

Abstract
Expression of the pre-B cell receptor (pre-BCR) leads to activation of the adaptor molecule SLP-65 and the cytoplasmic kinase Btk. Mice deficient for one of these signaling proteins have an incomplete block in B cell development at the stage of large cycling pre-BCR+CD43+ pre-B cells. Our recent findings of defective SLP-65 expression in ∼50% of childhood pre-B acute lymphoblastic leukemias and spontaneous pre-B cell lymphoma development in SLP-65−/− mice demonstrate that SLP-65 acts as a tumor suppressor. To investigate cooperation between Btk and SLP-65, we characterized the pre-B cell compartment in single and double mutant mice, and found that the two proteins have a synergistic role in the developmental progression of large cycling into small resting pre-B cells. We show that Btk/SLP-65 double mutant mice have a dramatically increased pre-B cell tumor incidence (∼75% at 16 wk of age), as compared with SLP-65 single deficient mice (<10%). These findings demonstrate that Btk cooperates with SLP-65 as a tumor suppressor in pre-B cells. Furthermore, transgenic low-level expression of a constitutive active form of Btk, the E41K-Y223F mutant, prevented tumor formation in Btk/SLP-65 double mutant mice, indicating that constitutive active Btk can substitute for SLP-65 as a tumor suppressor.
Keywords: Btk, lymphoma, precursor-B cell, SLP-65/BLNK, tumor suppressor
Introduction
B lymphocytes develop in the BM through distinct stages that are characterized by differential expression of various cell surface markers and the ordered rearrangement of Ig heavy (H) and light (L) chain gene segments (1, 2). In pro-B cells, productive V(D)J recombination of the Ig H chain gene leads to surface expression of the pre-B cell receptor (pre-BCR),* which acts as a checkpoint in early B cell development to monitor the expression of a functional Ig μ H chain. The pre-BCR is transiently expressed on the cell surface and is essential for the proliferative expansion of cytoplasmic μ H chain positive pre-B cells and for the induction of developmental progression into small pre-B cells in which Ig L chain rearrangement occurs (1, 2). The pre-BCR complex is comprised of μ H chain, the nonrearranging VpreB and λ5 surrogate light chain (SLC) proteins and the Igα/CD79a and Igβ/CD79b signaling components. The importance of the pre-BCR checkpoint function is evidenced by an arrest of B cell differentiation at the pro-B to pre-B cell transition both in agammaglobulinemia patients and in mice with mutations in any of these pre-BCR components (1–3).
Cell surface expression of the pre-BCR in the absence of a ligand appears to be sufficient to activate downstream signaling pathways (4, 5). This activation involves the formation of a lipid raft-associated calcium signaling module, composed of the tyrosine phosphorylated signaling molecules Lyn, Syk, SLP-65 (also known as B cell linker protein BLNK or BASH), phosphoinositide 3-kinase (PI3K), Bruton's tyrosine kinase (Btk), Vav, and phospholipase Cγ2 (PLCγ2). As a result, the activation of PLCγ2 induces calcium signaling and subsequently nuclear factor (NF)-κB activation (6–8). Additional checkpoints follow when functional L chain gene recombination in pre-B cells results in the expression of the B cell receptor (BCR) and when the resulting immature B cells progress to mature B cells. Mice deficient for any of the (pre)-BCR signaling proteins exhibit essentially similar immunological phenotypes, characterized by reduced numbers of mature peripheral B cells, absence of B-1 B cells, reduced levels of serum IgM and IgG3, lack of in vivo responses to T cell–independent type II antigens, and reduced in vitro responses to anti-IgM and LPS stimulation (9).
Several lines of evidence show that the adaptor molecule SLP-65 and the cytoplasmic kinase Btk are crucially involved in the regulation of the developmental program of pre-B cells, in particular by limiting pre-B cell expansion and promoting differentiation of large cycling to small resting pre-B cells. Both SLP-65–deficient and Btk-deficient mice show a partial block at the pre-B cell stage, characterized by an impaired developmental progression from large cycling CD43+ into small resting CD43− pre-B cells (10–14). In an analysis of the kinetics of pre-B cell differentiation in vivo, Btk-deficient cells manifested a specific developmental delay within the small pre-B cell compartment, when compared with WT cells (14). By introduction of a transgenic BCR into SLP-65-deficient mice, it was shown that in the absence of SLP-65 the production of κ light chain is decreased and cellular maturation of developing B cell is delayed (15). Reintroduction of SLP-65 into SLP-65-deficient pre-B cells led to pre-BCR down-regulation and enhanced differentiation (16). Furthermore, SLP-65−/− and Btk-deficient pre-B cells show enhanced proliferative expansion in vitro in the presence of IL-7, when compared with WT cells (14, 16–17). A synergistic role of SLP-65 and Btk in B cell development was demonstrated by the almost complete block in B cell development at the CD43+ pre-BCR+ preB cell stage in SLP-65/Btk double mutant mice (18).
We recently reported that SLP-65−/− mice spontaneously develop pre-B cell lymphomas expressing large amounts of pre-BCR on their cell surface (16). Moreover, also ∼50% of human childhood preB acute lymphoblastic leukemias (ALL) showed a complete loss or a drastic reduction of SLP-65 expression. Injection of murine SLP-65−/− pre-B cells into immunodeficient mice resulted in tumor development, while reconstitution of SLP-65 expression in these cells eliminated their tumorgenic capacity (19). Tyr96, which is the binding site for Btk, was identified as a crucial residue for the SLP-65 tumor suppressor function. Although Btk-deficient mice do not develop pre-B cell tumors, the possibility remains that Btk cooperates with SLP-65 as a tumor suppressor in pre-B cells. To investigate cooperation between Btk and SLP-65, we characterized the pre-B cell compartment in single and double mutant mice in detail. We show that Btk/SLP-65 double mutant mice have a high incidence of pre-B cell lymphoma and that transgenic expression of low levels of the constitutive active E41K-Y223F Btk mutant prevents tumor formation in SLP-65/Btk double mutant mice.
Materials and Methods
Mice.
Btk-deficient mice (20) were on the C57BL/6 background. Btk WT alleles were identified by an exon 9 forward primer (5′-CACTGAAGCTGAGGACTCCATAG-3′) and an exon 10 reverse primer (5′-GAGTCATGTGCTTGGAATACCAC-3′). For Btk KO alleles, primers were within the LacZ reporter (20), forward: 5′-TTCACTGGCCGTCGTTTTACAACGTCGTGA-3′, and reverse: 5′-ATGTGAGCGAGTAACAACCCGTCGGATTCT-3′. SLP-65-deficient mice (10) were on the Balb/c background and genotyped with the following primers: Neo2A 5′-CGGAGAACCTGCGTGCAATC-3′ and gxBr 5′-GAGTCCGAATGTTCATCTG-3′ (KO allele) and WtI 5′-TCAAACCTGGGTCTCAGAA-3′ and gxBr (WT allele). The presence of the BtkAct transgene was evaluated by PCR, using the following primers: CD19prom: 5′-TGCAATTAGTGGTGAACAAC-3′ and hmBtk.65R: 5′-AGATGCCAGGACTTGGAAGG-3′.
The BtkAct transgene consist of a ∼6.3 kb genomic fragment containing the CD19 promoter region, a 0.3 kb fragment with the first three exons of human Btk as cDNA sequence, as well as a 27.1 kb genomic DNA fragment, encompassing the Btk exons 3–19 (21). Using double stranded site-directed mutagenesis (Stratagene) the Y223F mutation, the replacement of AT by TC in exon 8, was introduced into the construct that was previously used to generate CD19-BtkE41K mice. The ∼34 kb MluI-NotI insert from the E41K-Y223F-Btk construct was excised from the vector, gel-purified and micro-injected into the pronuclei of FVB fertilized oocytes. Transgenic founder mice were identified by Southern blotting of BamHI digests using a partial human Btk cDNA probe (bp 133–1153), as described previously (20, 21), and crossed with Btk null mice. All mice were bred and maintained at the Erasmus MC animal care facility under specific pathogen free conditions. Statistical analyses of Kaplan-Meier tumor-free survival estimates of the various mouse groups were performed using SPSS 10.1.0 (SPSS Inc.).
Cell Culture and Flow Cytometry.
IL-7 driven BM cultures and determination of IL-7–dependent proliferative responses of total BM cells have been described previously (14, 22). Preparations of single-cell suspensions, standard and intracellular flow cytometry, and conjugated monoclonal antibodies (Becton Dickinson) have been described previously (14, 20). The anti-SLC hybridoma LM34 (23) was kindly provided by A. Rolink (University of Basel, Basel, Switzerland); antibodies were purified using protein G columns and conjugated to biotin according to standard procedures.
Western Blotting Analysis.
For analysis of Btk expression and protein phosphorylation, single-cell suspensions from spleen were depleted of erythrocytes by NH4Cl lysis and enriched for B cells by AutoMACS purification, using biotinylated antibodies to Gr-1, Ter119, CD4, CD8, and CD11b and magnetic streptavidin MicroBeads (Miltenyi Biotec) for negative selection. B-lineage cells were purified from BM by AutoMACS using anti-B220 MicroBeads for positive selection. Splenic B cell fractions were stimulated with 10 μg/ml F(ab′)2 fragment of polyclonal goat-anti–mouse IgM (Jackson ImmunoResearch Laboratories) in RPMI1640 at 37°C for 5 min. Western blotting was performed as described (20), using anti-Btk C-20, anti-Erk1/2 SC-094 (Santa Cruz Biotechnology, Inc.), or anti-phosphotyrosine P-Tyr-100 (Cell Signaling Technology).
Results and Discussion
Pre-B Cell Maturation Defects in Btk, SLP-65, and Double Mutant Mice.
Btk or SLP-65 single mutant mice have a partial block, while double mutant mice have an almost complete arrest at the pre-B cell stage in the BM (10–14, 18). As a result, the reduction of the numbers of mature B cells in the spleen of double mutant mice is much more drastic, when compared with Btk or SLP-65 single mutant mice (reference 18; Fig. 1 A). We have previously shown that Btk-deficient cells fail to efficiently modulate the expression of developmentally regulated cell surface markers during the transition of large cycling into small resting cytoplasmic μ+ pre-B cells (14). In particular, in Btk-deficient small pre-B cells the down-regulation of the pro-B/large pre-B stage-specific markers SLC, the sialoglycoprotein CD43, and the metallopeptidase BP-1 and the up-regulation of CD2 and CD25/interleukin-2 receptor, which are first expressed on small preB cells, are impaired (14). To analyze the effect of SLP-65 inactivation or the concomitant deficiency of SLP-65 and Btk on pre-B cell maturation, we compared the expression of these developmentally regulated markers in WT, Btk-deficient, SLP-65-deficient, and double mutant mice by flow cytometry. As MHC class II expression is also initiated at the pre-B cell stage (24), we included surface expression of MHC class II of cytoplasmic μ+ pre-B cells in the analyses.
Figure 1.
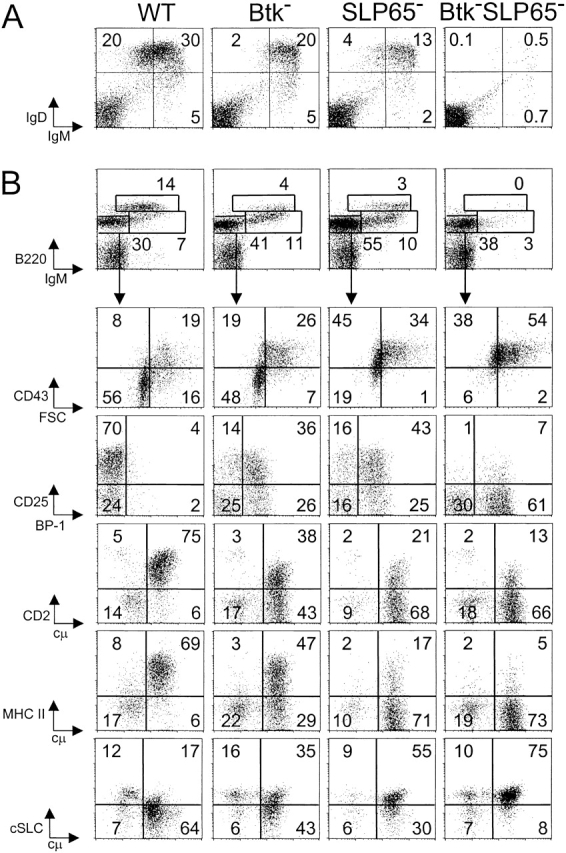
Impaired pre-B cell maturation in Btk-deficient, SLP-65-deficient, and Btk/SLP-65 double mutant mice. (A) Flow cytometric analysis of surface IgM/IgD expression on total lymphoid cells in the spleen. (B) Expression profiles of B220 and IgM on total lymphoid cells in the BM (top). The B220+IgM− pro-/pre-B cell fraction was gated and analyzed for the indicated markers (bottom). Data are displayed as dot plots and the percentages of cells within the indicated quadrants or gates are given. Data shown are representative of four mice examined within each group.
When compared with Btk-deficient cells, SLP-65−/− pre-B cells manifested a slightly more pronounced defect in the down-regulation of CD43, BP-1, and SLC and in the up-regulation of CD25, CD2, and MHC class II expression (Fig. 1 B). Consistent with the reported almost complete arrest at the pre-BCR+ stage (18), SLP-65/Btk double mutant pre-B cells had high forward scatter characteristics, were positive for CD43, BP-1, and SLC expression and showed very low surface expression of CD25, CD2, and MHCII (Fig. 1 B).
Collectively, these findings show that SLP-65 and Btk have a synergistic role in the developmental progression of large cycling into small resting cytoplasmic μ+ pre-B cells.
Deficient Differentiation of Btk and SLP-65 Mutant Pre-B Cells In Vitro.
Cytoplasmic μ+ pre-B cells undergo rapid cell division in response to IL-7 in vitro, whereby subsequent removal of IL-7 strongly induces exit from cell cycle and further differentiation into surface IgM+ B cells (25). We previously reported that Btk and SLP-65 single mutant pre-B cells manifest an enhanced proliferative response to IL-7 (14, 16). When we compared total BM cells from Btk and SLP-65 single mutants and the double mutant, these three groups of mice showed similar [3H]thymidine incorporation values after 5 d of culture in the presence of 100 U/ml IL-7 (Fig. 2 A). When total BM cell suspensions from WT mice were cultured in the presence of IL-7 for 7 d, the majority of cells consisted of B220+ cytoplasmic μ+ pre-B cells that were surface μ− or μlow, while a significant fraction (∼20–30%) performed productive κ L chain rearrangements and matured to surface IgM+IgD− or IgM+IgD+ B cell stages (Fig. 2 B, thin lines). In contrast, the IL-7 driven BM cultures from Btk and SLP-65 single or double mutant mice consisted almost exclusively of large μ+ SLC+ pre-B cells which did not express κ L chains in their cytoplasm (Fig. 2 B, thin lines).
Figure 2.


Analysis of IL-7 driven BM cultures from Btk and SLP-65 mutant mice. (A) Proliferative response to 100 U/ml IL-7, as determined by [3H]thymidine incorporation after 5 d of culture. Bars represent mean cpm and SEM of triplicate cultures. (B) Forward scatter (FSC) values and expression profiles of IgM, IgD, SLC, and cytoplasmic κ L chain of IL-7 driven BM cultures from the indicated mice. Data are displayed as histogram overlays of B220+ cells, either cultured under proliferating conditions (with 100 U/ml IL-7 for 7 d, thin lines) or under differentiating conditions (after 5 d of culture with IL-7 and subsequently without IL-7 for 2 d, bold lines). The percentages shown represent the fractions of the cells that are within the indicated marker under the two different culture conditions. (C) Percentage of surface IgM+ cells within the fraction of small FSC B220+ cells after 7 d of culture in the presence of the indicated concentrations of IL-7. Data are representative of four mice per group.
To analyze the differentiation capacity of pre-B cells, BM cells were cultured in the presence of IL-7 for 5 d and subsequently for 2 d on S17 stroma cells in the absence of IL-7. Under these conditions, WT cells acquired low FSC characteristics and ∼50% of the cells showed productive κ L chain rearrangement and differentiated into IgM+IgD+ cells. In contrast, the cultures from Btk-deficient mice contained no IgD-positive cells, a smaller fraction (∼25%) of cytoplasmic κ+ cells, and a significant proportion of SLC+ cells. The defects were more pronounced in the cultures from SLP-65−/− and Btk/SLP-65 double mutant mice, which were characterized by an inefficient exit from cell cycle and a dominance of SLC+κ− pre-B cells. In this respect, these two groups of mice were not significantly different.
Only when BM cells were cultured in the presence of limiting concentrations of IL-7 for 7 d, a difference between SLP-65 single mutant and SLP-65/Btk double mutant mice was noticed (Fig. 2 C). At low IL-7 concentrations, preB cell division was limited and cells rapidly differentiated into surface IgM+ B cells. Within the population of small noncycling B220+ cells, the percentage of surface IgM+ cells was lower in SLP-65/Btk double mutant mice, when compared with WT, Btk, or SLP-65 single mutant mice. These results indicate that, in the absence of SLP-65, Btk functions to enhance differentiation to sIgM+ B cells at low IL-7 concentrations.
Taken together, these findings show that in the presence of IL-7 Btk, SLP-65, and double mutant pre-B cells show increased proliferation and enhanced pre-BCR expression, when compared with WT pre-B cells. The ability to down-regulate SLC expression and to differentiate into IgM+ B cells upon IL-7 withdrawal, is mildly affected in Btk-deficient mice, more severely in SLP-65−/− mice, and even more so in SLP-65/Btk double mutant mice.
Btk/SLP-65 Double Mutant Mice Have a High Incidence of Pre-B Cell Lymphoma.
SLP-65−/− mice develop spontaneous pre-B cell tumors, with a frequency of <10% at 16 wk of age (16). When we followed a panel of SLP-65 single mutant mice up to 16 wk, ∼5% (3 out of 66) developed a lymphoma. Remarkably, when SLP-65/Btk double mutant mice were followed for 16 wk, a significantly enhanced frequency of tumor development was noticed, because 75% of these mice (12 out of 16) developed a pre-B cell lymphoma. In these mice external examination revealed either directly the presence of solid tumors, mainly close to the scapula, or indirectly as the mice displayed pareses of the hind limbs. In general, the mice also developed splenomegaly, and enlargement of the lymph nodes. Comparable tumors were not found in WT or Btk-deficient mice (less than 1/4,000 mice). The Kaplan-Meier tumor-free survival curves for the SLP-65 single and SLP-65/Btk double mutant mice are shown in Fig. 3 A.
Figure 3.

(A) Kaplan-Meier tumor-free survival estimates for Btk-deficient (dotted line), SLP-65−/− (thin line), and Btk-SLP-65−/− mice (bold line). Tumor-free survival in Btk-SLP-65−/− mice was significantly reduced (P < 0.0001 by log-rank) compared with SLP-65−/− mice. (B) Kaplan-Meier tumor-free survival estimates for SLP-65−/− (thin line) and BtkActSLP-65−/− mice (dotted line). Tumor-free survival in BtkActSLP-65−/− mice was significantly enhanced (P = 0.04 by log-rank) compared with SLP-65−/− mice.
These results demonstrate that deficiency of Btk strongly increases the frequency of tumor formation in SLP-65−/− mice, indicating that Btk cooperates with SLP-65 as a tumor suppressor in preB cells.
Constitutive Active BtkAct Prevents Pre-B Cell Tumor Formation in Btk/SLP-65 Double Mutant Mice.
The finding that Btk apparently cooperated with SLP-65 as a tumor suppressor prompted us to investigate whether transgenic expression of a constitutive active Btk mutant could prevent tumor development in SLP-65-deficient mice. The PH domain gain-of-function mutant E41K shows increased membrane localization and phosphorylation in quiescent cells, independent of PI3K activity and induces transformation of 3T3 fibroblasts in soft agar cultures (26, 27). This capacity is augmented by mutation of the main autophosphorylation site in the Btk SH3 domain (Y223F; reference 28). In Ramos B cells expression of E41K-Btk enhances the sustained increase in intracellular [Ca2+] after BCR cross-linking (29). Thus, the E41K mutant and the E41K-Y223F double mutant represent activated forms of Btk. When E41K-Btk was expressed at physiological levels in transgenic mice under the control of the B cell–specific CD19 promoter, B cell development was arrested at the immature B cells in the BM (probably because the E41K-Btk mutant mimics BCR occupancy by auto-antigens), while residual B cells were efficiently driven into IgM plasma cell differentiation (21). We recently generated a panel of 7 independent E41K-Btk (n = 3) or E41K-Y223F-Btk (n = 4) transgenic mouse lines, which were crossed onto the Btk null background. Expression of the two different mutants resulted in parallel phenotypes, whereby the deletion at the immature B cell stage was dose-dependent (unpublished data). From this panel we selected a low-copy E41K-Y223F-Btk transgenic mouse strain (BtkAct), in which the extent of the B cell arrest was limited, while the finding of enhanced protein tyrosine phosphorylation in splenic B cells and significantly increased serum IgM levels provided evidence for the constitutive active nature of BtkAct in vivo (see below; Fig. 4, A–C).
Figure 4.
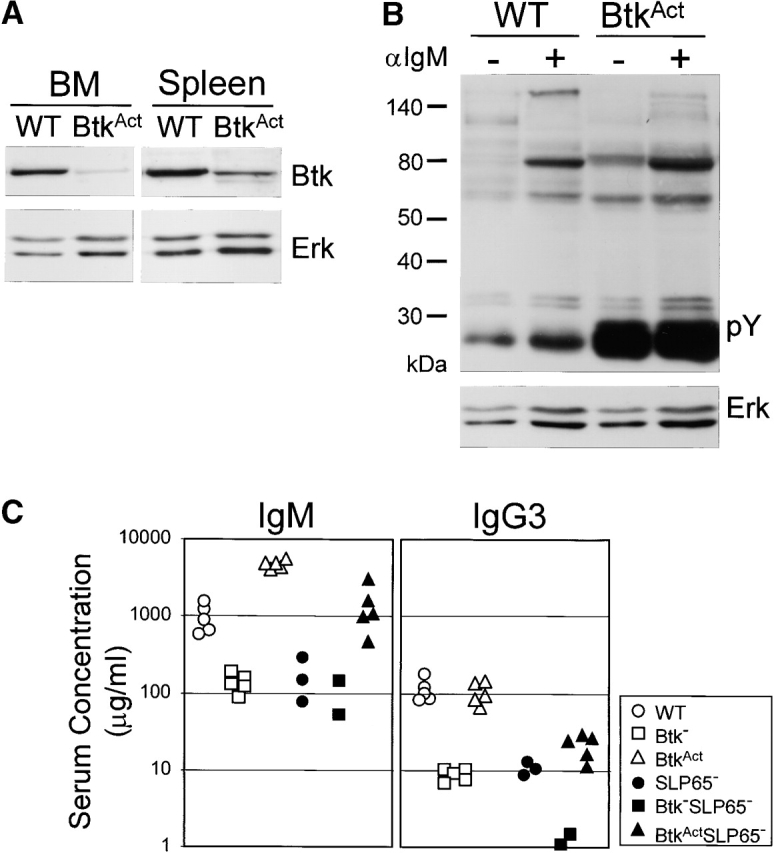

Low-level BtkAct expression can partially substitute for the absence of Btk and SLP-65. (A) Western blotting analysis of Btk expression in WT and BtkAct B cells from BM and spleen. Membrane was reblotted with anti-Erk. (B) Protein tyrosine phosphorylation in extracts of untreated and anti-IgM stimulated WT or BtkAct splenic B cells, analyzed by immunoblotting with a phosphotyrosine (pY)-specific antibody. Membrane was reblotted with anti-Erk. (C) Serum concentrations of IgM and IgG3 in the indicated mutant mouse strains. Mice were 2 mo of ages and each symbol represents an individual animal. (D) Flow cytometric analysis of surface IgM/IgD expression on total lymphoid cells in the spleen of the indicated mice. (E) Expression profiles of B220 and IgM on total lymphoid cells in the BM of the indicated mice (top). The B220+IgM- pro-/pre-B cell fractions were gated and analyzed for CD43/FSC and cytoplasmic SLC and μ H chain (bottom). Data are displayed as dot plots and the percentages of cells within the indicated quadrants or gates are given. Data shown are representative of four mice examined within each group.
BtkAct mice were crossed onto the Btk/SLP-65 double mutant background and a panel of 20 transgenic mice was followed for 8 mo. At this age, the fraction of SLP-65−/− mice that developed a preB cell lymphoma increased to 18% (12 out of 66; Fig. 3 B). In contrast, none of the BtkAct transgenic mice on the Btk/SLP-65 double mutant background developed lymphoma (Fig. 3 B). When these BtkAct mice were killed at 8 mo, no evidence for ongoing pre-B cell proliferation was found: pre-B cell numbers in the BM were not increased and splenomegaly or pre-B cell infiltrations into the spleen were absent.
These findings show that substitution of endogenous Btk by low levels of constitutively activated Btk prevents tumor formation in SLP-65–deficient mice. Therefore, we conclude that BtkAct can substitute for SLP-65 as a tumor suppressor in pre-B cells.
Low Level BtkAct Expression Can Partially Substitute for the Absence of Btk and SLP-65.
BtkAct protein expression was analyzed by Western blotting of purified B cell fractions from BM and spleen, in which Btk protein was visible as a single ∼77 kD band (Fig. 4 A). Quantification of Btk signals using Erk1/2 as a loading control showed that BtkAct expression was low (up to ∼20% of the physiological levels). This was confirmed in intracellular flow cytometry experiments by comparison of median fluorescence intensities of transgenic BtkAct and endogenous Btk (unpublished data). The analysis of unstimulated and anti-IgM stimulated splenic B cells by immunoblotting with a phosphotyrosine-specific antibody revealed that BtkAct B cells exhibit enhanced protein tyrosine phosphorylation in vivo (Fig. 4 B).
To evaluate the capacity of BtkAct to drive peripheral B cells into plasma cell differentiation, we determined serum IgM and IgG3 levels in 2-mo-old BtkAct mice, both on a Btk-deficient and on a Btk/SLP-65 double-deficient background, with nontransgenic WT, Btk−, SLP-65−/−, and Btk/SLP-65 double-deficient mice as controls. Consistent with previous reports, the serum levels of IgM and IgG3 in mice deficient for Btk or SLP-65 were significantly decreased (10–12, 30). In Btk/SLP-65 double mutant mice IgM levels equally low and IgG3 levels were even more reduced (Fig. 4 C). When BtkAct was expressed on the Btk-deficient background, the levels of IgM in the serum were increased with a factor of ∼5–10 and IgG3 levels were in the normal range, when compared with WT mice. Also the frequencies of IgM-producing cells in spleen and BM, as determined in an ELISPOT assay, were increased in these BtkAct mice (unpublished data). Expression of BtkAct completely corrected IgM levels and significantly restored IgG3 levels in Btk/SLP-65 double mutant mice (Fig. 4 C).
Flow cytometric analyses of the spleen revealed that low-level expression of BtkAct did not rescue the decrease in splenic B cell numbers in Btk-deficient mice (Fig. 4 D). On the other hand, BtkAct expression also did not result in significant deletion of peripheral B cells, in contrast to our previous findings in mice expressing physiological levels of the E41K-Btk mutant (21). Expression of BtkAct partially restored the almost complete absence of splenic B cells in Btk/SLP-65 double mutant mice, whereby their surface IgM/IgD profiles were similar to those in SLP-65 single mutant mice (see Figs. 1 A and 4 D). In the bone marrow, expression of the BtkAct transgene corrected the maturation defects of Btk-deficient cμ+ pre-B cells, i.e., the down-regulation of CD43 and SLC expression and the up-regulation of CD2, CD25, and MHC class II expression (unpublished data; shown for CD43 and SLC in Fig. 4 E). In SLP-65−/− mice, substitution of endogenous Btk by very low levels of constitutively activated Btk was associated with a reduction of the percentages of SLC+ and CD43+ cμ+ pre-B cells (Fig. 4 E).
Taken together, comparison of SLP-65−/− and BtkAct SLP-65−/− mice with respect to serum Ig concentration and the maturation of pre-B cells in vivo indicate that low-level BtkAct expression can partially substitute for the absence of SLP-65.
Characterization of Pre-B Cell Tumors.
All pre-B cell tumors characterized expressed high levels of SLC and Ig μ H chain in their cytoplasm, irrespective of the Btk genotype of the SLP-65−/− mice (Fig. 5 A). The two Btk+/− SLP-65−/− mice present in our panel of mutant mice also rapidly developed pre-B cell tumors. Because of the presence of a LacZ reporter in the targeted Btk allele, we could evaluate the X-chromosome inactivation status of the lymphoma cells (20). As a result of the process of random X-chromosome inactivation in Btk+/− heterozygous females, each X chromosome is active in about half of the pre-B cells. Similar to findings in lymphoma cells from male Btk−SLP-65−/− mice, we observed that the majority of lymphoma cells from Btk+/−SLP-65−/− mice were LacZ +. These results indicate that the Btk+/− lymphoma cells carried the disrupted Btk allele on the active X chromosome and therefore were functionally Btk-deficient (Fig. 5 B).
Figure 5.
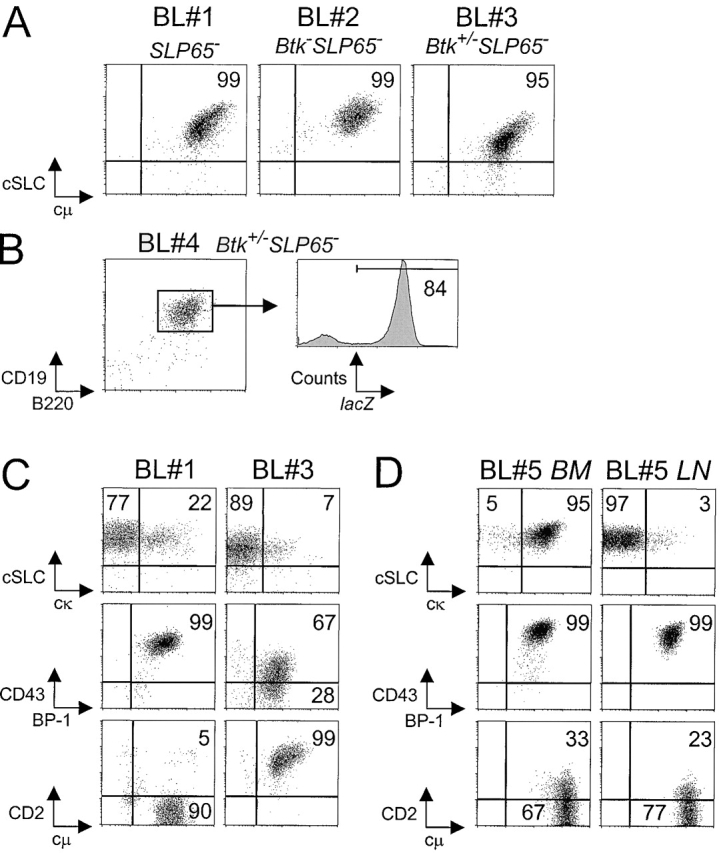
Characterization of pre-B cell tumors by flow cytometry. (A) Dot plots for cytoplasmic SLC and μ H chain in gated B220+ cells from tumor samples from the indicated mice, grown for 1 to 3 wk in the presence of IL-7. (B) Flow cytometric analysis of lacZ expression in gated CD19+B220+ pre-B lymphoma cells in a lymph node from a Btk+/−SLP-65−/− mouse. (C) Phenotype of two pre-B cell lymphoma cultures, showing variable expression of cytoplasmic κ L chain, CD43, and CD2. Cell suspensions were stained for the indicated markers in combination with B220, and the results are displayed as dot plots of gated B220+ cells. (D) Phenotype of two separate tumor cell suspensions derived from BM and mesenteric lymph node from a single mouse, which were cultured in the presence of Il-7 for 7 d.
As both Btk and SLP-65 are crucially involved in the modulation of pre-B cell surface makers and the initiation of Ig L chain gene rearrangement during the transition of large cycling into small resting cytoplasmic μ+ pre-B cells, we were interested in the phenotype of pre-B cell lymphoma cells. Most pre-B cell lymphomas contained a (minor) fraction of cells that coexpressed κ L chain and SLC in their cytoplasma and showed variable surface expression of the developmentally regulated markers, such as CD43 and CD2 (Fig. 5 C). In general, the expression of SLC and cell surface markers remained stable when the tumor cells were cultured in the presence of IL-7. Some of these cell lines became IL-7 independent or lost expression of the IL-7R, as detectable by flow cytometry (unpublished data). When we cultured pre-B lymphoma cells derived from different tissues from a single Btk/SLP-65 double mutant mouse in vitro, we found that a BM-derived cell culture mainly consisted of κ L chain positive cells, while a lymph node–derived cell culture was essentially κ L chain negative. Nevertheless, both cultures expressed high levels of CD43 and low levels of CD2 (Fig. 5 D).
Collectively, these results show that in the tumors the ordered differential expression of stage-specific pre-B cell markers (CD43, SLC, and BP-1 on large cycling pre-B cells but CD2, CD25, and MHCII on small resting pre-B cells) is lost. Furthermore, the finding of variable percentages of κ L chain expressing cells in the pre-B cell lymphomas suggests that in the absence of SLP-65 Ig L chain locus rearrangements are initiated in large cycling pre-BCR+ cells.
Btk and SLP-65 Cooperate as Tumor Suppressors.
By crossing SLP-65−/− mice with mice that were either Btk-deficient or expressed a constitutive active form of Btk we have demonstrated in this report that (a) Btk and SLP-65 have synergistic roles in the developmental progression of large cycling into small resting pre-B cells, (b) the concomitant deficiency of Btk significantly enhanced tumor formation in SLP-65−/− mice, and (c) expression of BtkAct prevents tumor formation in Btk/SLP-65 double mutant mice. We therefore conclude that BtkAct compensates for loss of SLP-65 tumor suppressor function, either by promoting pre-B cell differentiation or limiting pre-B cell expansion independent of SLP-65.
In contrast to SLP-65−/− mice, Btk/SLP-65 double mutant mice exhibit an almost complete arrest at the large cycling pre-BCR+ pre-B cell stage. Thus, it is conceivable that the increased frequency of malignant transformation in Btk/SLP-65 double mutant mice reflects the increased pool size of proliferating pre-B cells with a reduced ability to progress into CD43− small resting pre-B cells, when compared with SLP-65 single mutant mice. Alternatively, the absence of Btk may alter the proliferative capacities of SLP-65−/− pre-B cells in a signaling pathway different from the pre-BCR. In this respect, the IL-7R pathway would be an attractive candidate, as Btk-deficient RAG-1−/− pro-B cells were shown to have an increased responsiveness to IL-7, when compared with Btk+ RAG-1−/− pro-B cells (14). A third possibility would be that the concomitant absence of Btk and SLP-65 precludes the efficient down-regulation of RAG enzyme levels upon pre-BCR signaling (1, 2). The finding of SLC and κ L chain coexpression in the pre-B cell tumors suggests that Ig L chain rearrangements have occurred in large pre-BCR+ cycling pre-B cells. This implies that the V(D)J recombination machinery is active in SLP-65−/− cycling pre-B cells, which may cause DNA damage contributing to the induction of secondary mutations required for malignant transformation. Further experiments comparing Btk+ and Btk− SLP-65-deficient preB cell tumor cells should identify the nature of the cooperation between Btk and SLP-65 as tumor suppressors. In this context, we found that Btk expression levels vary considerably between different pre-B cell lines established from SLP-65−/− mice. Btk was not detectable in 1 out of 6 SLP-65−/− pre-B cell lines by Western blotting, suggesting that Btk could be a target for secondary mutations in SLP-65−/− mice (unpublished data). As SLP-65 is implicated in childhood pre-B ALL (19), the finding of cooperation between Btk and SLP-65 may also have important implications for our understanding of the etiology of this malignancy in humans.
Acknowledgments
We thank H. Dronk, H. Diepstraten, and P. Molenbeek from the Erasmus MC animal facility and M. van der Zee for their assistance.
Supported by Netherlands Organization for Scientific Research Grants and the Deutsche Forschungsgesellschaft (SFB 620).
Footnotes
Abbreviations used in this paper: BCR, B cell receptor; Btk, Bruton's tyrosine kinase; SLC, surrogate light chain.
References
- 1.Meffre, E., R. Casellas, and M.C. Nussenzweig. 2000. Antibody regulation of B cell development. Nat. Immunol. 1:379–385. [DOI] [PubMed] [Google Scholar]
- 2.Melchers, F., E. ten Boekel, T. Seidl, X.C. Kong, T. Yamagami, K. Onishi, T. Shimizu, A.G. Rolink, and J. Andersson. 2000. Repertoire selection by pre-B-cell receptors and B-cell receptors, and genetic control of B-cell development from immature to mature B cells. Immunol. Rev. 175:33–46. [PubMed] [Google Scholar]
- 3.Conley, M.E., J. Rohrer, L. Rapalus, E.C. Boylin, and Y. Minegishi. 2000. Defects in early B-cell development: comparing the consequences of abnormalities in pre-BCR signaling in the human and the mouse. Immunol. Rev. 178:75–90. [DOI] [PubMed] [Google Scholar]
- 4.Shaffer, A.L., and M.S. Schlissel. 1997. A truncated heavy chain protein relieves the requirement for surrogate light chains in early B cell development. J. Immunol. 159:1265–1275. [PubMed] [Google Scholar]
- 5.Rolink, A.G., T. Winkler, F. Melchers, and J. Andersson. 2000. Precursor B cell receptor-dependent B cell proliferation and differentiation does not require the bone marrow or fetal liver environment. J. Exp. Med. 191:23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guo, B., R.M. Kato, M. Garcia-Lloret, M.I. Wahl, and D.J. Rawlings. 2000. Engagement of the human pre-B cell receptor generates a lipid raft-dependent calcium signaling complex. Immunity. 13:243–253. [DOI] [PubMed] [Google Scholar]
- 7.Kouro, T., K. Nagata, S. Takaki, S. Nisitani, M. Hirano, M.I. Wahl, O.N. Witte, H. Karasuyama, and K. Takatsu. 2001. Bruton's tyrosine kinase is required for signaling the CD79b-mediated pro-B to pre-B cell transition. Int. Immunol. 13:485–493. [DOI] [PubMed] [Google Scholar]
- 8.Schebesta, M., P.L. Pfeffer, and M. Busslinger. 2002. Control of pre-BCR signaling by Pax5-dependent activation of the BLNK gene. Immunity. 17:473–485. [DOI] [PubMed] [Google Scholar]
- 9.Fruman, D.A., A.B. Satterthwaite, and O.N. Witte. 2000. Xid-like phenotypes: a B cell signalosome takes shape. Immunity. 13:1–3. [DOI] [PubMed] [Google Scholar]
- 10.Jumaa, H., B. Wollscheid, M. Mitterer, J. Wienands, M. Reth, and P.J. Nielsen. 1999. Abnormal development and function of B lymphocytes in mice deficient for the signaling adaptor protein SLP-65. Immunity. 11:547–554. [DOI] [PubMed] [Google Scholar]
- 11.Pappu, R., A.M. Cheng, B. Li, Q. Gong, C. Chiu, N. Griffin, M. White, B.P. Sleckman, and A.C. Chan. 1999. Requirement for B cell linker protein (BLNK) in B cell development. Science. 286:1949–1954. [DOI] [PubMed] [Google Scholar]
- 12.Hayashi, K., R. Nittono, N. Okamoto, S. Tsuji, Y. Hara, R. Goitsuka, and D. Kitamura. 2000. The B cell-restricted adaptor BASH is required for normal development and antigen receptor-mediated activation of B cells. Proc. Natl. Acad. Sci. USA. 97:2755–2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu, S., J.E. Tan, E.P. Wong, A. Manickam, S. Ponniah, and K.P. Lam. 2000. B cell development and activation defects resulting in xid-like immunodeficiency in BLNK/SLP-65-deficient mice. Int. Immunol. 12:397–404. [DOI] [PubMed] [Google Scholar]
- 14.Middendorp, S., G.M. Dingjan, and R.W. Hendriks. 2002. Impaired precursor B cell differentiation in Bruton's tyrosine kinase-deficient mice. J. Immunol. 168:2695–2703. [DOI] [PubMed] [Google Scholar]
- 15.Xu, S., and K.P. Lam. 2002. Delayed cellular maturation and decreased immunoglobulin kappa light chain production in immature B lymphocytes lacking B cell linker protein. J. Exp. Med. 196:197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Flemming, A., T. Brummer, M. Reth, and H. Jumaa. 2003. The adaptor protein SLP-65 acts as a tumor suppressor that limits pre-B cell expansion. Nat. Immunol. 4:38–43. [DOI] [PubMed] [Google Scholar]
- 17.Hayashi, S., P.L. Witte, and P.W. Kincade. 1989. The xid mutation affects hemopoiesis in long term cultures of murine bone marrow. J. Immunol. 142:444–451. [PubMed] [Google Scholar]
- 18.Jumaa, H., M. Mitterer, M. Reth, and P.J. Nielsen. 2001. The absence of SLP65 and Btk blocks B cell development at the preB cell receptor-positive stage. Eur. J. Immunol. 31:2164–2169. [DOI] [PubMed] [Google Scholar]
- 19.Jumaa, H., L. Bossaller, K. Portugal, B. Storch, M. Lotz, A. Flemming, M. Schrappe, V. Postila, P. Riikonen, J. Pelkonen, et al. 2003. Deficiency of the SLP-65 in pre-B-cell acute lymphoblastic leukemia. Nature. 423:452–456. [DOI] [PubMed] [Google Scholar]
- 20.Hendriks, R.W., M.F. de Bruijn, A. Maas, G.M. Dingjan, A. Karis, and F. Grosveld. 1996. Inactivation of Btk by insertion of lacZ reveals defects in B cell development only past the pre-B cell stage. EMBO J. 15:4862–4872. [PMC free article] [PubMed] [Google Scholar]
- 21.Maas, A., G.M. Dingjan, F. Grosveld, and R.W. Hendriks. 1999. Early arrest in B cell development in transgenic mice that express the E41K Bruton's tyrosine kinase mutant under the control of the CD19 promoter region. J. Immunol. 162:6526–6533. [PubMed] [Google Scholar]
- 22.Dingjan, G.M., S. Middendorp, K. Dahlenborg, A. Maas, F. Grosveld, and R.W. Hendriks. 2001. Bruton's tyrosine kinase regulates the activation of gene rearrangements at the lambda light chain locus in precursor B cells in the mouse. J. Exp. Med. 193:1169–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karasuyama, H., A. Rolink, Y. Shinkai, F. Young, F.W. Alt, and F. Melchers. 1994. The expression of Vpre-B/lambda 5 surrogate light chain in early bone marrow precursor B cells of normal and B cell-deficient mutant mice. Cell. 77:133–143. [DOI] [PubMed] [Google Scholar]
- 24.Tarlinton, D. 1994. B-cell differentiation in the bone marrow and the periphery. Immunol. Rev. 137:203–229. [DOI] [PubMed] [Google Scholar]
- 25.Rolink, A., U. Grawunder, D. Haasner, A. Strasser, and F. Melchers. 1993. Immature surface Ig+ B cells can continue to rearrange kappa and lambda L chain gene loci. J. Exp. Med. 178:1263–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li, T., S. Tsukada, A. Satterthwaite, M.H. Havlik, H. Park, K. Takatsu, and O.N. Witte. 1995. Activation of Bruton's tyrosine kinase (BTK) by a point mutation in its pleckstrin homology (PH) domain. Immunity. 2:451–460. [DOI] [PubMed] [Google Scholar]
- 27.Varnai, P., K.I. Rother, and T. Balla. 1999. Phosphatidylinositol 3-kinase-dependent membrane association of the Bruton's tyrosine kinase pleckstrin homology domain visualized in single living cells. J. Biol. Chem. 274:10983–10989. [DOI] [PubMed] [Google Scholar]
- 28.Park, H., M.I. Wahl, D.E. Afar, C.W. Turck, D.J. Rawlings, C. Tam, A.M. Scharenberg, J.P. Kinet, and O.N. Witte. 1996. Regulation of Btk function by a major autophosphorylation site within the SH3 domain. Immunity. 4:515–525. [DOI] [PubMed] [Google Scholar]
- 29.Fluckiger, A.C., Z. Li, R.M. Kato, M.I. Wahl, H.D. Ochs, R. Longnecker, J.P. Kinet, O.N. Witte, A.M. Scharenberg, and D.J. Rawlings. 1998. Btk/Tec kinases regulate sustained increases in intracellular Ca2+ following B-cell receptor activation. EMBO J. 17:1973–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khan, W.N., F.W. Alt, R.M. Gerstein, B.A. Malynn, I. Larsson, G. Rathbun, L. Davidson, S. Muller, A.B. Kantor, L.A. Herzenberg, et al. 1995. Defective B cell development and function in Btk-deficient mice. Immunity. 3:283–299. [DOI] [PubMed] [Google Scholar]