on the border of avidity necessary for negative selection undergo “altered negative selection” which results in a suppressor population (60).
In conclusion, a TCR transgenic mouse line specific for a known β cell autoantigen, GAD65, is protective and not diabetogenic. Determining the mechanism by which T cells from G286 mice protect may identify targets for immunomodulatory therapy. In addition, these results add to the accumulating evidence that many GAD65-specific responses may be protective and not pathogenic in diabetes. In the design of antigen-specific immunotherapy, this knowledge will be critical in deciding whether to manipulate the immune response for a particular antigen toward amplification of a preexisting protective response, or toward dampening or modulation of a pathogenic response.
Acknowledgments
We would like to thank Jon Toma and Mary Vadeboncoeur for technical assistance, and Dr. Valerie Mallet-Designe for producing I-Ag7-p286 monomers.
This work was supported by National Institutes of Health grants DK51667, DK55037, and AG04342 and Juvenile Diabetes Research Foundation file number 1-2001-162.
Footnotes
Abbreviations used in this paper: BCG, bacillus Calmette-Guerin; CTLA, CTL-associated antigen; CY, cyclophosphamide; GAD, glutamic acid decarboxylase; MFI, mean fluorescence intensity NOD, nonobese diabetic; p286, peptide 286-300 of GAD65; G286, mice expressing p286-specific TCR-α and -β transgenes.
References
- 1.Tisch, R., and H. McDevitt. 1996. Insulin-dependent diabetes mellitus. Cell. 85:291–297. [DOI] [PubMed] [Google Scholar]
- 2.Castano, L., and G.S. Eisenbarth. 1990. Type-I diabetes: a chronic autoimmune disease of human, mouse, and rat. Annu. Rev. Immunol. 8:647–679. [DOI] [PubMed] [Google Scholar]
- 3.Bendelac, A., C. Carnaud, C. Boitard, and J.F. Bach. 1987. Syngeneic transfer of autoimmune diabetes from diabetic NOD mice to healthy neonates. Requirement for both L3T4+ and Lyt-2+ T cells. J. Exp. Med. 166:823–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miller, B.J., M.C. Appel, J.J. O'Neil, and L.S. Wicker. 1988. Both the Lyt-2+ and L3T4+ T cell subsets are required for the transfer of diabetes in nonobese diabetic mice. J. Immunol. 140:52–58. [PubMed] [Google Scholar]
- 5.Andre, I., A. Gonzalez, B. Wang, J. Katz, C. Benoist, and D. Mathis. 1996. Checkpoints in the progression of autoimmune disease: lessons from diabetes models. Proc. Natl. Acad. Sci. USA. 93:2260–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gottlieb, P.A., and G.S. Eisenbarth. 1996. Mouse and man: multiple genes and multiple autoantigens in the aetiology of type I DM and related autoimmune disorders. J. Autoimmun. 9:277–281. [DOI] [PubMed] [Google Scholar]
- 7.Boitard, C., R. Yasunami, M. Dardenne, and J.F. Bach. 1989. T cell-mediated inhibition of the transfer of autoimmune diabetes in NOD mice. J. Exp. Med. 169:1669–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sakaguchi, S., M. Toda, M. Asano, M. Itoh, S.S. Morse, and N. Sakaguchi. 1996. T cell-mediated maintenance of natural self-tolerance: its breakdown as a possible cause of various autoimmune diseases. J. Autoimmun. 9:211–220. [DOI] [PubMed] [Google Scholar]
- 9.Salomon, B., D.J. Lenschow, L. Rhee, N. Ashourian, B. Singh, A. Sharpe, and J.A. Bluestone. 2000. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 12:431–440. [DOI] [PubMed] [Google Scholar]
- 10.Tisch, R., B. Wang, M.A. Atkinson, D.V. Serreze, and R. Friedline. 2001. A glutamic acid decarboxylase 65-specific Th2 cell clone immunoregulates autoimmune diabetes in nonobese diabetic mice. J. Immunol. 166:6925–6936. [DOI] [PubMed] [Google Scholar]
- 11.Groux, H., A. O'Garra, M. Bigler, M. Rouleau, S. Antonenko, J.E. de Vries, and M.G. Roncarolo. 1997. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature. 389:737–742. [DOI] [PubMed] [Google Scholar]
- 12.Serreze, D.V., H.D. Chapman, C.M. Post, E.A. Johnson, W.L. Suarez-Pinzon, and A. Rabinovitch. 2001. Th1 to Th2 cytokine shifts in nonobese diabetic mice: sometimes an outcome, rather than the cause, of diabetes resistance elicited by immunostimulation. J. Immunol. 166:1352–1359. [DOI] [PubMed] [Google Scholar]
- 13.Lipes, M.A., A. Rosenzweig, K.N. Tan, G. Tanigawa, D. Ladd, J.G. Seidman, and G.S. Eisenbarth. 1993. Progression to diabetes in nonobese diabetic (NOD) mice with transgenic T cell receptors. Science. 259:1165–1169. [DOI] [PubMed] [Google Scholar]
- 14.Serreze, D.V., E.A. Johnson, H.D. Chapman, R.T. Graser, M.P. Marron, T.P. DiLorenzo, P. Silveira, Y. Yoshimura, S.G. Nathenson, and S. Joyce. 2001. Autoreactive diabetogenic T-cells in NOD mice can efficiently expand from a greatly reduced precursor pool. Diabetes. 50:1992–2000. [DOI] [PubMed] [Google Scholar]
- 15.Haskins, K., and M. McDuffie. 1990. Acceleration of diabetes in young NOD mice with a CD4+ islet-specific T cell clone. Science. 249:1433–1436. [DOI] [PubMed] [Google Scholar]
- 16.Katz, J.D., B. Wang, K. Haskins, C. Benoist, and D. Mathis. 1993. Following a diabetogenic T cell from genesis through pathogenesis. Cell. 74:1089–1100. [DOI] [PubMed] [Google Scholar]
- 17.Schmidt, D., J. Verdaguer, N. Averill, and P. Santamaria. 1997. A mechanism for the major histocompatibility complex-linked resistance to autoimmunity. J. Exp. Med. 186:1059–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roep, B.O. 1996. T-cell responses to autoantigens in IDDM. The search for the Holy Grail. Diabetes. 45:1147–1156. [DOI] [PubMed] [Google Scholar]
- 19.Atkinson, M.A., and N.K. Maclaren. 1993. Islet cell autoantigens in insulin-dependent diabetes. J. Clin. Invest. 92:1608–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Verge, C.F., R. Gianani, E. Kawasaki, L. Yu, M. Pietropaolo, R.A. Jackson, H.P. Chase, and G.S. Eisenbarth. 1996. Prediction of type I diabetes in first-degree relatives using a combination of insulin, GAD, and ICA512bdc/IA-2 autoantibodies. Diabetes. 45:926–933. [DOI] [PubMed] [Google Scholar]
- 21.Daniel, D., R.G. Gill, N. Schloot, and D. Wegmann. 1995. Epitope specificity, cytokine production profile and diabetogenic activity of insulin-specific T cell clones isolated from NOD mice. Eur. J. Immunol. 25:1056–1062. [DOI] [PubMed] [Google Scholar]
- 22.Wong, F.S., J. Karttunen, C. Dumont, L. Wen, I. Visintin, I.M. Pilip, N. Shastri, E.G. Pamer, and C.A. Janeway, Jr. 1999. Identification of an MHC class I-restricted autoantigen in type 1 diabetes by screening an organ-specific cDNA library. Nat. Med. 5:1026–1031. [DOI] [PubMed] [Google Scholar]
- 23.Baekkeskov, S., H.J. Aanstoot, S. Christgau, A. Reetz, M. Solimena, M. Cascalho, F. Folli, H. Richter-Olesen, P. DeCamilli, and P.D. Camilli. 1990. Identification of the 64K autoantigen in insulin-dependent diabetes as the GABA-synthesizing enzyme glutamic acid decarboxylase. Nature. 347:151–156. [DOI] [PubMed] [Google Scholar]
- 24.Tisch, R., X.D. Yang, S.M. Singer, R.S. Liblau, L. Fugger, and H.O. McDevitt. 1993. Immune response to glutamic acid decarboxylase correlates with insulitis in non-obese diabetic mice. Nature. 366:72–75. [DOI] [PubMed] [Google Scholar]
- 25.Kaufman, D.L., M. Clare-Salzler, J. Tian, T. Forsthuber, G.S. Ting, P. Robinson, M.A. Atkinson, E.E. Sercarz, A.J. Tobin, and P.V. Lehmann. 1993. Spontaneous loss of T-cell tolerance to glutamic acid decarboxylase in murine insulin-dependent diabetes. Nature. 366:69–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu, L., M. Rewers, R. Gianani, E. Kawasaki, Y. Zhang, C. Verge, P. Chase, G. Klingensmith, H. Erlich, J. Norris, and G.S. Eisenbarth. 1996. Antiislet autoantibodies usually develop sequentially rather than simultaneously. J. Clin. Endocrinol. Metab. 81:4264–4267. [DOI] [PubMed] [Google Scholar]
- 27.Daniel, D., and D.R. Wegmann. 1996. Protection of nonobese diabetic mice from diabetes by intranasal or subcutaneous administration of insulin peptide B-(9-23). Proc. Natl. Acad. Sci. USA. 93:956–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tian, J., M.A. Atkinson, M. Clare-Salzler, A. Herschenfeld, T. Forsthuber, P.V. Lehmann, and D.L. Kaufman. 1996. Nasal administration of glutamate decarboxylase (GAD65) peptides induces Th2 responses and prevents murine insulin-dependent diabetes. J. Exp. Med. 183:1561–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yoon, J.W., C.S. Yoon, H.W. Lim, Q.Q. Huang, Y. Kang, K.H. Pyun, K. Hirasawa, R.S. Sherwin, and H.S. Jun. 1999. Control of autoimmune diabetes in NOD mice by GAD expression or suppression in beta cells. Science. 284:1183–1187. [DOI] [PubMed] [Google Scholar]
- 30.Zekzer, D., F.S. Wong, O. Ayalon, I. Millet, M. Altieri, S. Shintani, M. Solimena, and R.S. Sherwin. 1998. GAD-reactive CD4+ Th1 cells induce diabetes in NOD/SCID mice. J. Clin. Invest. 101:68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chao, C.C., and H.O. McDevitt. 1997. Identification of immunogenic epitopes of GAD 65 presented by I-Ag7 in non-obese diabetic mice. Immunogenetics. 46:29–34. [DOI] [PubMed] [Google Scholar]
- 32.Chao, C.C., H.K. Sytwu, E.L. Chen, J. Toma, and H.O. McDevitt. 1999. The role of MHC class II molecules in susceptibility to type I diabetes: identification of peptide epitopes and characterization of the T cell repertoire. Proc. Natl. Acad. Sci. USA. 96:9299–9304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kouskoff, V., K. Signorelli, C. Benoist, and D. Mathis. 1995. Cassette vectors directing expression of T cell receptor genes in transgenic mice. J. Immunol. Methods. 180:273–280. [DOI] [PubMed] [Google Scholar]
- 34.Coligan, J.E., A.M. Kruisbeek, D.H. Margulies, E.M. Shevach, and W. Strober. 1995. Curr. Protocols in Immunol. Vol. 1. National Institutes of Health. pp.3.1.3–3.1.5.
- 35.Stratmann, T., V. Apostolopoulos, V. Mallet-Designe, A.L. Corper, C.A. Scott, I.A. Wilson, A.S. Kang, and L. Teyton. 2000. The I-Ag7 MHC class II molecule linked to murine diabetes is a promiscuous peptide binder. J. Immunol. 165:3214–3225. [DOI] [PubMed] [Google Scholar]
- 36.Pearson, C.I., W. van Ewijk, and H.O. McDevitt. 1997. Induction of apoptosis and T helper 2 (Th2) responses correlates with peptide affinity for the major histocompatibility complex in self-reactive T cell receptor transgenic mice. J. Exp. Med. 185:583–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zal, T., S. Weiss, A. Mellor, and B. Stockinger. 1996. Expression of a second receptor rescues self-specific T cells from thymic deletion and allows activation of autoreactive effector function. Proc. Natl. Acad. Sci. USA. 93:9102–9107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Olivares-Villagomez, D., Y. Wang, and J.J. Lafaille. 1998. Regulatory CD4+ T cells expressing endogenous T cell receptor chains protect myelin basic protein-specific transgenic mice from spontaneous autoimmune encephalomyelitis. J. Exp. Med. 188:1883–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Akkaraju, S., W.Y. Ho, D. Leong, K. Canaan, M.M. Davis, and C.C. Goodnow. 1997. A range of CD4 T cell tolerance: partial inactivation to organ-specific antigen allows nondestructive thyroiditis or insulitis. Immunity. 7:255–271. [DOI] [PubMed] [Google Scholar]
- 40.Yasunami, R., and J.F. Bach. 1988. Anti-suppressor effect of cyclophosphamide on the development of spontaneous diabetes in NOD mice. Eur. J. Immunol. 18:481–484. [DOI] [PubMed] [Google Scholar]
- 41.Hammerling, G.J., G. Schonrich, I. Ferber, and B. Arnold. 1993. Peripheral tolerance as a multi-step mechanism. Immunol. Rev. 133:93–104. [DOI] [PubMed] [Google Scholar]
- 42.Hoffmann, M.W., W.R. Heath, D. Ruschmeyer, and J.F. Miller. 1995. Deletion of high-avidity T cells by thymic epithelium. Proc. Natl. Acad. Sci. USA. 92:9851–9855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sadelain, M.W., H.Y. Qin, J. Lauzon, and B. Singh. 1990. Prevention of type I diabetes in NOD mice by adjuvant immunotherapy. Diabetes. 39:583–589. [DOI] [PubMed] [Google Scholar]
- 44.McInerney, M.F., S.B. Pek, and D.W. Thomas. 1991. Prevention of insulitis and diabetes onset by treatment with complete Freund's adjuvant in NOD mice. Diabetes. 40:715–725. [DOI] [PubMed] [Google Scholar]
- 45.Chen, W., W. Jin, and S.M. Wahl. 1998. Engagement of cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) induces transforming growth factor β (TGF-β) production by murine CD4+ T cells. J. Exp. Med. 188:1849–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nakamura, K., A. Kitani, and W. Strober. 2001. Cell contact-dependent immunosuppression by CD4+CD25+ regulatory T cells is mediated by cell surface-bound transforming growth factor β. J. Exp. Med. 194:629–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Takahashi, T., T. Tagami, S. Yamazaki, T. Uede, J. Shimizu, N. Sakaguchi, T.W. Mak, and S. Sakaguchi. 2000. Immunologic self-tolerance maintained by CD25+CD4+ regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J. Exp. Med. 192:303–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dahlen, E., G. Hedlund, and K. Dawe. 2000. Low CD86 expression in the nonobese diabetic mouse results in the impairment of both T cell activation and CTLA-4 up-regulation. J. Immunol. 164:2444–2456. [DOI] [PubMed] [Google Scholar]
- 49.Chen, Y., V.K. Kuchroo, J. Inobe, D.A. Hafler, and H.L. Weiner. 1994. Regulatory T cell clones induced by oral tolerance: suppression of autoimmune encephalomyelitis. Science. 265:1237–1240. [DOI] [PubMed] [Google Scholar]
- 50.Muir, A., A. Peck, M. Clare-Salzler, Y.H. Song, J. Cornelius, R. Luchetta, J. Krischer, and N. Maclaren. 1995. Insulin immunization of nonobese diabetic mice induces a protective insulitis characterized by diminished intraislet interferon-γ transcription. J. Clin. Invest. 95:628–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wegmann, D.R., N. Shehadeh, K.J. Lafferty, M. Norbury-Glaser, R.G. Gill, and D. Daniel. 1993. Establishment of islet-specific T-cell lines and clones from islet isografts placed in spontaneously diabetic NOD mice. J. Autoimmun. 6:517–527. [DOI] [PubMed] [Google Scholar]
- 52.Haskins, K., and D. Wegmann. 1996. Diabetogenic T-cell clones. Diabetes. 45:1299–1305. [DOI] [PubMed] [Google Scholar]
- 53.Tisch, R., B. Wang, D.J. Weaver, B. Liu, T. Bui, J. Arthos, and D.V. Serreze. 2001. Antigen-specific mediated suppression of β cell autoimmunity by plasmid DNA vaccination. J. Immunol. 166:2122–2132. [DOI] [PubMed] [Google Scholar]
- 54.Weaver, D.J., Jr., B. Liu, and R. Tisch. 2001. Plasmid dnas encoding insulin and glutamic acid decarboxylase 65 have distinct effects on the progression of autoimmune diabetes in nonobese diabetic mice. J. Immunol. 167:586–592. [DOI] [PubMed] [Google Scholar]
- 55.Geng, L., M. Solimena, R.A. Flavell, R.S. Sherwin, and A.C. Hayday. 1998. Widespread expression of an autoantigen-GAD65 transgene does not tolerize non-obese diabetic mice and can exacerbate disease. Proc. Natl. Acad. Sci. USA. 95:10055–10060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Padovan, E., G. Casorati, P. Dellabona, S. Meyer, M. Brockhaus, and A. Lanzavecchia. 1993. Expression of two T cell receptor α chains: dual receptor T cells. Science. 262:422–424. [DOI] [PubMed] [Google Scholar]
- 57.Elliott, J.I., and D.M. Altmann. 1995. Dual T cell receptor α chain T cells in autoimmunity. J. Exp. Med. 182:953–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Heath, W.R., and J.F. Miller. 1993. Expression of two α chains on the surface of T cells in T cell receptor transgenic mice. J. Exp. Med. 178:1807–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sarukhan, A., C. Garcia, A. Lanoue, and H. von Boehmer. 1998. Allelic inclusion of T cell receptor alpha genes poses an autoimmune hazard due to low-level expression of autospecific receptors. Immunity. 8:563–570. [DOI] [PubMed] [Google Scholar]
- 60.Shevach, E.M. 2000. Regulatory T cells in autoimmunity. Annu. Rev. Immunol. 18:423–449. [DOI] [PubMed] [Google Scholar]