

Abstract
The predisposition of nonobese diabetic (NOD) mice to develop autoimmunity reflects deficiencies in both peripheral and central tolerance. Several defects have been described in these mice, among which aberrant antigen-presenting cell function and peroxynitrite formation. Prediabetes and diabetes in NOD mice have been targeted with different outcomes by a variety of immunotherapies, including interferon (IFN)-γ. This cytokine may be instrumental in specific forms of tolerance by virtue of its ability to activate immunosuppressive tryptophan catabolism. Here, we provide evidence that IFN-γ fails to induce tolerizing properties in dendritic cells from highly susceptible female mice early in prediabetes. This effect is associated with impaired tryptophan catabolism, is related to transient blockade of the Stat1 pathway of intracellular signaling by IFN-γ, and is caused by peroxynitrite production. However, the use of a peroxynitrite inhibitor can rescue tryptophan catabolism and tolerance in those mice. This is the first report of an experimental autoimmune disease in which defective tolerance is causally linked to impaired tryptophan catabolism.
Keywords: autoimmunity; indoleamine 2,3-dioxygenase; dendritic cells; tolerance; NOD mice
Introduction
The nonobese diabetic (NOD)* strain of mice has become a prototypic model of autoimmune disease (1, 2). A large proportion of female mice generally die of type 1 diabetes, reflecting the onset of severe insulitis at ∼4 wk of age and the T cell–mediated destruction of pancreatic β cells. The predisposition of NOD mice to develop autoimmunity is presumably due to defects in both peripheral and central tolerance mechanisms (3). Several abnormalities have been described in these mice, including aberrant APC function (4), IL-12 production (5), and peroxynitrite formation (6).
Among the immunotherapies that may oppose the onset or progression of autoimmune diabetes is the use of proinflammatory cytokines, such as TNF-α and IFN-γ. There are, however, remarkable differences in the modulation of susceptibility to diabetes by immunotherapeutic maneuvers, depending on timing of intervention. For example, TNF-α increases T cell autoreactivity to islet cells and exacerbates diabetes when administered from birth to 3 wk of age. In contrast, the administration of TNF-α to adult NOD mice will block progression of the autoreactive process (7). IFN-γ is protective when used in the form of recombinant cytokine administered in vivo to diabetic mice (8) or as a means of conditioning DCs in vitro before transfer into prediabetic recipients (9).
Although the protective effect of IFN-γ in NOD mice may appear paradoxical (8), we have proposed a model of transplantation tolerance that might help to explain the therapeutic efficacy of the recombinant cytokine in both transplantation and autoimmunity (10). According to this model, IFN-γ acts on tolerogenic DCs to activate the expression of the enzyme indoleamine 2,3-dioxygenase (IDO). This leads to the induction of immunosuppressive tryptophan catabolism and to the onset of specific tolerance (11, 12).
In the present study, we have investigated the possible interplay between the onset of specific tolerance and tryptophan catabolism in NOD mice. We provide evidence that IFN-γ is selectively unable to induce tolerizing properties in DCs from female mice early in prediabetes. This effect can be traced to peroxynitrite-induced blockade of IFN-γ signaling through Stat1, and results in impaired transcriptional activation of the IDO gene. Thus, in this model of autoimmune disease, an age-related defect is found in susceptible mice that affects DC function, tryptophan catabolism, and the induction of specific tolerance.
Materials and Methods
Mice and Reagents.
Female and male NOD/LtJ mice, 4 and 8 wk of age, were purchased from The Jackson Laboratory. The mice were housed and fed under specific pathogen-free conditions. Autoimmune diabetes develops in ∼80% of NOD/LtJ female mice by 1 yr of age. Female DBA/2J and C57BL/6 mice were also purchased from The Jackson Laboratory. The source and characteristics of the murine rIL-12 and murine rIFN-γ were described previously (13). Endotoxin was removed from all solutions containing recombinant cytokines with Detoxy-gel (Pierce Chemical Co.), resulting in endotoxin contamination below the detection limit (0.01 endotoxin U/ml) of the assay (Coatest Endotoxin; Chromogenix). NRP-A7 (KYNKANAFL), a synthetic peptide that acts as a mimotope for autoimmune diabetes in NOD mice (14, 15), was synthesized and purified as described (13). The enzyme inhibitors 1-methyl-dl-tryptophan (1-MT) and guanidinoethyl disulphide (GED) were purchased from Sigma-Aldrich. All in vivo studies were done in compliance with National and Perugia University Animal Care and Use Committee guidelines.
DC Preparations and Treatments and Immunization.
Splenic DCs were prepared from pools of at least five mice and fractionated according to CD11c/CD8α expression using positive selection columns in combination with CD11c and CD8α microbeads® and in the presence of EDTA to disrupt DC-T cell complexes, as described previously (13). The recovered cells were >98% CD11c+, >98% B7–2+, <0.1% CD3+, <0.5% B220+. In NOD mice of either sex, DCs consisted of 80–85% CD8− and 15–20% CD8+ cells. After cell fractionation, the recovered CD8− cells typically contained <0.5% contaminating CD8+ DCs, whereas the CD8+ fraction was made up of >95% CD8+ DCs. For cytokine activation, DCs were subjected to overnight exposure to 100 ng/ml rIL-12 (CD8− DCs) or 200 U/ml IFN-γ (CD8+ DCs) in the presence or absence of 2 μM 1-MT or 250 μM GED. For immunization, cells were washed between and after incubations before peptide loading (5 μM, 2 h at 37°C), irradiation, and intravenous injection into recipient hosts. Three × 105 CD8− DCs were injected either alone or in combination with 9 × 103 CD8+ DCs.
Skin Test Assay.
A skin test assay was used for measuring class I–restricted delayed-type hypersensitivity responses to synthetic peptides as previously described (16, 17), using 4-wk-old NOD female mice as recipients. Results were expressed as the increase in footpad weight of peptide-injected footpads over that of vehicle-injected counterparts. Data are the mean ± SD for at least six mice per group. The statistical analysis was performed using Student's paired t test by comparing the mean weight of experimental footpads with that of control counterparts.
Kynurenine Assay.
IDO functional activity was measured in vitro in terms of ability of DCs to metabolize tryptophan to kynurenine, whose concentrations were measured by HPLC as described (18).
Western Blot Analyses.
IDO expression was investigated as described (10, 18) using a specific Ab in CD8+ DCs, either untreated or exposed to IFN-γ in the presence or absence of GED. After incubation with medium or IFN-γ (200 U/ml for 18 h), cells were recovered and lysed in buffer containing 1% Nonidet P-40. After SDS-PAGE resolution, immunoblotting was performed with rabbit polyclonal IDO-specific antibody. Membranes were blocked in Tris-buffered saline containing 0.05% Tween 20, 5% nonfat dried milk, and 1% BSA, and incubated sequentially with the antibody (1:3,000) and HRP-conjugated anti–rabbit IgG (1:5,000). Controls consisted of IDO-expressing MC24 transfectants and mock-transfected MC22 cells. On studying Stat1 phosphorylation, CD8+ DCs were exposed overnight to 250 μM GED and then treated with IFN-γ (200 U/ml) for 10 min. After SDS-PAGE resolution, immunoblotting was performed with anti-Stat1 Abs. Membranes were blocked in Tris-buffered saline containing 0.05% Tween 20, 5% nonfat dried milk, and 1% BSA, and incubated sequentially with anti-phosphoStat1 (1:1,000; Cell Signaling Technology) and anti-Stat1 (1:1,000; Cell Signaling Technology), followed by HRP-conjugated anti-rabbit IgG (1:5,000).
Nitrotyrosine ELISA.
The assay was performed using a Nitrotyrosine ELISA kit (HyCult biotechnology b. v.) according to manufacturer's instructions. Briefly, ELISA plates coated with nitrotyrosine Ab were incubated with nitrated proteins as antigen (100 μl/well) for 1 h. The plates were then incubated with biotinylated Ab to nitrotyrosine in protein-stabilized buffer. Subsequently, plates were incubated with streptavidin peroxydase conjugate followed by specific substrate. The kit has a minimum detection level of 2 nM and a measurable concentration range of 2 to 1,500 nM.
Results
Failure of IFN-γ and CD8+ DCs from NOD Female Mice to Initiate IDO-dependent Tolerance in Prediabetes.
NRP-A7 is a synthetic nonapeptide that elicits the proliferation, cytokine secretion, differentiation, and cytotoxicity of a diabetogenic H-2Kd–restricted CD8+ T cell specificity that uses a TCRα rearrangement frequently expressed by CD8+ T cells propagated from the earliest insulitic lesions of NOD mice (14, 15). Cell populations in the spleens of conventional strains of mice contain variable proportions of mature CD8− and CD8+ DCs that mediate the respective immunogenic and tolerogenic presentation of NRP-A7. Upon transfer into recipient hosts, peptide-loaded CD8− DCs initiate immunity, and CD8+ DCs initiate anergy, when antigen-specific skin test reactivity is measured at 2 wk after cell transfer. The addition of as few as 3% CD8+ DCs to a population of CD8− DCs inhibits priming by the latter cells in this model of class I–restricted reactivity to the synthetic peptide (13, 17). We examined CD8− and CD8+ DCs from NOD mice for their patterns of presentation of NRP-A7 in vivo. Female mice of two different ages, 4 and 8 wk, were used as a source of splenic DCs that were fractionated, loaded with the peptide, and injected into recipient hosts to be assayed at 2 wk for NRP-A7-specific skin test reactivity. CD8− DCs were injected either alone or in combination with 3% CD8+ DCs, and each subset was used either as such or after exposure to IL-12 (for CD8− DCs) or IFN-γ (for CD8+ DCs; Fig. 1). Presentation by CD8− DCs resulted in effective priming, which effect was negated by the copresence of CD8+ DCs in the cell transfer. Similar to what observed in conventional mice (16), the suppressive effect of IFN-γ prevailed over the adjuvant activity of IL-12 acting on CD8− DCs when 8-wk-old mice were used as a source of DCs. The effect of IFN-γ was dependent upon effective tryptophan catabolism, as it was ablated by the addition of the IDO enzyme inhibitor, 1-MT, during CD8+ DC exposure to the cytokine. In contrast, IFN-γ was completely unable to induce suppressive properties in CD8+ DCs from 4-wk-old mice and thus overcome the stimulatory activity of IL-12. In experiments not reported here using male mice, we found that IFN-γ acted equally well in CD8+ DCs from donors of 4 or 8 wk of age. Thus, CD8+ DCs from early prediabetic female mice are characterized by specific unresponsiveness to modulation of activity by IFN-γ and by inability to counter the adjuvant activity of IL-12 acting on CD8− DCs.
Figure 1.

CD8+ DCs from early prediabetic NOD female mice are not activated by IFN-γ to suppress NRP-A7 priming by CD8− DCs. Induction of skin test reactivity to NRP-A7 was studied in 4-wk-old hosts transferred with DC subtypes with or without cytokine treatment. DCs from 8- or 4-wk-old mice were fractionated according to CD8 expression and were used as such (CD8−, CD8+) or after treatment with IL-12 (CD8−/IL-12) or IFN-γ (CD8+/IFN-γ). After peptide pulsing, the different fractions were injected either singly or in combination (indicated). Experimental groups included the use of CD8+ DCs treated with 2 μM 1-MT during exposure to IFN-γ. (*) P < 0.001, experimental versus control footpads. ND, not done. One experiment is reported representative of five.
Failure of IFN-γ to Induce Stat1-dependent Activation of IDO in Early Prediabetic NOD Female Mice.
The tolerogenic properties of CD8+ DCs are dependent on the activation of immunosuppressive tryptophan catabolism, which may affect the proliferation and survival of NRP-A7-specific T cells (13, 17). To gain insight into the mechanism underlying IFN-γ unresponsiveness in CD8+ DCs from 4-wk-old female mice, we measured IDO functional activity in terms of ability to metabolize tryptophan to kynurenine in vitro (Fig. 2 A). On comparing female and male mice of 4 or 8 wk of age, we found a selective inability of IFN-γ to induce tryptophan catabolism in female mice at 4 wk. Both NOD male mice and control C57BL/6 female mice were susceptible to the enhancing effect of IFN-γ at 4 and 8 wk. To investigate whether the defective IDO expression of female mice at 4 wk was due to inhibition of enzyme functional activity or to impaired gene transcription, we analyzed IDO expression by Western blot using rabbit polyclonal IDO-specific antibody (Fig. 2 B). Considerable expression of the enzyme protein was induced by activation with IFN-γ in CD8+ DCs from 4- or 8-wk-old NOD male mice as well as DBA/2 controls. In NOD females, IDO expression was induced by IFN-γ at 8 but not 4 wk of age. Thus, it appeared that IFN-γ would not cause IDO expression in 4-wk-old female mice. This could be due to a specific defect in the IFN-γ signaling mechanism regulating IDO expression in CD8+ DCs. Because Stat1 phosphorylation is required for IFN-γ-induced transcription of the IDO gene (10), we measured Stat1 phosphorylation in female versus male mice at 4 wk (Fig. 2 C). In contrast to NOD male mice and C57BL/6 controls, no Stat1 phosphorylation was observed in NOD females in response to IFN-γ. Thus, the failure of IFN-γ to induce IDO expression in CD8+ DCs from early prediabetic female mice appeared to be associated with defective phosphorylation of Stat1.
Figure 2.



Failure of IFN-γ to induce IDO expression and activity in early prediabetic NOD female mice. (A) Functional IDO activity in response to IFN-γ was measured in vitro in terms of the ability to metabolize tryptophan to kynurenine with the use of DCs recovered from NOD female and male mice of two different ages (4 or 8 wk) or control C57BL/6 female mice. Kynurenine levels in supernatants were measured by HPLC, and results are the mean ± SD of triplicate samples in one of three experiments. (B) IDO expression in DCs was assessed by Western blot. DCs were recovered from NOD female or male mice or control DBA/2 females and treated overnight with IFN-γ, and IDO expression was investigated with an IDO-specific antibody. The positive control consisted of IDO-expressing MC24 transfectants and the negative control consisted of mock-transfected MC22 cells. Loading controls (not shown in the figure) consisted of samples reprobed with β-actin-specific antibody. One experiments of three. (C) Stat1 phosphorylation in response to IFN-γ was studied in NOD female and male mice at 4 wk and in age-matched control C57BL/6 female mice. DC lysates were resolved on SDS-PAGE and immunoblotted with anti-phosphoStat1 and then reblotted with anti-Stat1, reblots showing equal loading of Stat1 in each set of lanes. One of three experiments with similar results.
Induction of Peroxynitrite by IFN-γ and Antagonistic Effect of GED on Peroxynitrite-induced Blockade of IFN-γ Signaling.
In NOD mice, activated macrophages produce high amounts of nitric oxide (NO), which is thought to contribute to the destructive phase of insulitis (19, 20). There is evidence that tyrosine nitration of Stat1 by endogenous NO may contribute to impaired response to IFN-γ in activated macrophages. This effect is negated by inhibition of NO synthase activity by concomitant treatment with l-NMMA, a competitive inhibitor of the enzyme (21). To investigate the possible involvement of NO in the defective Stat1 phosphorylation observed in CD8+ DCs from NOD females in response to IFN-γ, we exposed these cells to the cytokine in the presence of l-NMMA under conditions known to restore macrophage responsiveness to IFN-γ (21). Using an experimental setting as described above, we did not detect any effect of l-NMMA on Stat1 phosphorylation, IDO expression by Western blot analysis, or IDO functional activity as induced by IFN-γ (unpublished data). Peroxynitrite is a highly reactive oxidant resulting from the interaction of NO with superoxide, and is known to be produced in acutely diabetic NOD mice (6). Peroxynitrite may impair cell signaling via nitration of tyrosine residues (22). An inhibitor of NO synthase and scavenger of peroxynitrite (i.e., GED) prevents diabetes development in NOD mice (6). To investigate the possible production of peroxynitrites by DCs from NOD mice, we assayed IFN-γ–induced production of nitrotyrosine in CD8+ DCs from 4-wk-old female and male mice as well as control DBA/2 animals. NOD female mice were also assayed at 8 wk (Fig. 3). No nitrotyrosine formation was observed in cell supernatants or lysates of DCs unexposed to IFN-γ regardless of donor type (data not depicted). In contrast, IFN-γ induced high levels of peroxynitrite selectively in DCs from NOD female mice at 4 wk. In addition, GED prevented IFN-γ from inducing nitrotyrosine formation in these cells. In parallel, we examined the effect of GED on IFN-γ–induced Stat1 phosphorylation and IDO expression and function (Fig. 4). GED restored Stat1 phosphorylation and IDO protein expression (Fig. 4 A) as well as kynurenine production (Fig. 4 B) in CD8+ DCs from 4-wk-old NOD female mice treated with IFN-γ. Because NO is a precursor of peroxynitrite, the lack of activity of l-NMMA is not easily explained but could be related to the different relative potencies of l-NMMA and GED in inhibiting enzyme activity (21). Overall, these data demonstrate that CD8+ DCs from NOD female mice early in prediabetes produce peroxynitrite in response to IFN-γ. This oxidant species impairs the intracellular signal transduction pathways that lead to Stat1 phosphorylation and IDO expression in response to IFN-γ.
Figure 3.

Induction of peroxynitrite by IFN-γ in CD8+ DCs from early prediabetic NOD female mice. CD8+ DCs were recovered from NOD female and male mice at 4 wk or from age-matched control DBA/2 female mice to be incubated overnight with IFN-γ. NOD female mice were also assayed at 8 wk. Nitrotyrosine in cell supernatants or lysates was assayed by sandwich ELISA, the lower detection limit of the assay being 2 nM. Data are means ± SD of replicate samples in one of three experiments.
Figure 4.

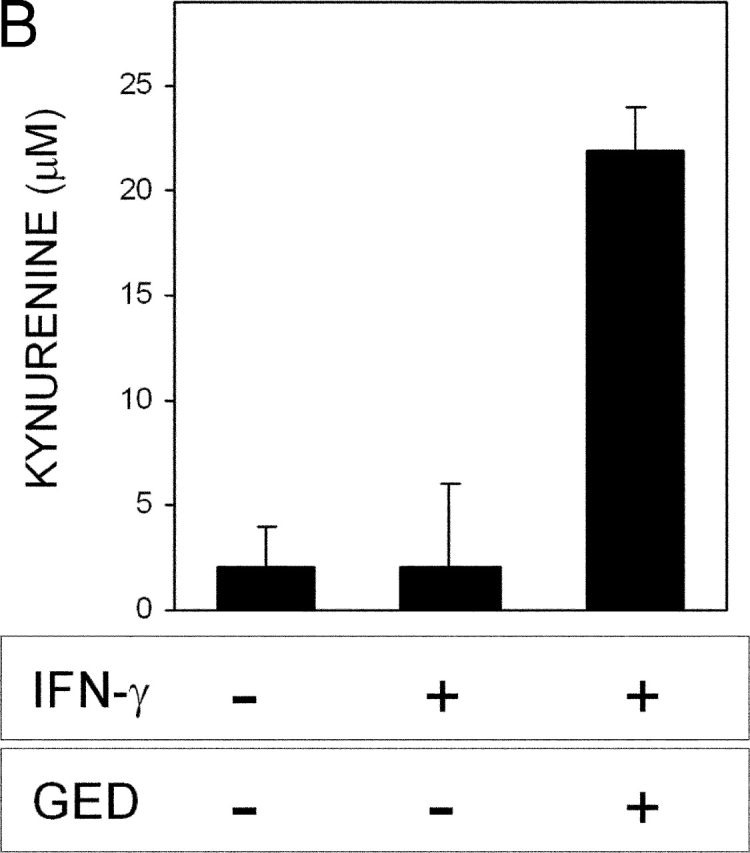
Ability of GED to restore IFN-γ responsiveness in CD8+ DCs from early prediabetic NOD female mice. (A) GED was examined for ability to restore IDO expression and Stat1 phosphorylation in response to IFN-γ. CD8+ DCs from 4-wk-old NOD female mice were treated overnight with IFN-γ and assayed for IDO expression or Stat1 phosphorylation. One experiment representative of three. (B) GED was also studied for ability to restore kynurenine production by CD8+ DCs from the same donors. One experiment representative of three.
Ability of GED to Rescue IDO-dependent Tolerance Induction in CD8+ DCs from Early Prediabetic NOD Female Mice.
Because of the ability of GED to restore IFN-γ responsiveness in vitro in CD8+ DCs from 4-wk-old NOD female mice, we wanted to investigate the effect of GED on the in vivo ability of the latter cells to modulate priming to NRP-A7 by CD8− DCs. We adopted an experimental model analogous to that of Fig. 1, in which a combination of CD8− and CD8+ DCs was injected into NOD mice to be assayed at 2 wk for skin test reactivity to the eliciting peptide. DCs from 4-wk-old NOD female mice were fractionated, treated with cytokines, loaded with the peptide, and injected into recipient hosts. IL-12 was used to enhance the priming ability of CD8− DCs and IFN-γ to induce tolerizing properties in CD8+ DCs, as illustrated above. Groups of CD8+ DCs were coexposed to 1-MT and/or GED during IFN-γ activation. As expected, IFN-γ treatment alone of CD8+ DCs did not enable these cells to prevent host priming to NRP-A7 (Fig. 5). However, the copresence of GED during IFN-γ activation rescued the ability of CD8+ DCs to suppress priming. The inhibitory effect was dependent on tryptophan catabolism as it was negated by the addition of 1-MT to IFN-γ and GED. These data demonstrate a role for peroxynitrite inhibition of tryptophan catabolism in the poor tolerogenic ability of DCs from NOD female mice in early prediabetes.
Figure 5.

Ability of GED to rescue the tolerizing properties of CD8+ DCs from early prediabetic NOD female mice. Combinations of CD8− and CD8+ DCs were loaded with NRP-A7 and injected into recipient mice to be assayed at 2 wk for skin test reactivity to the eliciting peptide. CD8− DCs were used as such or after activation with IL-12, whereas CD8+ DCs were exposed to IFN-γ. Groups of CD8+ DCs were coexposed in vitro to 1-MT and/or GED before peptide loading. (*) P < 0.001, experimental versus control footpads. One experiment is reported representative of three.
Discussion
Although IFN-γ has been implicated in the induction of tolerance in both autoimmunity (23) and transplantation (24), there is no clear evidence to date that IDO-based immunoregulation may be operative in the control of autoimmunity. In contrast, there is evidence in pregnancy (25) and transplantation (10) that, by locally depleting tryptophan and increasing apoptotic kynurenines, IDO expressed by DCs greatly affects T cell proliferation (26) and survival (27). IDO induction in DCs could be a common mechanism of deletional tolerance driven by regulatory T cells (12).
The NOD strain of mice has become a prototypic model of autoimmune disease (1, 2). These mice generally die from type 1 diabetes, reflecting the T cell–mediated destruction of pancreatic β cells, but also develop generalized autoimmune disease affecting multiple organs. There is evidence that disease susceptibility in NOD mice reflects a defect in peripheral and central tolerance (3), although the events linked to defective β cell tolerance within the T cell compartment remain largely ill defined (28). These events, however, are clearly influenced by multiple genetic and environmental factors (29). In this paper, we investigated whether impaired tryptophan catabolism might be a factor contributing to the breakdown of peripheral immunoregulation and subsequent development of autoreactivity in NOD mice.
Aberrant IL-12 production may predispose NOD mice to autoimmunity (5). This could be reflected in our model by the fact that the recombinant cytokine enhanced priming by CD8− DCs to NRP-A7, a peptide mimotope recognized by diabetogenic T cells in those mice (14, 15). However, in conventional strains of mice, the effect of IFN-γ acting on tolerogenic CD8+ DCs will prevail over the adjuvant properties of IL-12 acting on CD8− DCs (13, 16, 17). Under conditions of host priming with peptide-pulsed CD8− DCs whether or not activated by IL-12, not only will the copresence of IFN-γ–activated CD8+ DCs inhibit priming by the other subset, but it will also initiate a state of persistent unresponsiveness that has the characteristics of deletional tolerance. This is demonstrated by the fact that mice exposed to CD8+ DCs treated with IFN-γ will fail to develop specific immunity at a later time when treated with an otherwise immunogenic transfer of CD8− DCs (17).
Functional dichotomy of CD8− and CD8+ DCs, and contrasting effects of IL-12 and IFN-γ treatment, were also found in this study on examining NRP-A7 presentation by CD8− and CD8+ DCs from NOD male and 8-wk-old female mice (Fig. 1). Upon treatment with IFN-γ–treated CD8+ DCs, these mice developed a state of long-term unresponsiveness that made them unresponsive to subsequent, otherwise effective priming with CD8− DCs (unpublished data). In contrast, IFN-γ was unable to induce suppressive or tolerizing properties in DCs from NOD females in early prediabetes (Fig. 1). Two considerations appear to be important in this regard. First, the high incidence of diabetes observed in NOD female mice might be related to the reduced tolerogenic ability of DCs early in prediabetes. Second, the suppressive activity of CD8+ DCs from 8-wk-old female mice was contingent upon effective tryptophan catabolism in these cells. Thus, IDO activity might be critically involved in the induction or maintenance of tolerance to autoantigens in NOD mice, and IDO function might be defective in early prediabetic female mice.
To explore this possibility, we examined IDO expression and function in female versus male mice of 4 or 8 wk of age. Both IDO protein expression and function were severely impaired in NOD females at 4 wk (Fig. 2, A and B). Because transcriptional activation of the IDO gene is mediated by Stat1 (10), we examined the possible occurrence of defective Stat1 phosphorylation in response to IFN-γ in early prediabetic NOD female mice (Fig. 2 C). We obtained clear evidence that Stat1 phosphorylation in response to IFN-γ was impaired in these mice. Aberrant transcription factor activity is known to occur for nuclear factor (NF)-κB family members in NOD mice, with elevated NF-κB activation and aberrant production of IL-12 caused by intrinsic and unique patterns of NF-κB expression in DCs (5, 30), as well as defective proteasome production and NF-κB activation in other cell types (31). Because NF-κB is known to be activated by IFN-γ (32), these data suggested that in NOD mice, defective Stat1 signaling might skew DCs toward enhanced activity mediated transcriptionally by NF-κB, such as production of IL-12 (33) and NO (34).
NOD mice are characterized by abnormal production of NO, which might contribute to islet destruction by T cells (20, 35). Tyrosine nitration of Stat1 by endogenous NO may lead to impaired response to IFN-γ in activated macrophages (21). Peroxynitrite is a highly reactive oxidant species produced by the combination of the free radicals, superoxide and NO (36, 37). Peroxynitrite production has been observed in acutely diabetic NOD mice (38), and current evidence suggests that peroxynitrite is a more potent and cytotoxic mediator than superoxide or NO alone (37). An inhibitor of NO synthase and scavenger of peroxynitrite prevents diabetes in NOD mice (6). Tyrosine nitration, a footprint of peroxynitrite, has been demonstrated in the pancreatic islets as well as in the cardiovascular system of diabetic subjects (39, 40).
We examined nitrotyrosine formation and the effect of peroxynitrite inhibition in response to IFN-γ in early prediabetic NOD female mice (Fig. 3). On assaying CD8+ DCs from NOD female versus male or conventional mice, we found high-level nitrotyrosine formation in both cell supernatants and lysates of DCs from 4-wk-old NOD female mice. This effect was ablated by the copresence of a peroxynitrite scavenger during cell exposure to IFN-γ. Remarkably, the peroxynitrite scavenger also rescued Stat1 phosphorylation in response to IFN-γ as well as kynurenine production in CD8+ DCs from 4-wk-old NOD female mice (Fig. 4). These data strongly supported the contention that tyrosine nitration of Stat1 by peroxynitrite underlies the impaired response of the latter cells to IFN-γ, and may thus contribute to the failure to activate immunosuppressive tryptophan catabolism. Although the reason why significant peroxynitrite formation is only seen in NOD female mice in early prediabetes is unclear, it is possible that this production may be linked to the onset of insulitis in a systemic inflammatory context in which the redox status of APCs is abnormal (41). It is likely that the reduced activity of superoxide dismutase observed in NOD mice (42) results in increased peroxynitrite production and protein tyrosine nitration.
Peroxynitrite scavenger-induced inhibition of nitrotyrosine formation was also examined for possible effects on the tolerogenic properties of CD8+ DCs from NOD female mice upon treatment with IFN-γ in vitro. A combination of peptide-pulsed CD8− and CD8+ DCs from 4-wk-old female mice was injected into recipients to be assayed for development of peptide-specific reactivity. CD8+ DCs were treated either with IFN-γ alone or with IFN-γ in combination with 1-MT and/or the peroxynitrite scavenger. The results showed that treatment with GED fully restored the induction of tolerance to NRP-A7 by CD8+ DCs, and this effect was dependent upon effective tryptophan metabolism in these cells (Fig. 5). Rescue of the suppressive activity of CD8+ DCs by GED was associated with the induction of persistent unresponsiveness to NRP-A7, as induced by otherwise effective priming with CD8− DCs performed up to 90 d after primary cell transfer (unpublished data). Thus, the effect mediated by the CD8+ DCs exposed to IFN-γ and GED appears to be a true switch from immunity to tolerance rather than a switch between different types of immunity (43).
By locally depleting tryptophan and increasing apoptotic kynurenines, IDO expressed by DCs greatly affects T cell proliferation and survival. IDO induction in DCs could be a common mechanism of deletional tolerance driven by regulatory T cells (12). Because such responses can be expected to operate in all forms of physiological tolerance, tryptophan metabolism might be critically involved in the prevention of autoimmune disorders. However, despite much evidence for a role of IDO in mediating tolerance in both pregnancy (25) and transplantation (10), there is no evidence so far for a similar role of IDO in autoimmunity. Our present data in NOD mice, a prototypic model of a spontaneous autoimmune condition, provide evidence for a link between impaired tryptophan catabolism and development of autoreactivity. In NOD mice, several defects contribute to the predisposition to develop autoimmunity. Our data reveal a novel defect intrinsic to the DCs of highly susceptible mice that may be involved in the failure to initiate tolerance to self-antigens. Because DCs are currently regarded as the major discriminator of function in the induction of tolerance versus immunity (44), and are likewise considered to be a principal mediator of tolerance (45), our current data emphasize the critical role of these cells in maintaining an appropriate balance between tolerance and immunity. It is likely that the IDO-based pathway is only one of several alternative modes of tolerance (11). However, our current and previous data demonstrate that a unitary mechanism involving DCs and IDO operates in the induction of tolerance in both transplantation and autoimmunity.
Acknowledgments
We thank A. Mellor for the generous gift of the IDO-specific antibody.
This work was supported by a grant from the Juvenile Diabetes Research Foundation International.
Footnotes
Abbreviations used in this paper: GED, guanidinoethyl disulphide; IDO, indoleamine 2,3-dioxygenase; 1-MT, 1-methyl-dl-tryptophan; NF, nuclear factor; NO, nitric oxide; NOD, nonobese diabetic.
References
- 1.Delovitch, T.L., and B. Singh. 1997. The nonobese diabetic mouse as a model of autoimmune diabetes: immune dysregulation gets the NOD. Immunity. 7:727–738. [DOI] [PubMed] [Google Scholar]
- 2.Atkinson, M.A., and E.H. Leiter. 1999. The NOD mouse model of type 1 diabetes: as good as it gets? Nat. Med. 5:601–604. [DOI] [PubMed] [Google Scholar]
- 3.Kishimoto, H., and J. Sprent. 2001. A defect in central tolerance in NOD mice. Nat. Immunol. 2:1025–1031. [DOI] [PubMed] [Google Scholar]
- 4.Serreze, D.V., H.R. Gaskins, and E.H. Leiter. 1993. Defects in the differentiation and function of antigen presenting cells in NOD/Lt mice. J. Immunol. 150:2534–2543. [PubMed] [Google Scholar]
- 5.Liu, J., and D. Beller. 2002. Aberrant production of IL-12 by macrophages from several autoimmune-prone mouse strains is characterized by intrinsic and unique patterns of NF-κB expression and binding to the IL-12 p40 promoter. J. Immunol. 169:581–586. [DOI] [PubMed] [Google Scholar]
- 6.Suarez-Pinzon, W.L., J.G. Mabley, K. Strynadka, R.F. Power, C. Szabo, and A. Rabinovitch. 2001. An inhibitor of inducible nitric oxide synthase and scavenger of peroxynitrite prevents diabetes development in NOD mice. J. Autoimmun. 16:449–455. [DOI] [PubMed] [Google Scholar]
- 7.Christen, U., T. Wolfe, U. Mohrle, A.C. Hughes, E. Rodrigo, E.A. Green, R.A. Flavell, and M.G. von Herrath. 2001. A dual role for TNF-α in type 1 diabetes: islet-specific expression abrogates the ongoing autoimmune process when induced late but not early during pathogenesis. J. Immunol. 166:7023–7032. [DOI] [PubMed] [Google Scholar]
- 8.Sobel, D.O., J. Han, J. Williams, J.W. Yoon, H.S. Jun, and B. Ahvazi. 2002. Gamma interferon paradoxically inhibits the development of diabetes in the NOD mouse. J. Autoimmun. 19:129–137. [DOI] [PubMed] [Google Scholar]
- 9.Shinomiya, M., S.M. Fazle Akbar, H. Shinomiya, and M. Onji. 1999. Transfer of dendritic cells (DC) ex vivo stimulated with interferon-gamma (IFN-γ) down-modulates autoimmune diabetes in non-obese diabetic (NOD) mice. Clin. Exp. Immunol. 117:38–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grohmann, U., C. Orabona, F. Fallarino, C. Vacca, F. Calcinaro, A. Falorni, P. Candeloro, M.L. Belladonna, R. Bianchi, M.C. Fioretti, and P. Puccetti. 2002. CTLA-4-Ig regulates tryptophan catabolism in vivo. Nat. Immunol. 3:1097–1101. [DOI] [PubMed] [Google Scholar]
- 11.Finger, E.B., and J.A. Bluestone. 2002. When ligand becomes receptor–tolerance via B7 signaling on DCs. Nat. Immunol. 3:1056–1057. [DOI] [PubMed] [Google Scholar]
- 12.Grohmann, U., F. Fallarino, and P. Puccetti. 2003. Tolerance, DCs and tryptophan: much ado about IDO. Trends Immunol. 24:242–248. [DOI] [PubMed] [Google Scholar]
- 13.Grohmann, U., F. Fallarino, R. Bianchi, M.L. Belladonna, C. Vacca, C. Orabona, C. Uyttenhove, M.C. Fioretti, and P. Puccetti. 2001. IL-6 inhibits the tolerogenic function of CD8α+ dendritic cells expressing indoleamine 2,3-dioxygenase. J. Immunol. 167:708–714. [DOI] [PubMed] [Google Scholar]
- 14.Anderson, B., B.J. Park, J. Verdaguer, A. Amrani, and P. Santamaria. 1999. Prevalent CD8+ T cell response against one peptide/MHC complex in autoimmune diabetes. Proc. Natl. Acad. Sci. USA. 96:9311–9316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Amrani, A., J. Verdaguer, P. Serra, S. Tafuro, R. Tan, and P. Santamaria. 2000. Progression of autoimmune diabetes driven by avidity maturation of a T-cell population. Nature. 406:739–742. [DOI] [PubMed] [Google Scholar]
- 16.Grohmann, U., R. Bianchi, M.L. Belladonna, S. Silla, F. Fallarino, M.C. Fioretti, and P. Puccetti. 2000. IFN-γ inhibits presentation of a tumor/self peptide by CD8α− dendritic cells via potentiation of the CD8α+ subset. J. Immunol. 165:1357–1363. [DOI] [PubMed] [Google Scholar]
- 17.Grohmann, U., F. Fallarino, S. Silla, R. Bianchi, M.L. Belladonna, C. Vacca, A. Micheletti, M.C. Fioretti, and P. Puccetti. 2001. CD40 ligation ablates the tolerogenic potential of lymphoid dendritic cells. J. Immunol. 166:277–283. [DOI] [PubMed] [Google Scholar]
- 18.Fallarino, F., C. Vacca, C. Orabona, M.L. Belladonna, R. Bianchi, B. Marshall, D.B. Keskin, A.L. Mellor, M.C. Fioretti, U. Grohmann, and P. Puccetti. 2002. Functional expression of indoleamine 2,3-dioxygenase by murine CD8α+ dendritic cells. Int. Immunol. 14:65–68. [DOI] [PubMed] [Google Scholar]
- 19.McDaniel, M.L., G. Kwon, J.R. Hill, C.A. Marshall, and J.A. Corbett. 1996. Cytokines and nitric oxide in islet inflammation and diabetes. Proc. Soc. Exp. Biol. Med. 211: 24–32. [DOI] [PubMed] [Google Scholar]
- 20.Gurlo, T., K. Kawamura, and H. von Grafenstein. 1999. Role of inflammatory infiltrate in activation and effector function of cloned islet reactive nonobese diabetic CD8+ T cells: involvement of a nitric oxide-dependent pathway. J. Immunol. 163:5770–5780. [PubMed] [Google Scholar]
- 21.Llovera, M., J.D. Pearson, C. Moreno, and V. Riveros-Moreno. 2001. Impaired response to interferon-γ in activated macrophages due to tyrosine nitration of STAT1 by endogenous nitric oxide. Br. J. Pharmacol. 132:419–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matata, B.M., and M. Galinanes. 2002. Peroxynitrite is an essential component of cytokines production mechanism in human monocytes through modulation of nuclear factor-κB DNA binding activity. J. Biol. Chem. 277:2330–2335. [DOI] [PubMed] [Google Scholar]
- 23.Serreze, D.V., H.D. Chapman, C.M. Post, E.A. Johnson, W.L. Suarez-Pinzon, and A. Rabinovitch. 2001. Th1 to Th2 cytokine shifts in nonobese diabetic mice: sometimes an outcome, rather than the cause, of diabetes resistance elicited by immunostimulation. J. Immunol. 166:1352–1359. [DOI] [PubMed] [Google Scholar]
- 24.Kishimoto, K., S. Sandner, J. Imitola, M. Sho, Y. Li, P.B. Langmuir, D.M. Rothstein, T.B. Strom, L.A. Turka, and M.H. Sayegh. 2002. Th1 cytokines, programmed cell death, and alloreactive T cell clone size in transplant tolerance. J. Clin. Invest. 109:1471–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Munn, D.H., M. Zhou, J.T. Attwood, I. Bondarev, S.J. Conway, B. Marshall, C. Brown, and A.L. Mellor. 1998. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science. 281:1191–1193. [DOI] [PubMed] [Google Scholar]
- 26.Mellor, A.L., D.B. Keskin, T. Johnson, P. Chandler, and D.H. Munn. 2002. Cells expressing indoleamine 2,3-dioxygenase inhibit T cell responses. J. Immunol. 168:3771–3776. [DOI] [PubMed] [Google Scholar]
- 27.Fallarino, F., U. Grohmann, C. Vacca, R. Bianchi, C. Orabona, A. Spreca, M.C. Fioretti, and P. Puccetti. 2002. T cell apoptosis by tryptophan catabolism. Cell Death Differ. 9:1069–1077. [DOI] [PubMed] [Google Scholar]
- 28.Rosmalen, J.G., W. van Ewijk, and P.J. Leenen. 2002. T-cell education in autoimmune diabetes: teachers and students. Trends Immunol. 23:40–46. [DOI] [PubMed] [Google Scholar]
- 29.Marrack, P., J. Kappler, and B.L. Kotzin. 2001. Autoimmune disease: why and where it occurs. Nat. Med. 7:899–905. [DOI] [PubMed] [Google Scholar]
- 30.Poligone, B., D.J. Weaver, Jr., P. Sen, A.S. Baldwin, Jr., and R. Tisch. 2002. Elevated NF-κB activation in nonobese diabetic mouse dendritic cells results in enhanced APC function. J. Immunol. 168:188–196. [DOI] [PubMed] [Google Scholar]
- 31.Hayashi, T., and D. Faustman. 1999. NOD mice are defective in proteasome production and activation of NF-κB. Mol. Cell. Biol. 19:8646–8659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deb, A., S.J. Haque, T. Mogensen, R.H. Silverman, and B.R. Williams. 2001. RNA-dependent protein kinase PKR is required for activation of NF-κB by IFN-γ in a STAT1-independent pathway. J. Immunol. 166:6170–6180. [DOI] [PubMed] [Google Scholar]
- 33.Grohmann, U., M.L. Belladonna, R. Bianchi, C. Orabona, E. Ayroldi, M.C. Fioretti, and P. Puccetti. 1998. IL-12 acts directly on DC to promote nuclear localization of NF-κB and primes DC for IL-12 production. Immunity. 9:315–323. [DOI] [PubMed] [Google Scholar]
- 34.Alderton, W.K., C.E. Cooper, and R.G. Knowles. 2001. Nitric oxide synthases: structure, function and inhibition. Biochem. J. 357:593–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thomas, H.E., R. Darwiche, J.A. Corbett, and T.W. Kay. 2002. Interleukin-1 plus γ-interferon-induced pancreatic β-cell dysfunction is mediated by β-cell nitric oxide production. Diabetes. 51:311–316. [DOI] [PubMed] [Google Scholar]
- 36.Beckman, J.S., T.W. Beckman, J. Chen, P.A. Marshall, and B.A. Freeman. 1990. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. USA. 87:1620–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pryor, W.A., and G.L. Squadrito. 1995. The chemistry of peroxynitrite: a product from the reaction of nitric oxide with superoxide. Am. J. Physiol. 268:L699–722. [DOI] [PubMed] [Google Scholar]
- 38.Suarez-Pinzon, W.L., C. Szabo, and A. Rabinovitch. 1997. Development of autoimmune diabetes in NOD mice is associated with the formation of peroxynitrite in pancreatic islet β-cells. Diabetes. 46:907–911. [DOI] [PubMed] [Google Scholar]
- 39.Delaney, C.A., B. Tyrberg, L. Bouwens, H. Vaghef, B. Hellman, and D.L. Eizirik. 1996. Sensitivity of human pancreatic islets to peroxynitrite-induced cell dysfunction and death. FEBS Lett. 394:300–306. [DOI] [PubMed] [Google Scholar]
- 40.Szabo, C., J.G. Mabley, S.M. Moeller, R. Shimanovich, P. Pacher, L. Virag, F.G. Soriano, J.H. Van Duzer, W. Williams, A.L. Salzman, and J.T. Groves. 2002. Part I: Pathogenetic role of peroxynitrite in the development of diabetes and diabetic vascular complications: studies with FP15, a novel potent peroxynitrite decomposition catalyst. Mol. Med. 8:571–580. [PMC free article] [PubMed] [Google Scholar]
- 41.Murata, Y., M. Amao, and J. Hamuro. 2003. Sequential conversion of the redox status of macrophages dictates the pathological progression of autoimmune diabetes. Eur. J. Immunol. 33:1001–1011. [DOI] [PubMed] [Google Scholar]
- 42.Papaccio, G., S. Frascatore, F.A. Pisanti, M.V. Latronico, and T. Linn. 1995. Superoxide dismutase in the nonobese diabetic (NOD) mouse: a dynamic time-course study. Life Sci. 56:2223–2228. [DOI] [PubMed] [Google Scholar]
- 43.Boonstra, A., C. Asselin-Paturel, M. Gilliet, C. Crain, G. Trinchieri, Y.J. Liu, and A. O'Garra. 2003. Flexibility of mouse classical and plasmacytoid-derived dendritic cells in directing T helper type 1 and 2 cell development: dependency on antigen dose and differential toll-like receptor ligation. J. Exp. Med. 197:101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shortman, K., and W.R. Heath. 2001. Immunity or tolerance? That is the question for dendritic cells. Nat. Immunol. 2:988–989. [DOI] [PubMed] [Google Scholar]
- 45.Steinman, R.M., and M.C. Nussenzweig. 2002. Avoiding horror autotoxicus: the importance of dendritic cells in peripheral T cell tolerance. Proc. Natl. Acad. Sci. USA. 99:351–358. [DOI] [PMC free article] [PubMed] [Google Scholar]