

Abstract
GD3 synthase is rapidly activated in different cell types after specific apoptotic stimuli. De novo synthesized GD3 accumulates and contributes to the apoptotic program by relocating to mitochondrial membranes and inducing the release of apoptogenic factors. We found that sialic acid acetylation suppresses the proapoptotic activity of GD3. In fact, unlike GD3, 9-O-acetyl-GD3 is completely ineffective in inducing cytochrome c release and caspase-9 activation on isolated mitochondria and fails to induce the collapse of mitochondrial transmembrane potential and cellular apoptosis. Moreover, cells which are resistant to the overexpression of the GD3 synthase, actively convert de novo synthesized GD3 to 9-O-acetyl-GD3. The coexpression of GD3 synthase with a viral 9-O-acetyl esterase, which prevents 9-O-acetyl-GD3 accumulation, reconstitutes GD3 responsiveness and apoptosis. Finally, the expression of the 9-O-acetyl esterase is sufficient to induce apoptosis of glioblastomas which express high levels of 9-O-acetyl-GD3. Thus, sialic acid acetylation critically controls the proapoptotic activity of GD3.
Keywords: sialic acid, 9-O-acetyl GD3, mitochondria, apoptosis, glioblastoma
Introduction
Lipid and glycolipid diffusible mediators are important components of the apoptotic program in many cell types. GD3 ganglioside is actively synthesized and transiently accumulates intracellularly within 30 min from the clustering of death receptors in hemopoietic cells undergoing apoptosis (1). De novo synthesis of GD3 induced by engagement of death receptors requires the previous accumulation of ceramide deriving from the hydrolysis of membrane sphingomyelin, by the action of acidic sphingomyelinase (2). The final step of de novo GD3 synthesis is catalyzed by the α-2,8-sialyltransferase (GD3 synthase or ST8Sia-I), an early Golgi-resident luminal enzyme which mediates the attachment of a second sialic acid to the sialic acid of GM3 ganglioside, its main substrate. Transient accumulation of GD3 in the early Golgi is followed by relocation of GD3 to mitochondrial membranes (3). GD3 directly affects mitochondrial physiology by inducing the opening of the permeability transition pore complex (PTPC),* the collapse of mitochondrial transmembrane potential, mitochondrial swelling, uncoupling of the respiratory chain, generation of reactive oxygen species (ROS), and the release of apoptogenic factors such as cytochrome c and apoptosis-inducing factor (AIF; references 3–6). GD3-induced mitochondrial changes result in the activation of caspase-9 and are synergized by bax and antagonized by bcl-2 (3, 7). Mitochondrial changes and apoptosis can be induced directly by exposing the cells to natural or synthetic GD3 (8), or by forcing the expression of the GD3 synthase (1). Accordingly, blocking the expression of the GD3 synthase significantly prevents death receptor–induced and ceramide-induced apoptosis.
In some cell types, GD3 ganglioside is therefore a powerful inducer of mitochondrial dysfunction and apoptosis. However, the molecular mechanisms which regulate GD3 activity are completely unknown.
GD3 contains two sialic acid residues, sequentially attached to the galactose residue. The most common postsynthetic modification of GD3 is O-acetylation at the C9 position of its terminal sialic acid (9–11). The ester bond is catalyzed by specific, yet elusive, acetyltransferases (12), which probably colocalize with the GD3 synthase in the same Golgi compartments. The resulting 9-O-acetyl GD3 is expressed in a developmentally regulated and tissue-specific manner (13), and is abundant in a variety of tumors (14–16). Specific O-acetylesterases are responsible for the hydrolysis of the ester bond (17), converting the 9-O-acetyl GD3 back to GD3 (18). The ratio between acetylated and nonacetylated forms of GD3, and their subcellular distribution, may vary among different cells (19), suggesting cell-specific regulatory mechanisms and compartmentalized functions. O-acetylation affects in fact the structural properties of sialic acid, changing binding and antigenic specificities, as well as biological activities (9).
We therefore investigated whether O-acetylation affects the proapoptotic activity of GD3. We found that 9-O-acetyl GD3 is completely ineffective in inducing mitochondrial changes and cellular apoptosis and that some cells convert GD3 into 9-O-acetyl GD3 to prevent GD3-induced apoptosis. By turning proapoptotic GD3 into 9-O-acetyl GD3, sialic acid acetylation may therefore cooperate to cell survival.
Materials and Methods
Cell Culture and Reagents.
HEK-293 and U87 cells were cultured in DMEM supplemented with 10% fetal bovine serum. U118 were cultured in minimum essential medium (MEM) with Earle's Salts supplemented with 10% fetal bovine serum. GD3 was purchased from Sigma-Aldrich or Qbiogene. 9-O-acetyl GD3 was purified from bovine buttermilk as described previously (20). Monoclonal antibody for cytochrome c was from BD Biosciences (clone 7H8.2C12). Polyclonal antibody against caspase-9 p18 subunit was from Santa Cruz Biotechnology, Inc. (clone H-170). The anti-GD3 mAb (IgG3) was protein G-purified from the R24 hybridoma supernatant. Anti-9-O-acetyl GD3 mAb (IgM) was obtained from the M-T6004 hybridoma supernatant (a gift from Dr. E. Rieber, University of Dresden, Dresden, Germany; reference 21). The anti-9-O-acetyl GD3 P-Path mAb (IgM) is a gift of Dr. Y. Hirabayashi (Riken Institute, Saitama, Japan; reference 22). The anti-9-O-acetyl GD3 mAb UM4D4 (Ancell), was used for FACS® staining (23).
DNA Constructs.
GD3 synthase plasmids: WT GD3 synthase fused to the GFP, pEGFPwtST8, and its inactive mutant pEGFPmST8 have been described previously (1). The plasmid GFP-spectrin was a kind gift from Dr. R. Kalejta (Princeton University, Princeton, NJ; reference 24). The plasmid pAIISVL2-CHA4 containing the viral 9-O-acetylesterase gene was a kind gift of Dr. P. Palese (Mount Sinai Medical Center, New York, NY). The cDNA was further cloned into the expression vector pcDNA3 (Invitrogen). The viral 9-O-acetylesterase dead mutant S57A was generated using the Quick Change Site-directed Mutagenesis kit (Stratagene) and WT viral 9-O-acetylesterase as a template. Each mutation was confirmed by sequencing.
TLC, Immunostaining, and FACS® Analysis.
Gangliosides were extracted as previously described with minor modifications (1). Briefly, cells were disrupted by three cycles of freezing and thawing. The aqueous pellet was extracted with chloroform/methanol (1:2, vol/vol) after 30 s sonication at 10 Watts. After 20 min vortex and 5 min centrifugation at 15,000 g, the upper phase was recovered and water was added to obtain a final ratio of chloroform/methanol/water (4:8:5, vol/vol/vol). After vortex and 5 min centrifugation at 15,000 g, the upper phase was recovered. Two volumes of methanol were added and evaporated to dryness under nitrogen gas. Gangliosides were then resuspended in chloroform/methanol (2:1, vol/vol) and loaded on a silica gel 60 HP-TLC plate (Merck) and chromatographed in chloroform/methanol/CaCl2 containing 0.2% (wt/vol; 2:1:0.2, vol/vol/vol). Plates were treated with 0.5% polyisobutyl-metacrylate in hexane and dried. To carry out immunostaining for GD3 and 9-O-acetyl GD3, the plate was first treated with PBS, 0.05% Tween 20, 1% BSA for 15 min to minimize nonspecific staining, incubated for 1 h with R24 monoclonal anti-GD3-antibody (1:500), or monoclonal anti 9-O-acetyl GD3 (P-Path or M-T6004, 1:500 or 1:200 respectively) followed by a horseradish peroxidase–conjugated anti-IgG or anti-IgM secondary antibody, respectively. Specific bands were detected by chemiluminescence (ECL; Amersham Biosciences), according to the manufacturer's instructions.
Cellular levels of 9-O-acetyl GD3 were also measured by FACS® analysis. Briefly, cells were fixed for 15 min at room temperature with 2% paraformaldehyde, permeabilized for 30 min at 37°C with 60 μM digitonine, and treated with 3% FCS at 37°C for 1 h to minimize nonspecific staining. Samples were then incubated for 60 min at 37°C with anti-9-O-acetyl GD3 mAb (Ancell), or isotype-matched control mAb, at a final concentration of 20 μg/ml. Cells were further stained with secondary RPE-conjugated goat anti–mouse IgM antibody (Oxford Biomarketing) for 30 min at room temperature and analyzed with a FACScan™ flow cytometer (Becton Dickinson).
Alkaline Treatment of Gangliosides.
0.1 mg of 9-O-acetyl GD3 or GD3 were evaporated to dryness under nitrogen gas and resuspended in 1 ml of aqueous 1 N NaOH, sonicated for 5 min in an ultrasonic bath, and incubated for 60 min at 37°C. After neutralization with 1 ml of 1 N acetic acid, the recovered glycolipids were purified with C18 column (Supelclean LC-18, Supelco), run on an HP-TLC plate, and visualized by α-naphtol/sulfuric acid staining.
Liver Mitochondria Purification and Release of Apoptogenic Factors.
Mitochondria were purified from mouse liver, as described (3), and resuspended in 300 mM sucrose, 0.2 mM EGTA, 5 mM TES, pH 7.2. For the induction of permeability transition (PT), mitochondria (5 mg protein/ml) were resuspended in PT buffer (200 mM sucrose, 10 mM Tris-MOPS, pH 7.2, 5 mM Tris-succinate, pH 7.2, 1 mM Tris-phosphate, pH 7.2, 10 μM EGTA-Tris, 2 μM rotenone, and 5 μM Ca2+) and after addition of 200 μM Ca2+, 10–40 μM GD3, or 9-O-acetyl GD3 for 30 min, supernatants from mitochondria were recovered by centrifugation (15,000 g for 20 min). Supernatants were then analyzed by SDS-PAGE, blotted onto nitrocellulose membranes and probed with specific antibodies. The following antibodies were used: mouse anti-cytochrome c mAb (1:500 in PBS, 0.1% Tween 20, 5% milk powder), rabbit anti-caspase-9 polyclonal antibody (1:200 in PBS, 0.1% Tween 20, 5% milk powder). Horseradish peroxidase–conjugated secondary antibodies were used and Western blots were revealed by ECL.
Transfections and Apoptosis Analysis.
HEK-293 cells were transfected with the calcium phosphate method. The day before transfection, 6.5 × 106 cells were plated in 10-cm dishes. The next day, 70–80% confluent cells were transfected with 8 μg of the different expression vectors. 40 h after transfection, cells were harvested for TLC analysis. 10-cm dishes were divided in two equal parts, one for GD3 analysis, the other one for 9-O-acetyl GD3. U87 and U118 cells were transfected with Lipofectamine 2000 according to manufacturer's instructions. Briefly, cells were cotransfected with pEGFP vector (CLONTECH Laboratories, Inc.) or GFP-spectrin and vector of interest in a 1:3 ratio. Mitochondrial damage was assessed using JC-1 (Molecular Probes) to analyze the loss of mitochondrial transmembrane potential (ΔΨm) and nuclear condensation/fragmentation was evaluated using Hoechst 33342 (Molecular Probes). Carbocyanine dye JC-1 exists as a monomer or as J-aggregate forms, depending on concentration and membrane potential. While polarized mitochondria exhibit J-aggregates, with an emission max of 590 nm, depolarized mitochondria exhibit monomers, with an emission max at 527 nm, thus permitting to discriminate the mitochondrial transmembrane potential with a fluorescence microscope by using a standard fluorescein longpass optical filter (25). Briefly, HEK-293 cells were stained 30 min with JC-1 (1 μg/ml) and Hoechst (500 ng/ml) and then analyzed with a fluorescence microscope (IX50; Olympus) with adequate filters. Typically, two independent investigators scored the apoptotic cells in blind, counting at least 200 cells from nine different microscopic fields.
To score the percentage of transfected (green) HEK-293 cells undergoing apoptosis, cells were harvested 16 h after transfection and replated onto coverslips. Cells were then stained with Hoechst 33342 (500 ng/ml for 30 min) and cell death was calculated counting the condensed and/or fragmented nuclei, as detailed above. For U87 and U118, cells were stained with Hoechst (500 ng/ml for 30 min) 24 h after transfection and cell death was calculated counting the condensed and/or fragmented nuclei of transfected (green) cells, as described above.
Results and Discussion
9-O-acetyl GD3 Is Unable to Induce Cytochrome c Release from Mitochondria and Activation of Caspase-9.
The interaction of GD3 with mitochondrial membranes causes cytochrome c release and caspase-9 activation, thereby triggering apoptosis (3–5). To verify whether 9-O-acetyl GD3 retains the proapoptotic functions of GD3, purified mitochondria were exposed to 9-O-acetyl GD3 and the amount of cytochrome c released in the supernatants quantitated by Western blot analysis and immunostaining. Fig. 1 B shows that, unlike GD3, purified 9-O-acetyl GD3 is completely unable to induce the release of cytochrome c from mitochondria. Accordingly, exposure of isolated mitochondria to purified 9-O-acetyl GD3 did not result in caspase-9 activation, unlike GD3, as revealed by the detection of the caspase-9 p35 fragment in the supernatants, by Western blot analysis and immunostaining (Fig. 1 C). To verify that the acetyl moiety is responsible for the loss of function of 9-O-acetyl GD3, we deacetylated 9-O-acetyl GD3 in vitro by alkaline treatment, in order to recover GD3 (Fig. 1 A). Deacetylated 9-O-acetyl GD3 is able to induce cytochrome c release from isolated mitochondria to the same extent as GD3 (Fig. 1 B).
Figure 1.
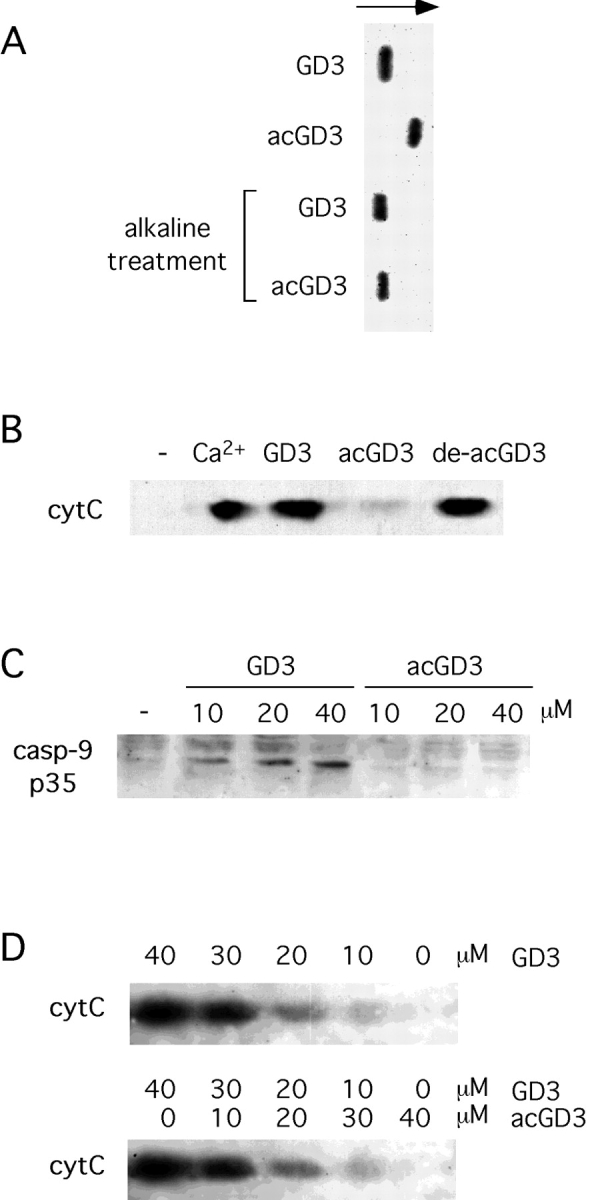
9-O-acetyl GD3 is unable to induce mitochondrial damage. (A) GD3 and 9-O-acetyl GD3 (acGD3) were subjected to alkaline treatment, and run on TLC for 50 min, along with untreated GD3 and 9-O-acetyl GD3. (B) Isolated mitochondria were treated for 40 min with 200 μM Ca2+, 30 μM GD3, 30 μM 9-O-acetyl GD3 (acGD3), 30 μM de-acetylated 9-O-acetyl GD3 (de-acGD3), then supernatants subjected to Western blotting with anti-cytC mAb. (C) Isolated mitochondria were treated for 30 min with the indicated concentrations of GD3 and 9-O-acetyl GD3, then supernatants subjected to Western blotting with anti-caspase-9 p35 mAb. Data are representative of three independent experiments. (D) Isolated mitochondria were treated for 40 min with the indicated concentrations of GD3/9-O-acetyl GD3 mixes or GD3 alone, then supernatants subjected to Western blotting with anti-cytC mAb. Data are representative of two independent experiments.
We also investigated whether 9-O-acetyl GD3 could inhibit GD3-induced mitochondrial changes. GD3 and 9-O-acetyl GD3 were mixed in different ratios within a fixed total amount of gangliosides (40 μM) which did not cause non specific, detergent-like effects. Isolated mitochondria were exposed to the different ganglioside mixes and the release of cytochrome c quantitated by Western blot analysis and immunostaining. Fig. 1 D shows that cytochrome c was similarly released by mitochondria exposed to the GD3/9-O-acetyl GD3 mixes or to GD3 alone, indicating that 9-O-acetyl GD3 does not inhibit GD3-induced changes.
9-O-acetyl GD3 Is Unable to Trigger Mitochondrial Changes and Apoptosis.
Exposure of intact cells to exogenous GD3 induces the collapse of mitochondrial transmembrane potential (ΔΨm) and triggers apoptosis. To investigate whether sialic acid acetylation affected the proapoptotic activity of GD3, HEK-293 cells were exposed to 9-O-acetyl GD3. As shown in Fig. 2, 9-O -acetyl GD3 could not affect the mitochondrial transmembrane potential of HEK-293 cells (Fig. 2 A), nor induce apoptosis (Fig. 2 B). In vitro deacetylation of sialic acid restored the ability of 9-O-acetyl GD3 to induce mitochondrial changes and apoptosis. Moreover, competition experiments indicated that 9-O-acetyl GD3 could not prevent GD3-induced apoptosis (unpublished data). Together these results strongly suggested that sialic acid 9-O-acetylation might represent a mechanism to inactivate GD3 in vivo. We therefore further investigated the functional implications of this finding.
Figure 2.

9-O-acetyl GD3 is unable to induce mitochondrial damage and apoptosis of HEK-293 cells. The percentage of HEK-293 cells with ΔΨm loss (A) or condensed nuclei (B) was analyzed at 30 h after stimulation with 200 μM of exogenous GD3, 9-O-acetyl GD3 (acGD3), and deacetylated 9-O-acetyl GD3 (de-ac-GD3). Data represent the mean ± 1 SD from three independent experiments.
Cells Which Are Resistant to Endogenous GD3 Accumulation Synthesize 9-O-acetyl GD3.
Several cell lines undergo massive mitochondrial damage and apoptosis upon acute intracellular GD3 accumulation, a condition which can be experimentally achieved by GD3 synthase overexpression. Selected cell lines, however, while still sensitive to exogenous GD3 exposure, fail to undergo apoptosis upon GD3 synthase overexpression and endogenous GD3 accumulation. As shown in Fig. 3 A, HEK-293 cells, which undergo apoptosis upon exposure to exogenous GD3 (Fig. 2 B), are completely resistant to GD3 synthase overexpression. To verify that GD3 synthase overexpression was indeed inducing GD3 accumulation in HEK-293 cells, the amount of GD3 was assessed by TLC and immunostaining. Fig. 3 B shows that HEK-293 cells transfected with GD3 synthase, but not with a catalytically dead GD3 synthase mutant which lacks part of the catalytic site (1), accumulate GD3 efficiently. Interestingly, a simultaneous immunostaining for 9-O-acetyl GD3 revealed that GD3 synthase-transfected HEK-293 were also accumulating 9-O-acetyl GD3, indicating that part of the de novo synthesized GD3 was converted to 9-O-acetyl GD3, by the concomitant activation of endogenous sialic acid acetyltransferases.
Figure 3.
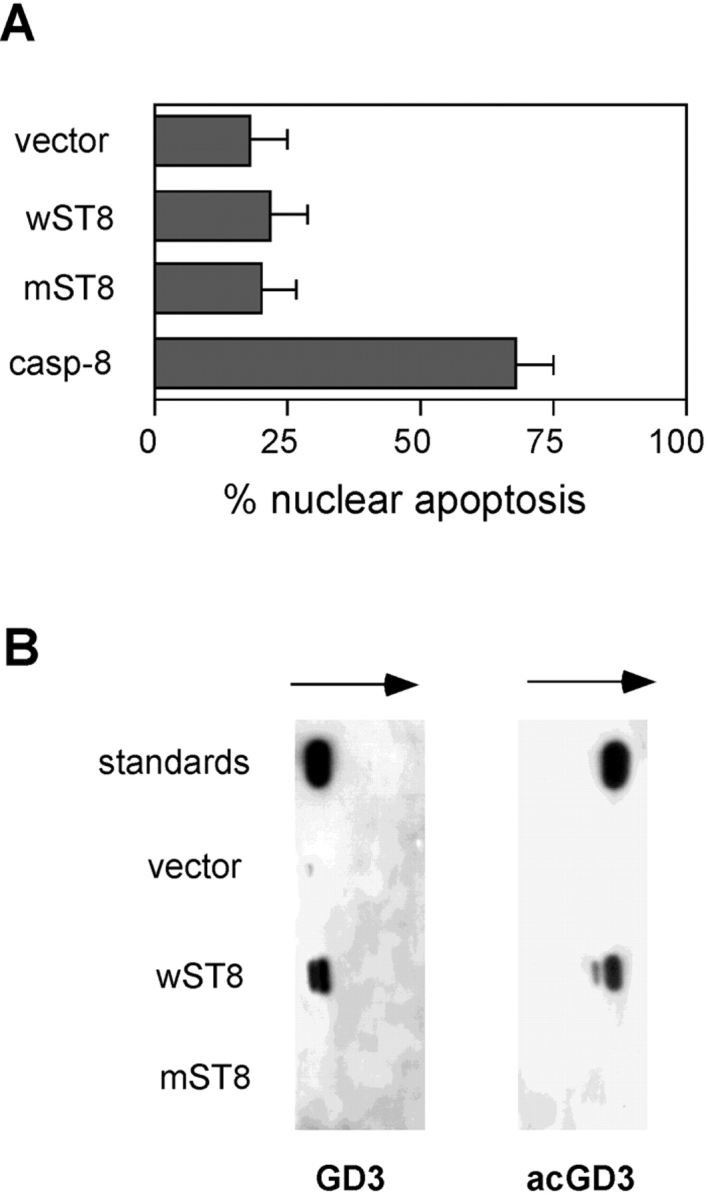
HEK-293 cells are resistant to endogenous GD3 accumulation and synthesize 9-O-acetyl GD3. (A) The percentage of apoptotic HEK-293 nuclei was analyzed 40 h after transfection with empty vector (vector), wild-type GD3 synthase (wST8), dead mutant GD3 synthase (mST8), and caspase-8 as a positive control. Data represent the mean ± 1 SD from five independent experiments. (B) GD3 and 9-O-acetyl GD3 content of HEK-293 cells was analyzed by TLC and immunostaining 40 h after transfection with the indicated constructs. Data are representative of six independent experiments.
In Vivo Deacetylation of De Novo–synthesized 9-O-acetyl GD3 Triggers Apoptosis.
The above mentioned findings suggested that GD3 to 9-O-acetyl GD3 conversion, mediated by endogenous sialic acid acetyltransferases, might represent an efficient cell survival strategy in response to stress-induced acute GD3 accumulation. To test this hypothesis, we first decided to force the in vivo deacetylation of de novo–synthesized 9-O-acetyl GD3 by coexpressing a specific sialic acid acetylesterase. We used the influenza type C viral 9-O-acetylesterase, known to efficiently de-acetylate 9-O-acetyl GD3 in mammalian cells (26). We also prepared an enzymatically dead version of the same viral acetylesterase by substituting a critical Ser57 residue with Ala, based on structural informations (27). We transiently coexpressed the wild-type 9-O-acetylesterase, or the dead 9-O-acetylesterase, together with the wild-type GD3 synthase, or the dead GD3 synthase in HEK-293 cells. The accumulation of GD3 and 9-O-acetyl GD3 in cotransfected cell cultures was monitored along with the induction of apoptosis. As shown in Fig. 4 A, the coexpression of both dead mutants, or the coexpression of the dead GD3 synthase together with the wild-type 9-O-acetylesterase, did not result in any GD3 or 9-O-acetyl GD3 accumulation. Moreover, the coexpression of the wild-type GD3 synthase together with the dead 9-O-acetylesterase, resulted in the accumulation of both GD3 and 9-O-acetyl GD3. This last condition parallels what already shown in Fig. 3, and no specific apoptosis can be induced in HEK-293 cells (Fig. 4 B). However, the coexpression of the wild-type forms of both enzymes resulted in enhanced accumulation of GD3 and in the disappearance of 9-O-acetyl GD3, due to the action of the 9-O-acetylesterase. Importantly, in this last condition, extensive apoptosis of HEK-293 could be induced (Fig. 4 B). Apoptosis is strictly dependent on specific deacetylation of 9-O-acetyl GD3, since the dead 9-O-acetylesterase is ineffective, and it does not occur in the absence of de novo 9-O-acetyl GD3 synthesis. These results strongly suggest that when the attempt to transform part of the accumulating GD3 into 9-O-acetyl GD3 is reversed by the action of the 9-O-acetylesterase, a critical GD3 threshold is exceeded and HEK-293 cells undergo apoptosis.
Figure 4.

Deacetylation of de novo synthesized 9-O-acetyl GD3 triggers apoptosis of HEK-293 cells. (A) GD3 and 9-O-acetyl GD3 content of HEK-293 cells was analyzed by TLC and immunostaining 40 h after transfection with wild-type GD3 synthase (ST8), wild-type 9-O-acetylesterase (Est), dead mutant GD3 synthase (mST8), and dead mutant 9-O-acetylesterase (mEst). Data are representative of three independent experiments. (B) The percentage of apoptotic HEK-293 nuclei was analyzed 40 h after transfection with the indicated constructs. Data represent the mean ± 1 SD from three independent experiments.
Deacetylation of Endogenous 9-O-acetyl GD3 Directly Induces Apoptosis of Human Tumor Cells.
To further support the notion that the ratio between GD3 and 9-O-acetyl GD3 might critically affect survival of tumor cells, we investigated tumor cell lines which constitutively synthesize and express both GD3 and 9-O-acetyl GD3. We tested the hypothesis that, by selectively breaking the endogenous GD3/9-O-acetyl GD3 balance in favor of GD3, we would induce apoptosis. The viral 9-O-acetylesterase is known to deacetylate 9-O-acetyl GD3 in neural tumor cells (18). Therefore, the viral 9-O-acetylesterase, and its catalytically dead mutant, were expressed in the human glioblastoma cells U118, which constitutively express both GD3 and 9-O-acetyl GD3, as assessed by TLC and immunostaining (Fig. 5 A). The 9-O-acetylesterase was also expressed in the human glioblastoma U87, which expresses GD3 but lacks detectable 9-O-acetyl GD3 (Fig. 5 A). As shown in Fig. 5 B, the 9-O-acetylesterase, but not its catalytically dead mutant, directly triggered apoptosis of U118 cells expressing 9-O-acetyl GD3. By contrast, the 9-O-acetylesterase, as well as its catalytically dead mutant, did not induce apoptosis in U87 cells lacking 9-O-acetyl GD3 (Fig. 5 C). To verify that the viral 9-O-acetylesterase was in fact active in glioblastoma cells, U118 cells were cotransfected with 9-O-acetylesterase and GFP-spectrin to allow the detection and electronic gating of transfected cells by FACS® analysis (24). Fig. 5 D shows that the levels of cellular 9-O-acetyl GD3 were significantly decreased in U118 cells transfected with the 9-O-acetylesterase, compared with vector-transfected cells. Together these results indicated that the 9-O-acetylesterase was specifically acting upon endogenous 9-O-acetyl GD3 and that the GD3/9-O-acetyl GD3 ratio may significantly control survival of tumor cells.
Figure 5.
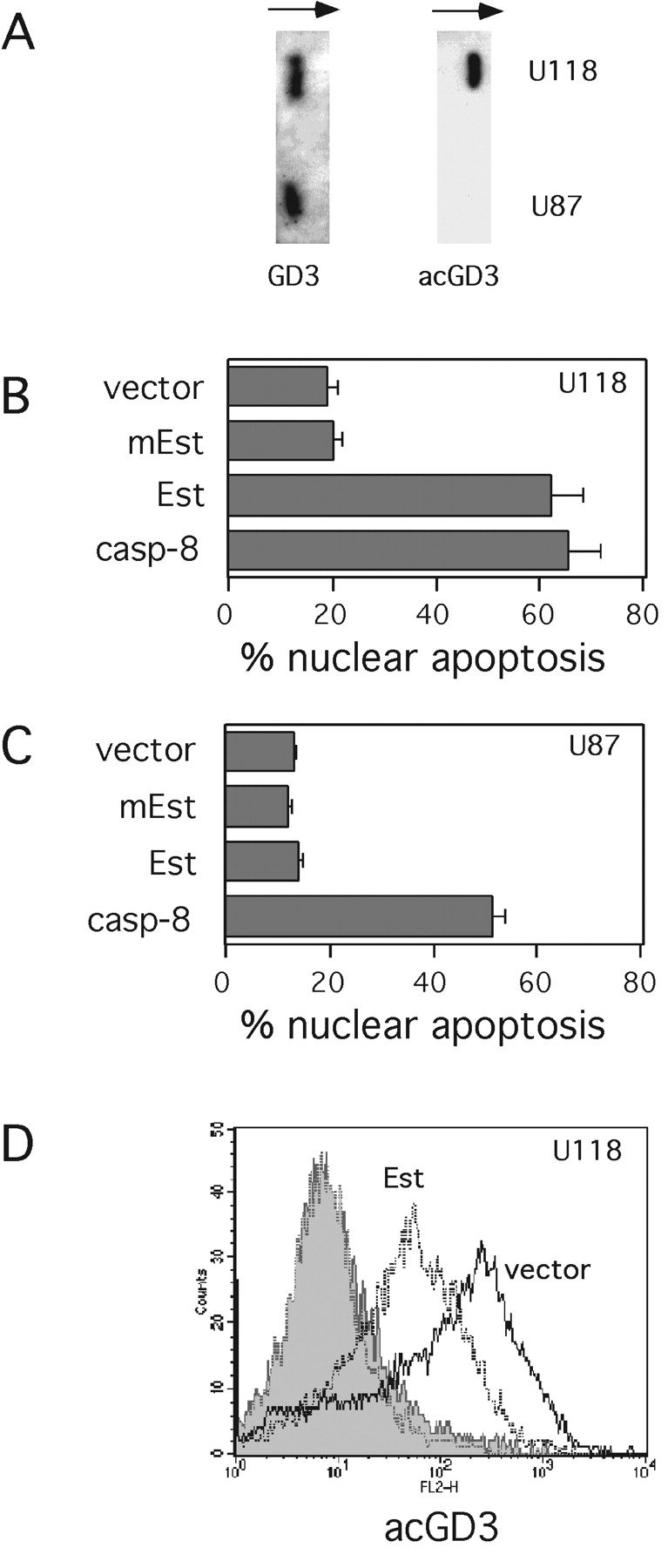
Deacetylation of endogenous 9-O-acetyl GD3 induces apoptosis of U118 glioblastoma cells. (A) The expression of GD3 and 9-O-acetyl GD3 from U118 and U87 glioblastoma cells was analyzed by TLC-immunostaining. The percentage of apoptotic U118 (B) and U87 (C) cells was analyzed 24 h after transfection with empty vector (vector), dead mutant 9-O-acetylesterase (mEst), wild-type 9-O-acetylesterase (Est), and caspase-8 as a positive control. The data represent the mean ± 1 SD from five independent experiments. (D) 24 h after cotransfection with GFP-spectrin and empty vector (vector), or 9-O-acetylesterase (Est), U118 glioma cells were stained with anti-9-O acetyl GD3 mAb (open areas) or control antibody (shaded areas), and secondary RPE-conjugated antibody. The expression of 9-O-acetyl GD3 within electronically gated transfected cells was analyzed by flow cytometry. Data are representative of three independent experiments.
Concluding Remarks.
Mitochondria act as “sensors” of stress-induced lipid and glycolipids, from simple unicellular organisms to metazoans (28–30). The stress mediator ceramide, generated by the action of sphingomyelinases, is largely converted to GD3 ganglioside. In different cell types, upon acute GD3 accumulation and relocalization to mitochondrial membranes, the mitochondrial PTPC opens and, concomitantly, cytochrome c is released, causing the activation of caspase-9 and downstream caspases (3–5). Some tumor cells, however, may actively O-acetylate the terminal sialic acid of GD3, generating 9-O-acetyl GD3, thus enhancing their in vivo metastatic potential. In fact, C6 rat glioma cells transfected with the GD3 synthase express both GD3 and 9-O-acetyl GD3 and show increased tumorigenicity and invasiveness (31). We found that 9-O-acetyl GD3 is completely unable to induce mitochondrial changes and cellular apoptosis. Sialic acid O-acetylation is likely to critically modify either hydrophobicity or steric conformation of GD3, so that putative interactions with PTPC elements or other components of the mitochondrial membrane are prohibited. By turning part of proapoptotic GD3 into the “harmless” 9-O-acetyl GD3, sialic acid O-acetylation acts as an effective antiapoptotic mechanism. Sialic acid O-acetylation may therefore represent an important strategy adopted by tumor cells to escape acute GD3 accumulation during cellular stress and to enhance survival.
Acetylation is a covalent modification of biological molecules, which may profoundly affect their stability, specificity, and function (32, 33). Specific acetyltransferases and deacetylases are therefore involved in multiple biological processes, including apoptosis. It has been shown, in fact, that N-acetylation of critical lysines in the COOH terminus of tumor suppressor p53, controlled by histone acetyltransferases and specific deacetylases (34), modulates p53 function, contributing to the regulation of the apoptotic program (35). Similarly, N-acetylation may regulate the proapoptotic activity of p73 and Rb proteins (36, 37). Here we show that sialic acid O-acetylation of GD3 ganglioside controls its ability to induce mitochondrial changes and to trigger apoptosis, providing the first example of gain/loss of proapoptotic function regulated by acetylation in a lipid mediator. Importantly, the ratio between GD3 and 9-O-acetyl GD3 may critically affect survival of tumor cells, as shown here in glioblastoma cells. Little molecular information is available at the moment on human GD3 acetyltransferases and acetylesterases. Further characterization of the enzymes involved in the control of GD3 acetylation in humans are expected to provide interesting new targets for the pharmacological manipulation of the apoptotic program.
Acknowledgments
We thank Dr. E. Rieber, (University of Dresden, Germany), Dr. Y. Hirabayashi (Riken Institute, Japan), Dr. P. Palese, (Mount Sinai Medical Center, New York), and Dr. R. Kalejta (Princeton University, NJ) for reagents.
This work has been supported by the Associazione Italiana Ricerca sul Cancro, the Agenzia Spaziale Italiana, the Ministero Istruzione Universita' e Ricerca, the European Commission, and by a grant from the Deutsche Forschungsgemeinschaft (Kn 442/1-3). I. Condò is a recipient of a Fondazione Italiana Ricerca sul Cancro fellowship. M.R. Rippo is currently at the Institute of Experimental Pathology, University of Ancona, Italy.
Footnotes
Abbreviation used in this paper: PTPC, permeability transition pore complex.
References
- 1.De Maria, R., M.L. Lenti, F. Malisan, F. d'Agostino, B. Tomassini, A. Zeuner, M.R. Rippo, and R. Testi. 1997. Requirement for GD3 ganglioside in CD95- and ceramide-induced apoptosis. Science. 277:1652–1655. [DOI] [PubMed] [Google Scholar]
- 2.De Maria, R., M.R. Rippo, H.E. Schuchman, and R. Testi. 1998. Acidic sphingomyelinase (ASM) is necessary for Fas-induced GD3 ganglioside accumulation and efficient apoptosis of lymphoid cells. J. Exp. Med. 187:897–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rippo, M.R., F. Malisan, L. Ravagnan, B. Tomassini, I. Condò, P. Costantini, S.A. Susin, A. Rufini, M. Todaro, G. Kroemer, and R. Testi. 2000. GD3 ganglioside directly targets mitochondria in a bcl-2-controlled fashion. FASEB J. 14:2047-2054. [DOI] [PubMed] [Google Scholar]
- 4.Scorrano, L., V. Petronilli, F. Di Lisa, and P. Bernardi. 1999. Commitment to apoptosis by GD3 ganglioside depends on opening of the mitochondrial permeability transition pore. J. Biol. Chem. 274:22581–22585. [DOI] [PubMed] [Google Scholar]
- 5.Kristal, B.S., and A.M. Brown. 1999. Apoptogenic ganglioside GD3 directly induces the mitochondrial permeability transition. J. Biol. Chem. 274:23169–23175. [DOI] [PubMed] [Google Scholar]
- 6.Garcia-Ruiz, C., A. Colell, R. Paris, and J.C. Fernandez-Checa. 2000. Direct interaction of GD3 ganglioside with mitochondria generates reactive oxygen species followed by mitochondrial permeability transition, cytochrome c release, and caspase activation. FASEB J. 14:847–858. [DOI] [PubMed] [Google Scholar]
- 7.Pastorino, J.G., M. Tafani, R.J. Rothman, A. Marcineviciute, J.B. Hoek, and J.L. Farber. 1999. Functional consequences of the sustained or transient activation by bax of the mitochondrial permeability transition pore. J. Biol. Chem. 274:31734–31739. [DOI] [PubMed] [Google Scholar]
- 8.Castro-Palomino, J.C., B. Simon, O. Speer, M. Leist, and R.R. Schmidt. 2001. Synthesis of ganglioside GD3 and its comparison with bovine GD3 with regard to oligodendrocyte apoptosis mitochondrial damage. Chemistry. 7:2178–2184. [DOI] [PubMed] [Google Scholar]
- 9.Varki, A. 1997. Sialic acids as ligands in recognition phenomena. FASEB J. 11:248–255. [DOI] [PubMed] [Google Scholar]
- 10.Klein, A., and P. Roussel. 1998. O-Acetylation of sialic acids. Biochimie. 80:49–57. [DOI] [PubMed] [Google Scholar]
- 11.Schauer, R. 2000. Achievements and challenges of sialic acid research. Glycoconj. J. 17:485–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shen, Y., J. Tiralongo, M. Iwersen, B. Sipos, H. Kalthoff, and R. Schauer. 2002. Characterization of the sialate-7(9)-O-acetyltransferase from the microsomes of human colonic mucosa. Biol. Chem. 383:307–317. [DOI] [PubMed] [Google Scholar]
- 13.Varki, A. 1992. Diversity in the sialic acids. Glycobiology. 2:25–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheresh, D.A., R.A. Reisfeld, and A.P. Varki. 1984. O-acetylation of disialoganglioside GD3 by human melanoma cells creates a unique antigenic determinant. Science. 225:844–846. [DOI] [PubMed] [Google Scholar]
- 15.Fuentes, R., R. Allman, and M.D. Mason. 1997. Ganglioside expression in lung cancer cell lines. Lung Cancer. 18:21–33. [DOI] [PubMed] [Google Scholar]
- 16.Fahr, C., and R. Schauer. 2001. Detection of sialic acids and gangliosides with special reference to 9-O-acetylated species in basaliomas and normal human skin. J. Invest. Dermatol. 116:254–260. [DOI] [PubMed] [Google Scholar]
- 17.Takematsu, H., S. Diaz, A. Stoddart, Y. Zhang, and A. Varki. 1999. Lysosomal and cytosolic sialic acid 9-O-acetylesterase activities can be encoded by one gene via differential usage of a signal peptide-encoding exon at the N terminus. J. Biol. Chem. 274:25623–25631. [DOI] [PubMed] [Google Scholar]
- 18.Ariga, T., G.M. Blaine, H. Yoshino, G. Dawson, T. Kanda, G.C. Zeng, T. Kasama, Y. Kushi, and R.K. Yu. 1995. Glycosphingolipid composition of murine neuroblastoma cells: O-acetylesterase gene downregulates the expression of O-acetylated GD3. Biochemistry. 34:11500–11507. [DOI] [PubMed] [Google Scholar]
- 19.Gocht, A., A. Gadatsch, G. Rutter, and B. Kniep. 2000. CDw60: an antigen expressed in many normal tissues and in some tumours. Histochem. J. 32:447–456. [DOI] [PubMed] [Google Scholar]
- 20.Kniep, B., C. Claus, J. Peter-Katalinic, D.A. Monner, W. Dippold, and M. Nimtz. 1995. 7-O-acetyl-GD3 in human T-lymphocytes is detected by a specific T-cell-activating monoclonal antibody. J. Biol. Chem. 270:30173–30180. [DOI] [PubMed] [Google Scholar]
- 21.Claus, C., A. Gocht, R. Schwartz-Albiez, H. Lünsdorf, and B. Kniep. 2002. CD60: Specificity of the antibodies, distribution of the antigens, and the functional aspects. In Leucocyte Typing VII. D. Mason, P. André, A. Bensussan, C. Buckley, C. Civin, E. Clark, M. de Haas, S. Goyert, M. Hadam, D. Hart, et al., editors. Oxford University Press, Oxford, UK. 187–188.
- 22.Leclerc, N., G.A. Schwarting, K. Herrup, R. Hawkes, and M. Yamamoto. 1992. Compartmentation in mammalian cerebellum: Zebrin II and P-path antibodies define three classes of sagittally organized bands of Purkinje cells. Proc. Natl. Acad. Sci. USA. 89:5006–5010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kniep, B., J. Peter-Katalinic, W. Flegel, H. Northoff, and E.P. Rieber. 1992. CDw 60 antibodies bind to acetylated forms of ganglioside GD3. Biochem. Biophys. Res. Commun. 187:1343–1349. [DOI] [PubMed] [Google Scholar]
- 24.Kalejta, R.F., T. Shenk, and A.J. Beavis. 1997. Use of a membrane-localized green fluorescent protein allows simultaneous identification of transfected cells and cell cycle analysis by flow cytometry. Cytometry. 29:286–291. [DOI] [PubMed] [Google Scholar]
- 25.Reers, M., T.W. Smith, and L.B. Chen. 1991. J-aggregate formation of a carbocyanine as a quantitative fluorescent indicator of membrane potential. Biochemistry. 30:4480–4486. [DOI] [PubMed] [Google Scholar]
- 26.Varki, A., F. Hooshmand, S. Diaz, N.M. Varki, and S.M. Hedrick. 1991. Developmental abnormalities in transgenic mice expressing a sialic acid-specific 9-O-acetylesterase. Cell. 65:65–74. [DOI] [PubMed] [Google Scholar]
- 27.Rosenthal, P.B., X. Zhang, F. Formanowski, W. Fitz, C.H. Wong, H. Meier-Ewert, J.J. Skehel, and D.C. Wiley. 1998. Structure of the haemagglutinin-esterase-fusion glycoprotein of influenza C virus. Nature. 396:92–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kolesnick, R.N., F.M. Goni, and A. Alonso. 2000. Compartmentalization of ceramide signaling: physical foundations and biological effects. J. Cell Physiol. 184:285–300. [DOI] [PubMed] [Google Scholar]
- 29.Hannun, Y. 2000. Ceramide in the eukaryotic stress response. Trends Cell Biol. 10:73–80. [DOI] [PubMed] [Google Scholar]
- 30.Tomassini, B., and R. Testi. 2002. Mitochondria as sensors of sphingolipids. Biochimie. 84:123–129. [DOI] [PubMed] [Google Scholar]
- 31.Sottocornola, E., I. Colombo, V. Vergani, G. Taraboletti, and B. Berra. 1999. Increased tumorigenicity and invasiveness of C6 rat glioma cells transfected with the human alpha-2,8 sialyltransferase cDNA. Invasion Metastasis. 18:142–154. [DOI] [PubMed] [Google Scholar]
- 32.Kouzarides, T. 2000. Acetylation: a regulatory modification to rival phosphorylation? EMBO J. 19:1176–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baker, R.R. 2000. Lipid acetylation reactions and the metabolism of platelet-activating factor. Neurochem. Res. 25:677–683. [DOI] [PubMed] [Google Scholar]
- 34.Vaziri, H., S.K. Dessain, E. Ng-Eaton, S.I. Imai, R.A. Frye, T.K. Pandita, L. Guarente, and R.A. Weinberg. 2001. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 107:149–159. [DOI] [PubMed] [Google Scholar]
- 35.Luo, J., F. Su, D. Chen, A. Shiloh, and W. Gu. 2000. Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature. 408:377–381. [DOI] [PubMed] [Google Scholar]
- 36.Costanzo, A., P. Merlo, N. Pediconi, M. Fulco, V. Sartorelli, P.A. Cole, G. Fontemaggi, M. Fanciulli, L. Schiltz, G. Blandino, et al. 2001. DNA damage-dependent acetylation of p73 dictates the selective activation of apoptotic target genes. Mol. Cell. 9:175–186. [DOI] [PubMed] [Google Scholar]
- 37.Chan, H.M., M. Krstic-Demonacos, L. Smith, C. Demonacos, and N.B. La Thangue. 2001. Acetylation control of the retinoblastoma tumour-suppressor protein. Nat. Cell Biol. 3:667–674. [DOI] [PubMed] [Google Scholar]