

Abstract
Allergic asthma is an inflammatory lung disease initiated and directed by T helper cells type 2 (Th2). The mechanism involved in generation of Th2 responses to inert inhaled antigens, however, is unknown. Epidemiological evidence suggests that exposure to lipopolysaccharide (LPS) or other microbial products can influence the development and severity of asthma. However, the mechanism by which LPS influences asthma pathogenesis remains undefined. Although it is known that signaling through Toll-like receptors (TLR) is required for adaptive T helper cell type 1 (Th1) responses, it is unclear if TLRs are needed for Th2 priming. Here, we report that low level inhaled LPS signaling through TLR4 is necessary to induce Th2 responses to inhaled antigens in a mouse model of allergic sensitization. The mechanism by which LPS signaling results in Th2 sensitization involves the activation of antigen-containing dendritic cells. In contrast to low levels, inhalation of high levels of LPS with antigen results in Th1 responses. These studies suggest that the level of LPS exposure can determine the type of inflammatory response generated and provide a potential mechanistic explanation of epidemiological data on endotoxin exposure and asthma prevalence.
Keywords: asthma, Toll-like receptor, T cell, dendritic cell, lung
Introduction
Asthma is a pulmonary inflammatory disease believed to be due to aberrant Th2 immune responses to commonly inhaled antigens (1). Only a subset of people exposed to these aeroallergens, however, develop pathological Th2 responses, and this process is not well understood. In particular, the role of adjuvants and the innate immune system in the induction of Th2 responses is unclear.
Respiratory infections have been linked to asthma in both a preventative and facilitating role, implicating Toll-like receptor (TLR) signaling in regulation of Th2-driven airway disease (2). Of particular interest is LPS, a cell wall component of Gram-negative bacteria that is ubiquitous in the environment, including household dusts. LPS activates cells through TLR4 with the accessory proteins CD14 and LPS binding protein (3), signaling through a common adaptor protein MyD88. This results in the transcription of several activation markers including MHC II and B7 molecules and the production of IL-1, IL-12, and TNF-α (3). The role of endotoxin exposure in asthma development in children has been controversial, with studies indicating either a protective role through Th1 induction or an exacerbating effect on asthma severity (1, 4, 5). It has been speculated that the opposing roles of LPS might be explained by differences in exposure levels (6). However, these studies did not address whether the association of household LPS levels with asthma severity is a result of enhanced allergen sensitization or direct irritant effects of LPS on previously sensitized individuals (4, 6). Our objective was to assess if LPS affects Th2 sensitization to aeroallergens and if the amount of LPS exposure affects the disease phenotype.
It is now clear that Th1 adaptive immune responses require TLR signals (7). However, Th2 priming is thought to occur either as a default pathway in the absence of TLR signaling or by a currently unidentified Th2-type activating receptor(s) (3). Therefore, the role a microbial adjuvant such as LPS plays in Th2 aeroallergen sensitization at the site of natural exposure, namely the lung, is unknown.
To directly address the role of LPS as an adjuvant for Th2 sensitization in the induction of allergic airway responses, we used a murine model of Th2 pulmonary inflammation in which priming occurs after antigen inhalation without the use of alum. We show that Th2 sensitization occurs only if inhaled allergens are encountered with LPS, signaling through TLR4. Furthermore, different doses of LPS induce distinct subsets of Th cells and therefore distinct types of inflammatory responses.
Materials and Methods
Animals.
BALB/cJ (WT) and C.C3H-Tlr4 Lps-d (TLR4d) mice were purchased from The Jackson Laboratory. BALB/cAnNCr mice were purchased from the National Cancer Institute. 6–10-wk-old female mice were used in all experiments with three or four mice per group.
Sensitization Protocols.
Mice were anesthetized with methoxyflurane (Metofane) and then sensitized intranasally with 100 μg OVA (Grade V; Sigma-Aldrich) in 50 μl PBS on days 0, 1, and 2 as previously described (8). For Fig. 3, we sensitized WT or TLR4d mice intraperitoneally with 100 μg OVA in 2 mg aluminum hydroxide (Pierce Chemical Co.) in a total volume of 0.25 ml.
Figure 3.


TLR4d mice sensitized intraperitoneally with the adjuvant aluminum hydroxide are capable of generating Th2 responses to OVA. (A) WT and TLR4d were primed either intranasally with OVA or intraperitoneally with OVA in alum and BAL was evaluated on day 21 after standard intranasal challenge. Stacked bars of cell differential are shown. Total BAL cell number is represented by height of stacked bars and standard error is based on total BAL number. *, P < 0.005 (intranasally primed TLR4d vs. WT mice). Mice immunized intraperitoneally with alum alone did not respond. (B) Cytokine production in pg/ml from DLN of intranasally or intraperitoneally primed WT (solid bars) or TLR4d (open bars) mice. ND, not detectable. IFN-γ was not detectable from cultures of WT or TLR4d mice primed intranasally or intraperitoneally with OVA containing a low dose of LPS. One representative experiment of two is shown.
Airway Challenge.
Mice were challenged on days 14, 15, 18, and 19 intranasally with 25 μg OVA and killed on day 21. We confirmed that TLR4d and WT mice inhaled the antigen solution equally by administering Evan's Blue (Sigma-Aldrich) intranasally (9).
LPS Depletion and Measurement.
Endotoxin Detoxi-Gel™ (Pierce Chemical Co.) was used according to the manufacturer's instructions to remove >99% of the contaminating LPS in the administered OVA solution (resulting in a total dose <0.001 μg LPS during priming), which was measured by limulus amebocyte assay (BioWhittaker).
Analysis of Bronchoalveolar Lavage (BAL).
Mice were killed and BAL inflammatory cells were obtained as previously described (10). We determined statistical significance using an unpaired Student's t test.
Lung Histology.
Paraffin-embedded coronal lung sections were prepared as previously described (8) and stained with hematoxylin and eosin (H&E) or periodic acid-Schiff (PAS). All images are at 100×.
Determination of Serum Antibody Concentration.
Serum was obtained on day 21 for measurement of OVA-specific IgE (11), IgG1, and IgG2a (8) antibodies by ELISA as previously described. Hyperimmune serum from OVA/alum immunized BALB/c mice was used for IgE standard and set at 500 U/ml. Levels of detection were 125 ng/ml (IgG1), 16 U/ml (IgE), and 81 U/ml (IgG2a).
Lymph Node Cytokine Production.
Mice were sensitized and challenged with either OVA (WT or TLR4d) or PBS (WT) and on day 21, mediastinal LN cells were isolated and stimulated in vitro with 200 μg/ml OVA and syngeneic T cell–depleted splenocytes. Cytokines in culture supernatants were measured using commercially available ELISA kits (R&D Systems). Levels of detection were 25.0 pg/ml (IL-4), 125.0 pg/ml (IL-5), and 1.9 ng/ml (IFN-γ).
Serum and Bone Marrow–derived Dendritic Cell (BMDC) IL-12 Detection and BMDC Activation Markers.
Serum from mice was obtained 4 h after the third inhalation of OVA with high or low dose LPS and measured p70 levels using commercially available ELISA kits (R&D Systems). For in vitro studies, BMDCs were cultured as previously described (12) from TLR4d and WT mice. On day 9 of culture, we added 100 μg/ml OVA, 100 ng/ml TNF-α, or 50 ng/ml LPS and harvested cells and supernatant at 12 h. p70 level of detection was 7.8 pg/ml. After Fc receptor blocking with 24G2, CDllchi (HL3) cells were evaluated by FACS® for MHC II (2G9) and B7.2 (GL1; BD Biosciences).
Dendritic Cell (DC) Migration Studies.
0.5 mg FITC-OVA (Molecular Probes) was administered intranasally with low dose (0.1 μg) LPS on days 0, 1, and 2 with or without 2 μg TNF-α on day 1. On day 3, we harvested and pooled the draining LNs in each group and blocked Fc receptors and then stained them with anti-CD11c fluorochrome.
Results
Dose of LPS Determines Type of Immune Response Generated to Inhaled Antigen.
We have previously shown that sensitization of mice by exposure to inhaled OVA leads to robust pulmonary Th2 responses (8). To test the role of LPS in these responses, we sensitized mice by intranasal exposure to OVA depleted of contaminating LPS (<0.001 μg) or OVA with a high (100 μg) or low (0.1 μg) dose of LPS. These low and high doses of LPS are analogous to reported endotoxin levels of samples from homes of atopic versus nonatopic children, respectively (5). Mice exposed to LPS-depleted OVA showed no airway inflammatory responses after challenge with inhaled antigen (Fig. 1 A) and had total BAL cell numbers equivalent to PBS controls. In contrast, mice sensitized with OVA containing low dose LPS demonstrated significant increases in total BAL cell numbers as well as lung tissue infiltrates and airway mucus secretion (Fig. 1, A and B). Both airway and tissue infiltrates were dominated by eosinophils, consistent with Th2-mediated inflammation. Draining lymph node (DLN) IL-5 and IL-13 production confirmed the Th2 nature of the inflammatory response (Fig. 1 C). Mice exposed to PBS or low dose LPS alone did not generate pulmonary inflammation after OVA challenge (Fig. 2 A).
Figure 1.

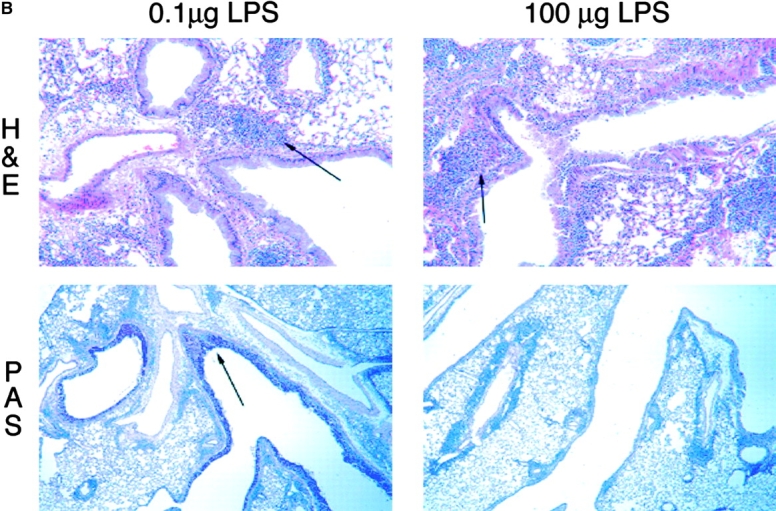


The dose of LPS inhaled with antigen determines the nature of the immune response generated. (A) BAL inflammatory cells of BALB/c mice exposed to LPS-depleted OVA (open bars), OVA with low dose LPS (gray bars), or OVA with high dose Escherichia coli LPS (solid bars; Sigma-Aldrich) after challenge. Monocytes constitute the remainder of BAL cells (not depicted). Bars depict the mean ± standard deviation. *, P < 0.01 (eosinophils in depleted vs. low LPS groups); **, P < 0.01 (eosinophils in high vs. low LPS groups); ***, P < 0.01 (number of neutrophils in high vs. low LPS groups). One representative experiment of six is shown. (B) Representative lung sections stained with H&E or PAS at 100×. Arrows indicate areas or peribronchiolar cellular infiltrate (H&E) or positive mucus staining (PAS). (C) Cytokine production from lung draining LNs in low (solid bars) and high (open bars) dose LPS groups. One representative experiment of four is shown. ND, not detectable. (D) Serum antibodies of low (▵) and high (○) dose LPS groups are compared with pooled sera from naive BALB/c mice (×). Line depicts the mean. P < 0.05 (LPS high vs. low dose) for IgG1, IgE, and IgG2a responses.
Figure 2.


TLR4 signaling is required for Th2 sensitization to inhaled OVA. (A) BAL inflammatory cells of WT or TLR4d mice sensitized intranasally with OVA with low dose LPS (0.1 μg), or WT primed with LPS alone, or PBS on day 21. Total bar height represents total cell number in BAL and error bars are based on total cell numbers. *, P < 0.04 (total BAL cell number from TLR4d vs. WT). One representative experiment of six is shown. (B) Serum antibody responses by ELISA on day 21 in WT (▴) and TLR4d (♦) mice compared with pooled naive serum (×). P < 0.05 (WT vs. TRL4d) for IgG1 and IgE responses.
As LPS is known to be a potent inducer of IL-12 production from APCs in vitro, it might be expected to preferentially stimulate Th1 responses. Therefore, we tested whether the surprising induction of Th2 responses was a result of the low dose of LPS exposure. Use of a high dose of LPS during intranasal OVA priming resulted in a Th1-associated response dominated by neutrophils and an absence of airway mucus production in the lung (Fig. 1, A and B; reference 10). IFN-γ production from DLNs confirmed the induction of a Th1 response in high dose LPS-exposed mice (Fig. 1 C). Serum antibody isotype patterns in groups sensitized with OVA containing low versus high dose LPS were also consistent with the generation of Th2 (high IgE and IgG1) versus Th1 (high IgG2a) immunity, respectively (Fig. 1 D). Thus, no airway inflammatory response was generated in mice that had been sensitized with LPS-depleted OVA, whereas antigen-specific immune responses were induced in the presence of LPS with low and high doses inducing Th2 or Th1 responses, respectively.
TLR4 Signaling Is Required for Th2 Priming to Inhaled Antigens.
The requirement for LPS in the generation of Th2 responses to inhaled antigen was confirmed in C.C3H-Tlr4 Lps-d mice (13) expressing a nonfunctional TLR4 (TLR4d). When compared with WT, TLR4d mice exposed to OVA in the presence of low dose LPS showed marked reduction in airway inflammation (Fig. 2 A) and DLN Th2 cytokine production (Fig. 3 B, I.N.). We obtained similar results using C3H/HeJ mice. Th1 responses initiated with high dose LPS were similarly abrogated in TLR4d mice (not depicted).
These data support the observation that LPS is required for the development of Th2 (and Th1) responses to inhaled antigen. However, because LPS signaling is absent during both sensitization and challenge in TLR4d mice, we next asked at what stage LPS was required (6). To address this question, Th2 cell–dependent OVA-specific antibody secretion was measured. TLR4d mice demonstrated significantly reduced OVA-specific IgG1 and no IgE or IgG2a antibody responses (Fig. 2 B). In addition, there was evidence of a reduced proliferative response in the lung DLN of TLR4d mice as the cellularity after intranasal priming was substantially diminished (5.9 ± 1.4 × 106 in WT vs. 2.3 ± 0.3 × 106 cells in TLR4d). Thus, there was evidence of abrogated Th2 priming in TLR4d mice by systemic antibody responses, DLN cellularity, lung inflammation, and cytokine responses, consistent with defective T cell priming in the absence of LPS signaling.
TLR4d Mice Are Capable of Mounting Th2 Responses Using the Adjuvant Aluminum Hydroxide.
To confirm that recruitment pathways were intact in the lungs of TLR4d mice, a TLR4 independent mechanism of Th2 priming was used. Alum is a potent Th2 adjuvant that does not contain microbial products and therefore should not involve TLR4 signaling to initiate immune responses. Therefore, TLR4d and WT mice were immunized intraperitoneally with OVA/alum or intranasally with OVA/LPS. 2 wk later, both groups were challenged with inhaled antigen. TLR4d mice were fully capable of initiating Th2 immunity in the presence of a non-TLR4 adjuvant as evidenced by eosinophilic BAL inflammation and Th2 cytokine responses in the lung DLNs (Fig. 3, A and B). Thus, circumventing deficient Th2 priming with the adjuvant alum results in equivalent pulmonary inflammation in TLR4d and WT mice, indicating that lung recruitment of eosinophils and lymphocytes is not impaired in TLR4d mice.
TNF-α Restores Pulmonary Inflammation in TLR4d Mice.
Adjuvants initiate adaptive immune responses by activating DCs to present antigen in the context of MHC and costimulatory molecules in the DLN (3). We hypothesized that if we could induce DC maturation and migration in the absence of LPS adjuvant signals in TLR4d mice, we could restore T cell priming to inhaled antigen. TNF-α is both a product of LPS-stimulated DCs and is known to activate DCs. Using this cytokine to circumvent deficient maturation signals by LPS, Th2 responses were completely restored in TLR4d mice with administered TNF-α during sensitization to inhaled antigen. This included airway inflammatory responses (Fig. 4 A) and antibody responses (not depicted). In addition, TNF-α administration restored DLN cytokine production in TLR4d mice (116 ± 22 vs. 1516 ± 590 pg/ml IL-5 and 524 ± 130 vs. 2225 ± 1186 pg/ml IL-13 in TLR4d vs. TLR4d with TNF-α, respectively). These data indicate that defective T cell priming can be overcome using the LPS/TLR-induced cytokine TNF-α, implicating a role for DC maturation and migration in the LPS adjuvant effect.
Figure 4.



Th2 pulmonary responses and DC activation in response to OVA with LPS are abrogated in TLR4d mice but can be restored with TNF-α. (A) We sensitized mice as before with half of groups receiving 2 μg recombinant murine TNF-α (R&D Systems) intranasally on day 1. The number of inflammatory cells recovered by BAL on day 21 is represented by the height of the stacked bars with error bars. *, P < 0.001 (WT vs. TLR4d); **, P = 0.001 (TLR4d vs. TLR4d with TNF-α). (B) MHC II and B7.2 FACS® analysis of CDllchi BMDCs from WT or TLR4d stimulated for 12 h with PBS, 100 μg/ml OVA/LPS, or 100 ng/ml TNF-α. (C) Number of FITC+ CDllc+ cells in mediastinal LNs on day 3 after intranasal administration of FITC-OVA with low dose (0.1 μg) LPS (gray bars) with (+) or without (−) 2 μg intranasal TNF-α (solid bars) on day 1. One representative experiment of three is shown. *, P = 0.01 (TLR4d + vs. − TNF-α).
DC Maturation and Migration to the DLN Are Diminished in TLR4d.
To test whether the role of low dose LPS with OVA inhalation is to induce DC maturation and migration resulting in Th2 priming, we examined BMDCs for up-regulation of MHC II and B7.2 in the presence or absence of OVA/LPS or TNF-α. Although both activation markers were up-regulated on DCs from WT mice in response to either OVA/LPS or TNF-α, only TNF-α activated TLR4d DCs in vitro (Fig. 4 B). We then used inhaled FITC-OVA with low dose LPS to track migration in vivo of antigen-containing DCs from the lung to the DLNs in WT versus TLR4d mice. Although migration of CD11c+ FITC+ DCs to DLNs was seen in WT mice, no significant antigen-loaded DC migration occurred in TLR4d mice (Fig. 4 C). Migration was restored in TLR4d mice upon the administration of TNF-α with FITC-OVA. Thus, Th2 sensitization is abrogated in the absence of TLR4-associated DC migration. When migration to the DLN is restored using TNF-α in TLR4d mice, Th2 responses are also restored.
DC IL-12 Production Differs after Exposure to Low and High Doses of LPS.
LPS is known to induce both cell surface DC maturation and the production of TNF-α, IL-1, and IL-12 (3). As IL-12 is a potent Th1 skewing cytokine, we hypothesized that differences in IL-12 production following high versus low dose LPS inhalation with OVA might explain the induction of Th1 versus Th2 responses, respectively. To test this, serum IL-12 levels were analyzed. In contrast to mice immunized with low dose LPS OVA, WT mice immunized with high dose LPS OVA had significantly higher levels of serum IL-12 (Fig. 5 A). In vitro evaluation of WT BMDC confirmed that only high dose LPS was capable of inducing IL-12 production, whereas OVA (containing low dose LPS) did not (Fig. 5 B). These data are consistent with the differential inflammatory response observed in vivo (Th1 vs. Th2) and implicate an LPS threshold requirement for IL-12 secretion. Interestingly, TNF-α, a cytokine capable of inducing DC maturation and Th2 sensitization, was unable to induce IL-12 in WT BMDCs. This is consistent with our observations that TNF-α administration during priming was capable of rescuing Th2 responses in TLR4d mice without the induction of Th1 immunity (Fig. 4 A). As expected, no IL-12 was detected from TLR4d serum or BMDCs stimulated with OVA, TNF-α, or LPS.
Figure 5.


Differential IL-12 production with high and low dose LPS. (A) Serum IL-12 (p70) levels on day 2 of priming with inhaled OVA containing either high (100 μg) or low (0.1 μg) levels of LPS. (B) IL-12 (p70) production from WT or TLR4d BMDCs after stimulation with 100 μg/ml OVA with low dose LPS, 100 ng/ml TNF-α, or high dose (50 ng/ml) LPS for 12 h. ND, not detectable.
Discussion
The results presented here support a model of sensitization to inhaled inert proteins that requires LPS and the TLR4 signaling pathway. In addition, the amount of LPS present during sensitization determines whether Th1 or Th2 immunity is observed. Although recent studies in MyD88-deficient mice support a role for TLRs in the generation of Th1 responses to proteins, Th2 responses were shown to be MyD88 independent, suggesting TLR signaling is not important for the induction of Th2 cells (7). However, recent work with MyD88-deficient DCs showed that LPS stimulation induced IL-4 production with normal up-regulation of costimulatory molecules resulting in a Th2 skewing bias (14), suggesting that a MyD88-independent pathway, TIRAP/MAL, is responsible for the observed response. We might speculate that the threshold of induction for these two signaling pathways of TLR4 requires distinct levels of signaling intensities, resulting in differential effects on the adaptive immune response. The results from this study demonstrate the importance of TLR-dependent adjuvants in the induction of Th2 responses and the LPS dose differential of Th1/Th2 activation.
Another study using crystalline OVA in alum intraperitoneally suggested that TLR4-defective mice could not recall Th2-type inflammation to the lung (15). However, the results presented here demonstrate that T cell priming using the adjuvant alum and cell recruitment to the lung are intact in TLR4d mice, as would be expected from an LPS-free, non-TLR–dependent adjuvant such as aluminum hydroxide (Fig. 3 A). This discrepancy may lie in the genetic variation that could occur between the substrains of mice used in their study.
The data reported here may help explain previously observed differences in the response to inhaled protein, where both tolerance and Th2 immunity have been seen (8, 9). It is plausible that these differences are a result of varying levels of LPS contamination and that one reason this protein has been an effective antigen in many asthma models relates to its inherent LPS contamination (16).
Various animal models indicate that exposure to microbial sequences such as LPS can down-regulate Th2 pulmonary responses (17). Epidemiological data in humans support a differential dose model with endotoxin exposure correlated with both increased and decreased incidence of lung disease and severity (1). Our data provide a model to explain these conflicting findings in that OVA exposure in the presence of high dose LPS fails to induce Th2 cells, but instead induces both IL-12 production and a Th1 response. By contrast, low dose LPS is not sufficient to induce Th1 cells but is required to induce Th2 inflammation. In the absence of LPS there is no significant lung response. Thus, different levels of LPS exposure resulting in different Th cell inflammatory responses might explain the discrepancies in human studies. Recently discovered missense mutations in human TLR4 could likewise provide an explanation for the variability in human sensitization to ubiquitous aeroallergens (18).
Respiratory syncytial virus (RSV) infections during childhood have also been identified as a major risk factor for the development of asthma (2). Although RSV is likely to have multiple pathways of influencing asthma, it was recently found that the innate immune response to RSV is mediated by CD14 and TLR4 (19). This raises the question of whether LPS has a unique role in asthma or if other TLR ligands could induce Th2 sensitization.
Acknowledgments
We thank R. Flavell and R. Medzhitov for critical review of the manuscript and discussion, and P. Ranney and L. Xu for technical assistance.
This work was supported by National Institutes of Health grants, AI26791, HL54450, and MSTP 5T32GM07205.
References
- 1.Liu, A.H. 2002. Endotoxin exposure in allergy and asthma: reconciling a paradox. J. Allergy Clin. Immunol. 109:379–392. [DOI] [PubMed] [Google Scholar]
- 2.Gern, J.E. 2000. Viral and bacterial infections in the development and progression of asthma. J. Allergy Clin. Immunol. 105:S497–S502. [DOI] [PubMed] [Google Scholar]
- 3.Medzhitov, R. 2001. Toll-like receptors and innate immunity. Nat. Rev. Immunol. 1:135–145. [DOI] [PubMed] [Google Scholar]
- 4.Schwartz, D.A. 2001. Does inhalation of endotoxin cause asthma? Am. J. Respir. Crit. Care Med. 163:305–306. [DOI] [PubMed] [Google Scholar]
- 5.Braun-Fahrlander, C., J. Riedler, U. Herz, W. Eder, M. Waser, L. Grize, S. Maisch, D. Carr, F. Gerlach, A. Bufe, et al. 2002. Environmental exposure to endotoxin and its relation to asthma in school-age children. N. Engl. J. Med. 347:869–877. [DOI] [PubMed] [Google Scholar]
- 6.Reed, C.E., and D.K. Milton. 2001. Endotoxin-stimulated innate immunity: a contributing factor for asthma. J. Allergy Clin. Immunol. 108:157–166. [DOI] [PubMed] [Google Scholar]
- 7.Schnare, M., G.M. Barton, A.C. Holt, K. Takeda, S. Akira, and R. Medzhitov. 2001. Toll-like receptors control activation of adaptive immune responses. Nat. Immunol. 2:947–950. [DOI] [PubMed] [Google Scholar]
- 8.Herrick, C.A., H. MacLeod, E. Glusac, R.E. Tigelaar, and K. Bottomly. 2000. Th2 responses induced by epicutaneous or inhalational protein exposure are differentially dependent on IL-4. J. Clin. Invest. 105:765–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tsitoura, D.C., R.H. DeKruyff, J.R. Lamb, and D.T. Umetsu. 1999. Intranasal exposure to protein antigen induces immunological tolerance mediated by functionally disabled CD4+ T cells. J. Immunol. 163:2592–2600. [PubMed] [Google Scholar]
- 10.Cohn, L., R.J. Homer, N. Niu, and K. Bottomly. 1999. T helper 1 cells and interferon gamma regulate allergic airway inflammation and mucus production. J. Exp. Med. 190:1309–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seymour, B.W., L.J. Gershwin, and R.L. Coffman. 1998. Aerosol-induced immunoglobulin (Ig)-E unresponsiveness to ovalbumin does not require CD8+ or T cell receptor (TCR)-gamma/delta+ T cells or interferon (IFN)-gamma in a murine model of allergen sensitization. J. Exp. Med. 187:721–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lutz, M.B., N. Kukutsch, A.L. Ogilvie, S. Rossner, F. Koch, N. Romani, and G. Schuler. 1999. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J. Immunol. Methods. 223:77–92. [DOI] [PubMed] [Google Scholar]
- 13.Vogel, S.N., J.S. Wax, P.Y. Perera, C. Padlan, M. Potter, and B.A. Mock. 1994. Construction of a BALB/c congenic mouse, C.C3H-Lpsd, that expresses the Lpsd allele: analysis of chromosome 4 markers surrounding the Lps gene. Infect. Immun. 62:4454–4459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaisho, T., K. Hoshino, T. Iwabe, O. Takeuchi, T. Yasui, and S. Akira. 2002. Endotoxin can induce MyD88-deficient dendritic cells to support T(h)2 cell differentiation. Int. Immunol. 14:695–700. [DOI] [PubMed] [Google Scholar]
- 15.Dabbagh, K., M.E. Dahl, P. Stepick-Biek, and D.B. Lewis. 2002. Toll-like receptor 4 is required for optimal development of Th2 immune responses: role of dendritic cells. J. Immunol. 168:4524–4530. [DOI] [PubMed] [Google Scholar]
- 16.Wan, G.H., C.S. Li, and R.H. Lin. 2000. Airborne endotoxin exposure and the development of airway antigen-specific allergic responses. Clin. Exp. Allergy. 30:426–432. [DOI] [PubMed] [Google Scholar]
- 17.Tulic, M.K., J.L. Wale, P.G. Holt, and P.D. Sly. 2000. Modification of the inflammatory response to allergen challenge after exposure to bacterial lipopolysaccharide. Am. J. Respir. Cell Mol. Biol. 22:604–612. [DOI] [PubMed] [Google Scholar]
- 18.Arbour, N.C., E. Lorenz, B.C. Schutte, J. Zabner, J.N. Kline, M. Jones, K. Frees, J.L. Watt, and D.A. Schwartz. 2000. TLR4 mutations are associated with endotoxin hyporesponsiveness in humans. Nat. Genet. 25:187–191. [DOI] [PubMed] [Google Scholar]
- 19.Kurt-Jones, E.A., L. Popova, L. Kwinn, L.M. Haynes, L.P. Jones, R.A. Tripp, E.E. Walsh, M.W. Freeman, D.T. Golenbock, L.J. Anderson, et al. 2000. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat. Immunol. 1:398–401. [DOI] [PubMed] [Google Scholar]