

Abstract
We evaluated the proposal that during microbial infection, dendritic cells (DCs) undergo maturation and present a mixture of peptides derived from the microbe as well as harmless environmental antigens. Mice were exposed to an aerosol of endotoxin free ovalbumin (OVA) in the absence or presence of influenza virus. In its absence, OVA failed to induce B and T cell responses and even tolerized, but with influenza, OVA-specific antibodies and CD8+ cytolytic T lymphocytes developed. With or without infection, OVA was presented selectively in the draining mediastinal lymph nodes, as assessed by the comparable proliferation of infused, CD8+ and CD4+, TCR transgenic T cells. In the absence of influenza, these OVA-specific T cells produced little IL-2, IL-4, IL-10, and IFN-γ, but with infection, both CD4+ and CD8+ T cells made high levels of IL-2 and IFN-γ. The OVA plus influenza-treated mice also showed accelerated recovery to a challenge with recombinant vaccinia OVA virus. CD11c+ DCs from the mediastinal lymph nodes of infected mice selectively stimulated both OVA- and influenza-specific T cells and underwent maturation, with higher levels of MHC class II, CD80, and CD86 molecules. The relatively slow (2–3 d) kinetics of maturation correlated closely to the time at which OVA inhalation elicited specific antibodies. Therefore respiratory infection can induce DC maturation and simultaneously B and T cell immunity to an innocuous antigen inhaled concurrently.
Keywords: influenza virus, dendritic cell, maturation, endotoxin free ovalbumin, airway
Introduction
The respiratory portal serves as an important entry site for pathogenic organisms, but it is also a site for effective innate and adaptive immune responses. An important constituent of the airways are antigen presenting DCs, which line the alveolar septae and airway epithelia (1–4) and are capable of capturing and presenting antigens to initiate immunity especially within the draining mediastinal or peribronchial lymph nodes (5–9). This defense mechanism imposes a risk however, which is that the respiratory tract also is chronically exposed to many innocuous but potentially immunogenic proteins. In fact, DCs appear to be capturing proteins from the airway constitutively (6, 8). As DCs may capture these harmless proteins together with a pathogen, there also is a potential for the development of unwanted immune responses and chronic reactivity to airway proteins (10). Can the DC arm of the immune system avoid the capture of harmless airway proteins during an infection?
We set out to investigate this question using OVA, as a model harmless protein, and influenza virus as a common respiratory pathogen. In planning the experiments, we considered the fact that the initiation of T cell–mediated immunity entails two broad groups of changes in antigen presenting DCs: the capture and successful processing of antigens to form MHC–peptide complexes, the ligands for T cell antigen receptors, and the maturation of the DCs to acquire costimulatory and other functions required for the induction of immunity (11–13). Influenza virus is associated with the maturation of DCs in tissue culture (14–16) and recently in vivo (17).
A number of prior studies have shown that respiratory virus infections can enhance the immune response to protein antigens (18–25). However, in an effort to establish an allergy model, many of these prior investigations used alum as an adjuvant to polarize the immune response toward the Th2 type that is associated with allergy. Another complicating feature was that the antigens used in prior studies were from sources that are now known to be contaminated with LPS. Eisenbarth et al. (26) recently reported on the immune response to intranasal OVA in the presence of low (0.1 μg) or high (100 μg) levels of LPS. They found antibody and T cell responses in both instances, but that the high dose of LPS converted these responses from a Th2 to a Th1 type. Akbari et al. (27) also followed the immune response to OVA applied intranasally. They noted a strong up-regulation of costimulatory molecules on the DCs in the draining lymph node, and that these DCs could elicit IL-10–producing regulatory T cells. The observed up-regulation of costimulatory molecules signifies that the OVA administration was somehow accompanied by a DC maturation stimulus. Therefore, it seemed important to use endotoxin free antigens to assess if immunity will develop to a harmless airway protein given before, or concurrently with, an influenza infection.
Here we show that endotoxin free preparations of OVA do not immunize either the B or T cell compartments when inhaled, and that the OVA is tolerogenic. Nevertheless, the OVA is presented efficiently by DCs in the mediastinal lymph nodes in the steady-state, at substantial levels to what is observed when OVA is given during an influenza virus infection. However, only in the latter instance do the mice develop antibodies as well as combined Th1 type CD4+ and CD8+ T cell immunity to OVA. We find that influenza virus infection of the respiratory tract, but not OVA alone, induces maturation of DCs in the draining mediastinal lymph nodes; this maturation correlates with the induction of immunity to the otherwise harmless protein OVA in the airway. These results emphasize that the mucosal immune system must have efficient tolerance mechanisms to avoid simultaneous immunization to resident foreign proteins that are later processed by DCs during infection.
Materials and Methods
Mice.
C57BL/6 (B6) female mice were purchased from Taconic and used at 8–12 wk of age. OT-I mice were provided by Dr. F. Carbone (University of Melbourne, Parkville, Victoria, Australia), and OT-II mice were obtained from the Trudeau Institute. CD45.1+ OT-I mice were produced by crossing OT-I with B6.SJL-Ptprc mice.
Antibodies.
Antibodies to CD80/16–10A1, CD86/GL1, MHC class II I-Ab/AF6–120.1, B220, and CD45.1/A20 were purchased from BD Biosciences. Hybridomas producing antibodies were obtained from the American Type Culture Collection including MHC class II (M5/114, TIB120), F4/80 (HB 198), NK1.1, B220 (RA3–6B2), CD4 (GK1.5, TIB 207), and CD8 (53.6.7, TIB 105).
Viruses.
Influenza virus A/HK X-31 (X-31), H3N2 subtype and B/Lee/40 were grown for 40 h in 9 d embryonated eggs at 37°C. Allantoic fluid was harvested and stored at −80°C. Virus stocks and virus-infected lungs were titrated by infection of Madin-Darby Canine Kidney cells (MDCK) as described elsewhere (28). Vaccinia-OVA virus was grown for 48h in CV-1 cells (kidney cells from African green monkey) in DMEM, 10% FCS at 37°C. The CV-1 cells containing the vaccinia-OVA virus were harvested and stored at −80°C. CV-1 cells were infected to titer virus stocks and lung extracts. CV1 cells were seeded at 3 × 105/ml in 96-well plates and when confluent, they were incubated with serial dilutions (25 μl) of virus stocks or lung extract for 1 h. Extra media was added and the plates incubated for 48 h at 37°C. The supernatant was discarded, the plates were stained with crystal violet solution, and plaque forming units were counted.
Infection of Mice with Influenza and Vaccinia Viruses.
Infections of mice with influenza A (X-31) and B (B/Lee) were performed in an infection chamber. For X-31 a 1:50 dilution of virus (106.9 TCID50/25 μl allantoic fluid) in sterile PBS was used. This infection resulted in the following lung virus titers expressed as TCID50/25 μl of lung extract: 20 h after infection, 103.91 ± 0.47; 45 h after infection, 106.3 ± 0.41; 72 h after infection, 106.4 ± 0.48; 96 h after infection, 106.4 ± 0.33; 7 d after infection, 105.1 ± 0.35; and 10 d post infection, below detection. For B/Lee a 1:20 dilution of virus (2 × 107 pfu/ml allantoic fluid) in sterile PBS was used. 7 ml virus solution was nebulized into the chamber resulting in infection of 100% of the mice. For infection with recombinant vaccinia OVA virus, mice were anaesthetized with avertin (Sigma-Aldrich) and inoculated with 105 pfu intranasally.
Administration of OVA and Measurement of Endotoxin Levels.
Administration of OVA was performed in an infection chamber. The mice were exposed for 15 min to a solution of 10 mg/ml egg white OVA (Charles River Laboratories) or commercial OVA (Calbiochem-Novabiochem) dissolved in sterile PBS. A control group receiving aerosolized PBS was always included in every experiment. For determination of endotoxin levels in the OVA preparations, the Limulus amebocyte Lysate assay (Biowhittaker Inc.) was used according to the manufacturer's protocol.
Detection of DC Maturation In Vivo.
X-31 virus or OVA was administered by aerosol into the airway as above. The draining lymph nodes were harvested at 20 h to 10 d after infection, and CD11c+ cells were isolated by staining with PE conjugated anti-CD11c (BD Biosciences) and anti-PE microbeads (Miltenyi Biotec) as described in the manufacturer's protocol. The CD11c+ or CD11c− cells were stained with FITC-labeled antibodies against F4/80 (Serotec), CD80, CD86, I-Ab (BD Biosciences), APC-labeled antibody against B220 (BD Biosciences), and PerCP-labeled antibody against CD11b (BD Biosciences) and were analyzed by flow cytometry (FACSCalibur™; BD Biosciences).
Purification and Proliferation of OT-I and OT-II, OVA-specific, TCR Transgenic T Cells.
To purify OT-I and OT-II T cells, as described elsewhere (29), spleen and lymph nodes were harvested and the single cell suspension incubated with hybridoma supernatants directed against MHC II, F4/80, B220, NK1.1 and CD4 or CD8 (depletion) and goat anti-rat Dynabeads (Dynal) to enrich for CD8+ or CD4+ T cells, respectively. For analysis of in vivo proliferation, the enriched OT-I and OT-II T cells were labeled with CFSE. In brief, 107 cells/ml were incubated with CFSE (Molecular Probes) at 5 μM for 10 min at 37°C. An equal volume of FCS was added, and the cells were washed twice in PBS, 0.1% BSA, and once with PBS before intravenous injection of 2–3 × 106 CFSE-labeled OT-I or OT-II T cells into B6 mice. The following day, the mice were infected with X-31 or PBS and then administered egg OVA for 3 consecutive days. 5 (OT-II) or 6 d (OT-I) after infection with X-31, the draining lymph nodes were harvested and stained for expression of CD45.1 and CD8 or CD4. The proliferation of OT-I and OT-II T cells was analyzed by flow cytometry.
In Vitro Proliferation Assay for OVA and Influenza Presentation.
B6 mice were infected with X-31 or given PBS as a control on day 1. On days 2, 3, and 4, they were administered egg OVA and on day 5 the draining lymph nodes were harvested and incubated with 1 mg/ml collagenase D (Roche) for 25 min at 37°C. CD11c+ cells were purified as above. The CD11c− fraction was further depleted for T cells using anti-CD5 beads (Miltenyi Biotec) according to the manufacturers protocol. Graded doses of CD11c+ or CD11c− cells were cultured at 37°C with 105 OT-I or OT-II T cells in round bottom 96-well plates, to which 3H-thymidine (1 μCi/well; Amersham Biosciences) was added at 48–72 h to detect incorporation into DNA. For evaluation of influenza peptide presentation by CD11c+ cells, B6 mice were infected with X-31 on day 1 and on day 12 bulk T cells were purified from the draining LNs and used as responder cells in a proliferation assay. In brief the LN cells were depleted from B cells, DCs, and macrophages by using anti-CD19, anti-CD11c, and anti-MHC class II microbeads (Miltenyi Biotec) according to the manufacturers protocol. For generation of APCs B6 mice were infected with X-31 or mock infected on day 1, and on day 5 CD11c+ and CD11c− cells were isolated from the draining LNs and cocultured with 105 bulk T cells from influenza virus infected animals to assess the T cell proliferative response.
Intracellular Cytokine Production from OT-I and OT-II T Cells.
B6 mice were injected with 106 OT-I or OT-II T cells intravenously. After aerosol administration of OVA with or without concomitant influenza infection as above, draining mediastinal lymph nodes were harvested at day 6 and 5 × 106 cells were cultured for 5 h in 24-well plates with OVA257–264 (OT-I) or OVA323–339 (OT-II) in the presence of 10 μg/ml Brefeldin A (Sigma-Aldrich). After harvesting the cells were stained for expression of CD45.1, CD4/CD8, and intracellular IL-2 or IFN-γ. For intracellular cytokine staining, the PharMingen Cytokine Staining Starter Kit was used. The expression of cytokines was evaluated by flow cytometry.
Detection of OVA-specific Antibodies.
X-31 influenza virus was used to infect B6 mice on day 1 and then egg OVA was aerosolized on days 2, 3, and 4 as above. 14 d after infection, the mice were bled and serum levels of OVA-specific antibodies determined by ELISA. Briefly, the plates were coated with 5 μg/ml OVA overnight and blocked with PBS plus 1% BSA. 10-fold serum dilutions were incubated for 2 h at room temperature. The plates were washed, incubated with peroxidase conjugated anti–mouse IgG1 or IgG2b for 1 h (Boehringer), washed, and bound antibodies detected by adding ABTS tablets (Boehringer). The OD was measured at 405 nm. For OVA-specific IgE detection, plates were coated overnight with 2.5 μg/ml rat anti–mouse IgE (BD Biosciences) followed by 10-fold serum dilutions for 2 h at room temperature. After washing, the plates were incubated with 5 μg/ml biotinylated OVA for 2 h, washed and incubated with streptavidin-POD (Roche Diagnostic Corporation) for 1 h. The plates were developed as above.
Cytotoxicity Assay.
B6 mice were infected with X-31 on day 1 and followed by OVA on days 2, 3, and 4. 8 d after infection, the mediastinal lymph nodes were harvested and 20 × 106 lymph node cells were stimulated in vitro with 20 × 106 spleen cells from naive B6 mice for 5 d at 37°C, 7% CO2. The spleen cells had been gamma irradiated and pulsed with SIINFEKL peptide (5 μg/ml final concentration) for 1 h at 37°C followed by washing to remove excess peptide. The cytotoxicity assay was performed as described elsewhere (30). Briefly cells from the 5 d cultures were mixed with 51Cr-labeled EL-4 target cells in V bottom 96-well plates. The target cells were prepared by pulsing 2 × 106 EL-4 cells with SIINFEKL peptide (5 μg/ml final concentration) or PBS for 1 h at 37°C and then labeled with 100 μCi sodium chromate (Dupont/New Life Science Products) for 1 h. Effector cells were added at various effector/target ratios and incubated 4 h at 37°C. The supernatants were harvested and released radioactivity was measured in a gamma-counter. Percent cytotoxicity was calculated using the formula: % cytotoxicity = (test release – spontaneous release)/(maximum release – spontaneous release) × 100. Maximum and spontaneous release was determined by incubating the cells with 0.5% NP-40 or media, respectively.
Statistical Analysis.
The Mann-Whitney test was used for data analysis.
Results
Identification of Endotoxin-free OVA That Is Nonimmunogenic via the Airway.
To identify an abundant source of an endotoxin free and harmless protein antigen, we used egg white as a source of OVA. We compared the endotoxin levels in egg white OVA and commercially available OVA using the Limulus Amebocyte Lysate assay. The egg white OVA was endotoxin free (below detection level <0.1 EU/ml) at a concentration as high as 10 mg/ml OVA. In contrast, the commercial source of OVA was contaminated with endotoxin, as 100 μg/ml OVA contained 2 EU/ml. When administered to B6 (Fig. 1) or BALB/C (unpublished data) mice via the airway, the commercial “LPS OVA” induced anti-OVA antibody responses primarily of the IgG1 and IgG2b isotypes, whereas the LPS-free “egg OVA” did not induce antibodies (Fig. 1, A and B). To assess the response to the two different forms of OVA at the T cell level, we measured the generation of CD8+ CTL, but neither preparation of OVA was immunogenic (Fig. 1 C). We therefore prioritized the use of egg OVA as a harmless protein that is by itself nonimmunogenic via the airway.
Figure 1.

Generation of antibody and CTL responses in naive animals following coadministration of OVA and influenza virus. (A and B) B6 mice were infected with aerosolized X-31 on day 1 followed by egg OVA (A) or LPS OVA (B) on days 2, 3, and 4. 14 d after infection, serum levels of OVA-specific antibodies were determined by ELISA (mean ± SD from four individual mice; representative of two or more experiments. (C) B6 mice were infected with aerosolized X-31 on day 1 and given LPS OVA or egg OVA on days 2, 3, and 4. 8 d after infection the mediastinal lymph nodes were harvested and the lymph node cells were restimulated with SIINFEKL in vitro for 5 d. Data are means ± SD from groups of four mice and representative of three experiments. (D) B6 mice were given egg OVA on days –5, −3, and –1. On day 1, the mice were infected with X-31 and on days 2, 3, and 4 they were administered LPS OVA (egg OVA, then influenza + LPS OVA). A positive control group which was infected with X-31 on day 1 and administered LPS OVA on day 2, 3, and 4 was also included (influenza + LPS OVA). Data are means ± SD from groups of four mice and representative of two experiments.
Coadministration of OVA and Influenza Virus Leads to OVA-specific Antibodies and CTLs.
To assess the effect of influenza virus infection on immune responses to OVA, B6 mice were infected with influenza virus or mock infected with PBS at day 1, and then either LPS OVA or egg OVA was administered by aerosol on three consecutive days, days 2, 3, and 4. 14 d after infection, the serum was analyzed for OVA-specific antibodies. Mice that received influenza virus and egg OVA now produced OVA-specific, IgG1 and IgG2b antibodies, while mice that received egg OVA alone did not (Fig. 1 A). We also detected small levels of OVA specific IgE in mice infected with influenza virus and administered LPS OVA, while mice administered influenza virus followed by LPS-free egg OVA failed to produce these antibodies (Fig. 1, A and B). To examine whether a cellular immune response was generated, we measured the OVA specific CTL response. B6 mice were treated as described above and 8 d after influenza virus infection the mediastinal lymph node cells were isolated and restimulated with the SIINFEKL peptide for 5 d in vitro. In mice that received influenza virus and egg OVA, OVA-specific CTLs were induced, while none of the control groups demonstrated any CTL activity (Fig. 1 C). Also, we were able to detect an effect of endotoxin in the commercial OVA, because the CTL response initiated using LPS OVA and virus infection was stronger than when egg OVA was used (Fig. 1 C). These results demonstrate that endotoxin-free OVA administered by aerosol into the respiratory tract fails to prime for an immune response, but if given during an influenza virus infection, OVA-specific humoral and cellular immune responses are generated.
LPS-free OVA Given before Infection Is Able to Induce Tolerance.
While LPS-free OVA was nonimmunogenic, the prior studies did not differentiate between immune ignorance or active immune tolerance as the underlying mechanism. To begin to examine this, we gave mice soluble egg OVA for 3 d by the airway, infected with influenza virus, and then followed by further doses of soluble LPS OVA as in the prior immunizing regimen of Fig. 1 C. The mice pretreated with soluble LPS-free OVA (Fig. 1 D, the group labeled egg OVA, then influenza + LPS OVA) now failed to develop CTLs in response to coadministered LPS-OVA. The tolerance was specific in that CTL responses to influenza developed comparably to those seen in mice that had not received OVA (unpublished data). Therefore while coadministration of a harmless protein with influenza leads to immunity, prior administration of that harmless protein is capable of inducing tolerance.
Airway OVA Is Presented in the Draining Lymph Nodes in the Steady State.
To investigate whether the lack of immune response toward endotoxin-free OVA, when administered in the absence of influenza virus infection, stemmed from a failure of OVA to reach the draining lymph nodes, we took advantage of transgenic CD8+ OT-I T cells (recognizing OVA257–264 [SIINFEKL] on Kb) and CD4+ OT-II T cells (recognizing OVA323–339 on Ab) to report on the presentation of MHC–peptide in the lymph nodes. These T cells were labeled with CFSE and infused into naive B6 mice. The mice then received influenza virus (day 1) and egg OVA (days 2, 3, 4), or the mice were mock infected (day 1) and given egg OVA (days 2, 3, 4). On day 6, we monitored T cell proliferation in the draining (mediastinal) and distal (cervical) lymph nodes by flow cytometry, looking for a progressive halving of the CFSE labeling per cell as an index of T cell proliferation and successful OVA presentation. As shown in Fig. 2, both OT-I and OT-II T cells proliferated in the mediastinal lymph nodes (Fig. 2 A), and to a much lower extent in the cervical nodes (Fig. 2 B), demonstrating that OVA was presented locally on both MHC class I and II products in the lymph nodes draining the airways. Interestingly, we observed comparable proliferation of transgenic T cells in mice receiving influenza virus and egg OVA, as well as mice that only received egg OVA. Thus, the OVA antigen was indeed transported to the draining LNs and presented as MHC–peptide complexes, both in the presence and absence of virus infection. We did not further assess if the OVA was transported by migrating DCs or if OVA was gaining direct access via the lymph to the DCs in the draining lymph node.
Figure 2.

Active presentation of airway OVA in the draining mediastinal lymph nodes in the presence or absence of influenza virus infection. 2–3 × 106 CFSE-labeled OT-I or OT-II T cells were injected intravenously into B6 mice at day 0. On day 1 the mice were infected with X-31 or mock infected and then administered egg OVA on day 2, 3, and 4. 5 (OT-II) or 6 d (OT-I) after infection with X-31 the draining LNs were harvested and stained for expression of CD45.1 and CD8/CD4. Shown is proliferation of OT-I and OT-II T cells in the draining mediastinal with and without egg OVA in the presence and absence of influenza virus (A) and nondraining cervical in the presence of egg OVA (B) lymph nodes. Data are representative of three experiments.
Coinfection with Influenza Virus Leads to OVA-specific, Effector T Cell Formation.
As we observed comparable proliferation of OT-I and OT-II T cells in mice given aerosol OVA without or with influenza virus (Fig. 2), we were interested in determining the differentiation of these T cells into effector cells in vivo. Mice were given the transgenic T cells at day 0, influenza or mock infected at day 1, and then given 3 doses of egg OVA on days 2, 3, and 4. On day 6, the lymph node cells were isolated, and stimulated 5 h with OVA peptides appropriate for the antigen presenting H-2Kb and I-Ab molecules. T cells were then studied by flow cytometry to monitor T cell numbers as well as the intracellular accumulation of IL-2 and IFN-γ. Administration of egg OVA, with or without influenza virus infection, expanded the numbers of T cells as expected from the prior CFSE experiments (Fig. 2), although the expansion in T cell numbers at day 6 was threefold higher if there was influenza coinfection (Fig. 3, A and B, top rows). More dramatic differences were noted at the level of cytokine production, as much higher proportions of OT-I and OT-II T cells producing IL-2 and IFN-γ were detected when mice were aerosolized with influenza virus and egg OVA, and virtually no cytokine production was seen in the absence of influenza virus (Fig. 3, A and B, middle and lower rows). OVA-specific, transgenic T cells from mice treated only with influenza virus or PBS did not produce significant cytokines, i.e., little IL-2 or IFN-γ was detected (Fig. 3) and no IL-4 or IL-10 (unpublished data). Therefore influenza virus infection has a striking effect on the functional outcome of the OVA specific T cell response, leading to IL-2 and IFN-γ production.
Figure 3.

Coadministration of OVA and influenza virus leads to formation of OVA specific cytokine producing effector cells. B6 mice were injected with 106 OT-I or OT-II T cells intravenously on day 0. Day 1 mice were infected with X-31 or mock infected and administered egg OVA on day 2, 3, and 4. On day 6 the draining lymph nodes were harvested and 5 × 106 cells were cultured for 5 h with SIINFEKL (OT-I) or OVA323–339 (OT-II) in the presence of Brefeldin A. After harvesting, the cells were stained for expression of CD45.1 and CD4/CD8 and intracellular IL-2 or IFN-γ. The response of OT-II T cells (A) and OT-I T cells (B) is shown for cell numbers (top rows) and IFN-γ/IL-2 production (middle and lower rows) in the mediastinal LNs. Data are representative of two experiments.
CD11c+ DCs Present OVA and Viral Antigens in the Draining Lymph Nodes.
To define the cell types involved in the presentation of OVA in the draining lymph nodes, we purified CD11c+ and CD11c− cells and added these to cultures of OVA-specific transgenic T cells to stimulate proliferation. Again, the mice received either aerosolized influenza virus or mock infection, followed by aerosolized egg OVA for three consecutive days. 1 d after the last OVA treatment, the antigen-presenting cells were purified from draining mediastinal lymph nodes and nondraining brachial lymph nodes and tested (Fig. 4). Only CD11c+ cells from the mediastinal lymph nodes could elicit OT-I T cell proliferation, indicating that DCs were the major cell involved in the presentation of OVA in the draining lymph nodes. CD11c+ cells from mice infected with influenza virus and egg OVA were much more efficient at presenting OVA to OT-I (and OT-II cells, unpublished data) than CD11c+ cells from mice receiving egg OVA only (Fig. 4 A), but the later did stimulate above the PBS background control. The DCs from influenza virus infected mice also stimulated influenza primed T cells (Fig. 4 B), demonstrating that DCs as a population are capable of both presenting OVA peptides and influenza virus peptides.
Figure 4.

Presentation of OVA and viral antigens by DCs from the draining mediastinal lymph nodes. B6 mice were infected with X-31 or mock infected on day 1. On day 2, 3, and 4, they were administered egg OVA. On day 4, CD11c+ and CD11c− cells were purified from the draining LNs (six mice/group) and cocultured with OT-I cells for 48–72 h and assessed for T cell proliferation by 3H-thymidine incorporation. (A) Proliferation of OT-I T cells in the presence of CD11c+ or CD11c− lymph node presenting cells purified from draining mediastinal lymph nodes and nondraining brachial lymph nodes. (B) Proliferation of bulk influenza virus-specific T cells in the presence of CD11c+ and CD11c− cells enriched from the draining lymph nodes in part A.
Influenza Virus Induces Maturation of DCs In Vivo.
Because influenza infection was associated with an immune response to coadministered OVA, we looked for the development of mature DCs in the mediastinal lymph nodes, using changes in cell surface phenotype as a criterion. Mice inhaled aerosolized OVA or influenza, and then we evaluated the expression of maturation markers on CD11c+ and CD11c− cells 72 h later. The expression of CD80, CD86, and MHC II were each up-regulated on CD11c+ cells from influenza virus–infected animals, when compared with cells from mice that were mock infected or received egg OVA only (Fig. 5). As Akbari et al. (9) recently described the induction of ICOS-L as well as CD80 and CD86 after intranasal OVA, we also checked for expression of ICOS-L on mediastinal lymph node DCs 3 d after inhalation of LPS-free OVA. While CD80 and CD86 expression increased markedly after influenza virus infection, ICOS-L was only slightly up-regulated and only in the presence of influenza virus (unpublished data). The influenza virus infection also had an impact on the number of CD11c+ cells in the draining lymph nodes, because the frequency of CD11c+ cells and the total number of mediastinal lymph node cells were each increased up to twofold compared with mock infected mice or mice administered egg OVA. Influenza infection also was accompanied by an expansion of CD11c− CD11b+ macrophages expressing or not expressing the F4/80 antigen (Fig. 5 C, arrows), but in contrast to DCs, B cells and macrophages did not show increased expression of CD80/86 (Figs. 5, B and D). Overall these results demonstrate that influenza virus leads to DC maturation without inducing T cell costimulatory molecules on B cells and macrophages.
Figure 5.
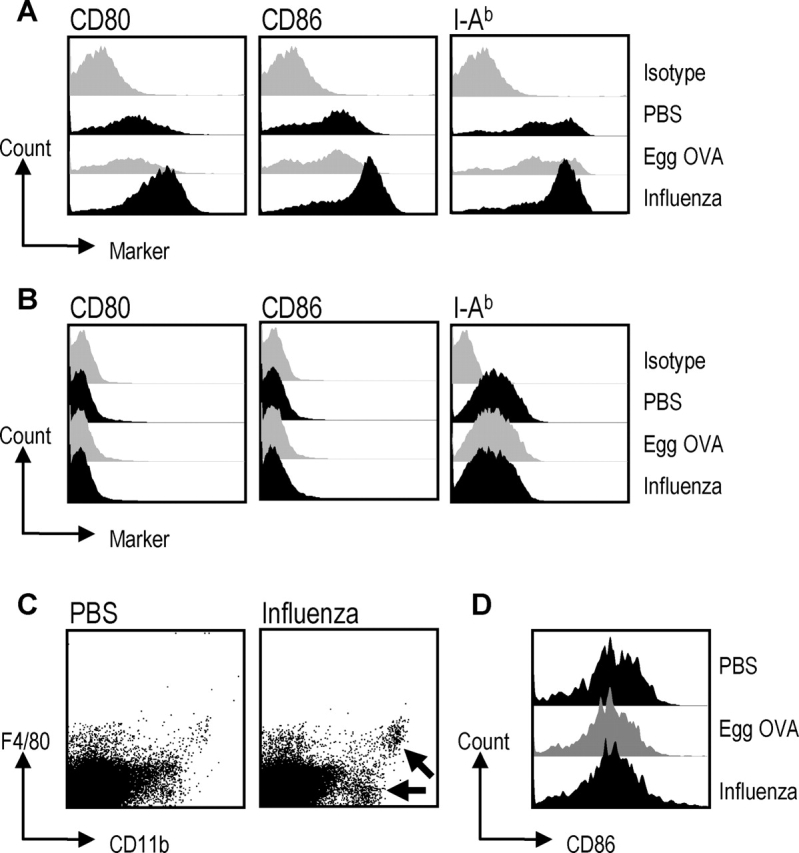
Influenza virus induces maturation of DCs from the draining mediastinal lymph nodes. Six B6 mice/group were administered X-31, PBS, or egg OVA at time point 0. The egg OVA group received OVA at 24 and 48 h also. 72 h after infection CD11c+ and CD11c− cells were purified from the draining lymph nodes and stained for expression of CD80, CD86, MHC II, B220, CD11b, and F4/80. (A) The DC population is gated on CD11c+ cells, (B) the B cell population is gated on B220+ cells and (C and D) the macrophages are gated as CD11c−, CD11b+, and F4/80+ or F4/80− (arrows to the two macrophage subsets). Data are representative of at least two experiments.
The Maturation Status of the DCs Correlates with the Ability to Induce Production of OVA-specific Antibodies.
To test if the maturation status of the DCs was correlated with the priming of OVA-specific immunity, we first assessed maturation at several time points after influenza virus infection. 20 h after infection, we did not observe any differences in the levels of costimulatory molecules (CD80 and CD86) and MHC II between influenza virus and mock-infected mice. However, by 45 h we detected an up-regulation of CD80, CD86, and MHC II in mice infected with influenza virus, and this effect was maximal at 72 h (Fig. 6 A). At 96 h, the expression of maturation markers was slightly reduced compared with the 72 h time point. At day 7, we observed a down-regulation of maturation markers compared with the mock-infected mice, and this down-regulation was even more pronounced at day 10 (unpublished data). At the same time points during the course of influenza infection (0, 20, 45, 72, 96 h, 7 d, 10 d), we aerosolized a single dose of egg OVA and evaluated the OVA-specific antibody response as a read-out for immune priming. As shown in Fig. 6 B, the production of OVA-specific antibodies correlated with the degree of maturation of the DCs. The best responses were obtained when the maturation of the DCs was peaking, whereas negligible OVA-specific antibody responses were induced when OVA was given at 0, 20 h, or days 7 and 10. These results indicate that the maturation of DCs in vivo correlates well with the induction of immune responses.
Figure 6.
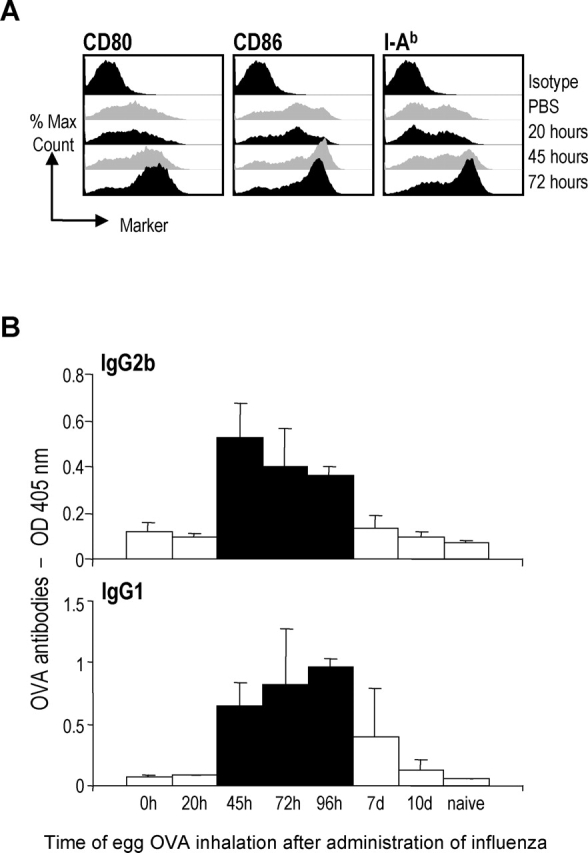
Influenza virus induced DCs maturation correlates with production of OVA-specific antibodies. (A) Six B6 mice/group were infected with X-31 or mock infected and CD11c+ cells were purified from the draining LNs 20, 45 and 72 h after infection and stained for expression of CD80, CD86, and MHC II. (B) Four mice/group were infected with X-31 at time point 0. At various time points post virus infection (0, 20, 45, 72, 96 h, 7 d, and 10 d), the mice received a single dose of egg OVA. 22 d after virus infection, the serum was analyzed for OVA-specific IgG1 and IgG2b antibodies. All the black bars are significantly different from the white bars (P < 0.05) except for day 7 in the IgG1 chart. Data are expressed as mean ± SD and shown at 1:10 serum dilutions. Data are representative of three experiments.
OVA-specific T Cell Immunity Is Associated with Protection against Vaccinia OVA Infection.
To test if the strong T cell responses to OVA in influenza infected mice also led to OVA-specific resistance to infection, a different recombinant OVA virus was used. A vaccinia virus carrying the full length OVA gene has been shown to present OVA-derived peptides on the surface of infected cells. We infected B6 mice with influenza virus and administered egg OVA on day 2, 3, and 4, then rested the mice for 3 wk before boosting with influenza B virus and 3 doses of egg OVA. Influenza B virus infects mice with similar kinetics to influenza A virus, but neither T cells nor antibodies elicited to influenza A cross react with the influenza B used to boost OVA immunity. The boost with a second heterologous virus was needed to induce significant protection to vaccinia-OVA challenge. 3 wk after the second boost, the mice were challenged with vaccinia-OVA intranasally. 7 d after infection the lungs were harvested and virus particles in the lungs were evaluated to assess the level of protection imparted by the OVA-specific T cell response.
As shown in Fig. 7 A, mice treated with both influenza and egg OVA had a significantly lower vaccinia virus titer compared with all the control groups. The influenza group, the most important control group, had 7 times higher vaccinia titers than influenza egg OVA groups, while the two other control groups had 40–60 times higher virus titers. Losses of body weight, as a manifestation of influenza infection, were also monitored (Fig. 7 B). The weight loss data correlated with the virus titers in the lungs, i.e., there was much less weight loss in protected animals with lower virus titers. We also assayed the production of IFN-γ from CD8+ T cells in the spleens of OVA-vaccinia challenged mice and found that mice administered egg OVA in the presence of influenza virus had a higher proportion of IFN-γ producing CD8+ T cells when restimulated with the OT-I peptide (SIINFEKL) relative to the other experimental groups (Fig. 7 C). In the same mice we analyzed the OVA specific antibody response after administration of egg OVA to follow the priming of the mice. As shown in Fig. 7 D, the level of OVA-specific antibodies increased from the first immunization to the second OVA exposure, and after challenge with vaccinia–OVA, antibody levels were further enhanced. Importantly, there was no demonstrable OVA-specific antibodies in mice administered egg OVA only, even after exposure to OVA seven times over a 7-wk period. These mice failed to make any detectable level of antibody, emphasizing that endotoxin free OVA is not immunogenic via the airway but induces protective OVA-specific immunity if inhaled during an influenza infection.
Figure 7.

Generation of protective immunity following coadministration of OVA and influenza virus. B6 mice were infected with aerosolized X-31 on day 1 and administered egg OVA on day 2, 3, and 4. 3 wk after infection the groups that received X-31 (influenza + egg OVA and influenza) were infected with B/Lee and boosted with egg OVA for three consecutive days (only influenza + egg OVA and egg OVA groups). 3 wk after infection with B/Lee all the groups were challenged with 105 pfu/mouse Vaccinia-OVA intranasally. 7 d after vaccinia-OVA infection the lungs were harvested and analyzed for virus and the spleen was assayed for IFN-γ–producing T cells. Also, the serum was collected after each administration of egg OVA to measure production of OVA specific antibodies. (A) Vaccinia virus titer in lungs. Data are expressed as mean ± SD from groups of five mice. The difference between the influenza + egg OVA and the influenza group was statistically significant (*P < 0.05). Data are representative of four independent experiments. (B) Weight loss in mice infected with vaccinia–OVA. (C) Production of IFN-γ from CD8 T cells from spleen cells restimulated with SIINFEKL. Data are representative of two independent experiments. (D) OVA specific IgG1 antibody production after primary immunization (influenza A + egg OVA), secondary immunization (influenza B + egg OVA), and tertiary immunization (vaccinia-OVA). Antibodies (IgG1 and IgG2b [unpublished data]) were observed only in the serum of animals given OVA and virus infection. The results shown are 1:100 serum dilutions but identical results were observed with a 1:10 dilution. Data are expressed as mean ± SD from groups of five mice.
Discussion
The Interaction of a Nonimmunogenic Airway Protein with DCs in the Steady-State.
The immature DCs within lymphoid organs are able to capture antigens by a variety of routes, e.g., as solutes in the fluid phase or bound to uptake receptors (29, 31, 32) and as particles during phagocytosis (33–35). Uptake can take place without ostensible perturbation of DC markers and function. Following uptake, the antigens are presented to T cells, but the T cells are not immunized, failing to develop effector functions and memory (29, 31, 33). In this paper, we have monitored the response of DCs and the immune system to inhalation of an endotoxin-free preparation of OVA. We first noted that our preparations of OVA were nonimmunogenic at the level of B and T cell responses (Fig. 1, A and C). In most prior studies, even those concluding that OVA could introduce tolerance after intranasal application (36), priming for IgG but not IgE antibody responses was observed. Nevertheless, DCs in the mediastinal lymph nodes that drain the lower airway, but not the cervical nodes that drain the upper airway, acquire and present LPS-free OVA on MHC class I and II products. The mediastinal (peribronchial) lymph node DCs otherwise were not perturbed relative to untreated mice with respect to several cell markers that characteristically increase during inflammation and infection, e.g., CD40, CD80, CD86, ICOS-L, and MHC class II (Fig. 5). This finding is also different from prior reports in which intranasal application of OVA led to marked increases in the expression of these molecules (9). Importantly, the immunologic outcome for T cells in both instances can be tolerance to OVA. In the seminal studies of Akbari et al. (27), the administration of OVA led to the induction of regulatory T cells that could produce high levels of IL-10 and suppress OVA-specific CD4+ T cell reactivity. In our studies with endotoxin free OVA, we did not detect IL-10 in responding T cells, but we did observe tolerance (Fig. 1 D). However, we did not investigate the underlying mechanism, e.g., to assess whether tolerance might involve peripheral T cell deletion, as has been observed with DCs presenting other antigens in the steady-state (29, 31, 33), anergy, or regulatory T cell induction. Nevertheless, our studies show that immature DCs in lymph nodes need not ignore nonimmunogenic proteins in the airway and can efficiently present these as MHC–peptide complexes in the obstensible absence of DC maturation, and B and T cell priming.
The Response to Endotoxin-free OVA When Inhaled during the Course of Airway Infection with Influenza Virus.
When DCs in lymphoid tissues are exposed in vivo to defined maturation stimuli, such as LPS (37), CpG deoxyoligonucleotides (38), NKT cells (39) and CD40 agonists (29, 31, 33), the DCs markedly up-regulate the expression of T cell costimulatory molecules and become effective inducers of immunity. In this paper, we find that DCs in the draining lymph nodes selectively undergo maturation over a period of 2–3 d after inhalation of influenza (Figs. 5 and 6 A). These kinetics are much slower than a recent report of maturation within 6 h of administration of influenza (17). We do not understand the basis for this difference in kinetics, since in our hands, influenza titres expand considerably between day 2 and 3, when the maturation of DCs becomes evident. In any case, when OVA was aerosolized after the onset of influenza infection, immunogenicity was observed simultaneous with the presence of maturing DCs, i.e., when the OVA was inhaled 2–3 d after the administration of influenza. The response to OVA in the presence of influenza virus coinfection was primarily of the Th1 type, based on the generation of IgG2a (BALB/c unpublished data) or IgG2b (B6) but no IgE antibodies, and the expansion of IFN-γ secreting CD4+ and CD8+ T cells, as well as lytic CD8+ T cells. Animals immunized with OVA during a virus infection also had improved recovery to a subsequent challenge with a recombinant vaccinia virus expressing full length OVA.
Prior studies in which OVA was coadministered with influenza virus also have observed immune responses to OVA alone but most of these experiments used adjuvants like alum, and very likely had LPS in the OVA. Also the OVA was administered intranasally, a practice which leads to fluid accumulation and potentially immunostimulating organisms into the lower respiratory tract. The exception is the report by O'Donnell, where OVA was administered by aerosol and OVA-specific antibodies were only observed if given during virus infection (25); the endotoxin contamination was not determined in this study, however. In the current study, we used an endotoxin free OVA preparation and gave the antigen as a small particle aerosol mist without any adjuvants. Under these circumstances, influenza virus coinfection and DC maturation were associated with the induction of strong OVA-specific immunity to the otherwise nonimmunogenic protein. Therefore, influenza-associated, DC maturation likely has a pivotal role in initiating immunity, although it remains important experimentally to develop the means to selectively block maturation in the presence of infection to certify that immunity will not develop.
The Danger of Infection-induced DC Maturation and a Solution via Immature DCs.
Our results demonstrate that respiratory virus infection is altering the response to a nonrelated harmless antigen, such as endotoxin-free OVA, to allow for the generation of a strong Th1 response. This illustrates the risk to DC maturation, which is that during infection, DCs, at least at the population level, should capture and present a mix of microbial antigens, innocuous self-antigens, and environmental proteins. This capacity of DCs to induce strong cell-mediated immunity to an otherwise harmless airway protein during infection was shown by the production of high levels of IFN-γ and IL-2 from OT-I and OT-II T cells infused into animals that received OVA protein in the presence of virus infection (but not when OVA alone was administered; Fig. 3). In light of the results showing that OVA peptides were presented in both instances, without or with influenza infection, the alterations in the functional T cell outcome likely results from the maturation of the DCs. The cells involved in presenting antigen in the draining lymph nodes were CD11c+ cells (Fig. 4) and as such, very likely to be DCs (40, 41). As mentioned above, when we did kinetic studies, CD11c+ DCs (but not B cells and macrophages, Fig. 5) collected from the draining lymph nodes of infected mice showed a strong up-regulation of MHC class II, as well as costimulatory molecules such as CD80 and CD86. This became detectable by 45 h after infection, peaked at 72 h and was no longer evident at 7 d, even though virus was still present in the lungs at this juncture. Interestingly, when a single dose of OVA was administered at various time points beginning at the time of virus infection and continuing until day 10, the ability of influenza virus infection to promote a response to OVA correlated exactly with the presence in the lymph nodes of mature CD11c+ cells. Several stimuli could contribute to influenza virus–induced maturation of DCs, including inflammatory cytokines like TNF and IL-1, type I interferons, and TLR3 signaling.
The DCs within lymphoid tissues in the steady-state seem able to deal with the dilemma that is inherent in DC maturation. These DCs can capture and process antigens to form MHC–peptide complexes (Figs. 2 and 3), but the T cells can be tolerized, e.g., by deletion (29, 31, 33). In the studies of Umetsu's group, intranasal OVA induced a limited form of tolerance in which IgE antibody responses were ablated (36) but IgG responses were not and IL-10 producing regulatory T cells were induced (27). These mechanistic possibilities were not directly analyzed here. What is emphasized here is the capacity of immature DCs in lymph nodes to capture and present LPS-free and normally nonimmunogenic proteins from the airway, so efficiently that the initial T cell proliferative response is substantial even when compared with the OVA–T cell responses in influenza virus infection. However, unless the animals are also infected with influenza virus, the OVA-specific T cells do not differentiate to produce cytokines, become killer cells, or help for antibody formation. The steady-state functions of DCs in lymphoid tissues seem critical for defining immunologic self and ensuring that the T cell repertoire is conditioned such that immunity to self and environmental proteins does not develop when DCs are called upon to mature and present microbial antigen. Otherwise, harmless proteins would induce combined B and T immunity when inhaled during an infection like influenza.
Acknowledgments
We thank P. Palese and A. Flandorfer for providing the B/Lee virus, J.W. Yewdell for the vaccinia virus expressing full length OVA, and J. Schulman for helpful discussions. We also thank D. Darguste for excellent technical assistance and D. Bonnyay and J. Adams for help with the illustrative material.
This work was supported by grants from the National Institutes of Health, AI41111 and AI48204 (to T. Moran) and AI13013 and AI051573 (to R.M. Steinman) and by a fellowship from the Instituto Mexicano del Seguro Social to L. Bonifaz.
M.K. Brimnes and L. Bonifaz contributed equally to this work.
References
- 1.Holt, P.G., M.A. Schon-Hegrad, J. Oliver, B.J. Holt, and P.G. McMenamin. 1990. A contiguous network of dendritic antigen-presenting cells within the respiratory epithelium. Int. Arch. Allergy Appl. Immunol. 91:155–159. [DOI] [PubMed] [Google Scholar]
- 2.Nelson, D.J., C. McMenamin, A.S. McWilliam, M. Brenan, and P.G. Holt. 1994. Development of the airway intraepithelial dendritic cell network in the rat from class II major histocompatibility (Ia)-negative precursors: differential regulation of Ia expression at different levels of the respiratory tract. J. Exp. Med. 179:203–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sertl, K., T. Takemura, E. Tschachler, V.J. Ferrans, M.A. Kaliner, and E.M. Shevach. 1986. Dendritic cells with antigen-presenting capability reside in airway epithelium, lung parenchyma, and visceral pleura. J. Exp. Med. 163:436–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gong, J.L., K.M. McCarthy, J. Telford, T. Tamatani, M. Miyasaka, and E.E. Schneeberger. 1992. Intraepithelial airway dendritic cells: a distinct subset of pulmonary dendritic cells obtained by microdissection. J. Exp. Med. 175:797–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Havenith, C.E., P.P. van Miert, A.J. Breedijk, R.H. Beelen, and E.C. Hoefsmit. 1993. Migration of dendritic cells into the draining lymph nodes of the lung after intratracheal instillation. Am. J. Respir. Cell Mol. Biol. 9:484–488. [DOI] [PubMed] [Google Scholar]
- 6.Holt, P.G., M.A. Schon-Hegrad, and J. Oliver. 1988. MHC class II antigen-bearing dendritic cells in pulmonary tissues of the rat. Regulation of antigen presentation activity by endogenous macrophage populations. J. Exp. Med. 167:262–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xia, W., C.E. Pinto, and R.L. Kradin. 1995. The antigen-presenting activities of Ia+ dendritic cells shift dynamically from lung to lymph node after an airway challenge with soluble antigen. J. Exp. Med. 181:1275–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vermaelen, K.Y., I. Carro-Muino, B.N. Lambrecht, and R.A. Pauwels. 2001. Specific migratory dendritic cells rapidly transport antigen from the airways to the thoracic lymph nodes. J. Exp. Med. 193:51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Akbari, O., G.J. Freeman, E.H. Meyer, E.A. Greenfield, T.T. Chang, A.H. Sharpe, G. Berry, R.H. DeKruyff, and D.T. Umetsu. 2002. Antigen-specific regulatory T cells develop via the ICOS-ICOS-ligand pathway and inhibit allergen-induced airway hyperreactivity. Nat. Med. 8:1024–1032. [DOI] [PubMed] [Google Scholar]
- 10.Steinman, R.M., and M.C. Nussenzweig. 2002. Avoiding horror autotoxicus: the importance of dendritic cells in peripheral T cell tolerance. Proc. Natl. Acad. Sci. USA. 99:351–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schuler, G., and R.M. Steinman. 1985. Murine epidermal Langerhans cells mature into potent immunostimulatory dendritic cells in vitro. J. Exp. Med. 161:526–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Inaba, K., G. Schuler, M.D. Witmer, J. Valinksy, B. Atassi, and R.M. Steinman. 1986. Immunologic properties of purified epidermal Langerhans cells. Distinct requirements for stimulation of unprimed and sensitized T lymphocytes. J. Exp. Med. 164:605–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Romani, N., S. Koide, M. Crowley, M. Witmer-Pack, A.M. Livingstone, C.G. Fathman, K. Inaba, and R.M. Steinman. 1989. Presentation of exogenous protein antigens by dendritic cells to T cell clones. Intact protein is presented best by immature, epidermal Langerhans cells. J. Exp. Med. 169:1169–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang, Q., D. Liu, P. Majewski, L.C. Schulte, J.M. Korn, R.A. Young, E.S. Lander, and N. Hacohen. 2001. The plasticity of dendritic cell responses to pathogens and their components. Science. 294:870–875. [DOI] [PubMed] [Google Scholar]
- 15.Cella, M., M. Salio, Y. Sakakibara, H. Langen, I. Julkunen, and A. Lanzavecchia. 1999. Maturation, activation, and protection of dendritic cells induced by double-stranded RNA. J. Exp. Med. 189:821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lopez, C.B., A. Garcia-Sastre, B.R.G. Williams, and T.M. Moran. 2003. The type 1 IFN induction pathway, but not released IFN, participates in the maturation of dendritic cells induced by negative strand RNA viruses. J. Infect. Dis. 187:1126–1136. [DOI] [PubMed] [Google Scholar]
- 17.Legge, K.L., and T.J. Braciale. 2003. Accelerated migration of respiratory dendritic cells to the regional lymph nodes is limited to the early phase of pulmonary infection. Immunity. 18:265–277. [DOI] [PubMed] [Google Scholar]
- 18.Yamamoto, N., S. Suzuki, Y. Suzuki, A. Shirai, M. Nakazawa, M. Suzuki, T. Takamasu, Y. Nagashima, M. Minami, and Y. Ishigatsubo. 2001. Immune response induced by airway sensitization after influenza A virus infection depends on timing of antigen exposure in mice. J. Virol. 75:499–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamamoto, N., S. Suzuki, A. Shirai, M. Suzuki, M. Nakazawa, Y. Nagashima, and T. Okubo. 2000. Dendritic cells are associated with augmentation of antigen sensitization by influenza A virus infection in mice. Eur. J. Immunol. 30:316–326. [DOI] [PubMed] [Google Scholar]
- 20.Sakamoto, M., S. Ida, and T. Takishima. 1984. Effect of influenza virus infection on allergic sensitization to aerosolized ovalbumin in mice. J. Immunol. 132:2614–2617. [PubMed] [Google Scholar]
- 21.Suzuki, S., Y. Suzuki, N. Yamamoto, Y. Matsumoto, A. Shirai, and T. Okubo. 1998. Influenza A virus infection increases IgE production and airway responsiveness in aerosolized antigen-exposed mice. J. Allergy Clin. Immunol. 102:732–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schwarze, J., E. Hamelmann, K.L. Bradley, K. Takeda, and E.W. Gelfand. 1997. Respiratory syncytial virus infection results in airway hyperresponsiveness and enhanced airway sensitization to allergen. J. Clin. Invest. 100:226–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leibovitz, E., J. Freihorst, P.A. Piedra, and P.L. Ogra. 1988. Modulation of systemic and mucosal immune responses to inhaled ragweed antigen in experimentally induced infection with respiratory syncytial virus implication in virally induced allergy. Int. Arch. Allergy Appl. Immunol. 86:112–116. [DOI] [PubMed] [Google Scholar]
- 24.Dakhama, A., A.M. Bramley, N.G. Chan, K.O. McKay, R.R. Schellenberg, and R.G. Hegele. 1999. Effect of respiratory syncytial virus on subsequent allergic sensitization to ovalbumin in guinea-pigs. Eur. Respir. J. 13:976–982. [DOI] [PubMed] [Google Scholar]
- 25.O'Donnell, D.R., and P.J. Openshaw. 1998. Anaphylactic sensitization to aeroantigen during respiratory virus infection. Clin. Exp. Allergy. 28:1501–1508. [DOI] [PubMed] [Google Scholar]
- 26.Eisenbarth, S.C., D.A. Piggott, J.W. Huleatt, I. Visintin, C.A. Herrick, and K. Bottomly. 2002. Lipopolysaccharide-enhanced, toll-like receptor 4-dependent T helper cell type 2 responses to inhaled antigen. J. Exp. Med. 196:1645–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Akbari, O., R.H. DeKruyff, and D.T. Umetsu. 2001. Pulmonary dendritic cells producing IL-10 mediate tolerance induced by respiratory exposure to antigen. Nat. Immunol. 2:725–731. [DOI] [PubMed] [Google Scholar]
- 28.Lopez, C.B., A. Fernandez-Sesma, S.M. Czelusniak, J.L. Schulman, and T.M. Moran. 2000. A mouse model for immunization with ex vivo virus-infected dendritic cells. Cell. Immunol. 206:107–115. [DOI] [PubMed] [Google Scholar]
- 29.Bonifaz, L., D. Bonnyay, K. Mahnke, M. Rivera, M.C. Nussenzweig, and R.M. Steinman. 2002. Efficient targeting of protein antigen to the dendritic cell receptor DEC-205 in the steady state leads to antigen presentation on major histocompatibility complex class I products and peripheral CD8+ T cell tolerance. J. Exp. Med. 196:1627–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lopez, C.B., A. Fernandez-Sesma, J.L. Schulman, and T.M. Moran. 2001. Myeloid dendritic cells stimulate both th1 and th2 immune responses depending on the nature of the antigen. J. Interferon Cytokine Res. 21:763–773. [DOI] [PubMed] [Google Scholar]
- 31.Hawiger, D., K. Inaba, Y. Dorsett, M. Guo, K. Mahnke, M. Rivera, J.V. Ravetch, R.M. Steinman, and M.C. Nussenzweig. 2001. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J. Exp. Med. 194:769–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Crowley, M., K. Inaba, and R.M. Steinman. 1990. Dendritic cells are the principal cells in mouse spleen bearing immunogenic fragments of foreign proteins. J. Exp. Med. 172:383–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu, K., T. Iyoda, M. Saternus, Y. Kimura, K. Inaba, and R.M. Steinman. 2002. Immune tolerance after delivery of dying cells to dendritic cells in situ. J. Exp. Med. 196:1091–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kamath, A.T., J. Pooley, M.A. O'Keeffe, D. Vremec, Y. Zhan, A.M. Lew, A. D'Amico, L. Wu, D.F. Tough, and K. Shortman. 2000. The development, maturation, and turnover rate of mouse spleen dendritic cell populations. J. Immunol. 165:6762–6770. [DOI] [PubMed] [Google Scholar]
- 35.Iyoda, T., S. Shimoyama, K. Liu, Y. Omatsu, Y. Akiyama, Y. Maeda, K. Takahara, R.M. Steinman, and K. Inaba. 2002. The CD8+ dendritic cell subset selectively endocytoses dying cells in culture and in vivo. J. Exp. Med. 195:1289–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsitoura, D.C., R.H. DeKruyff, J.R. Lamb, and D.T. Umetsu. 1999. Intranasal exposure to protein antigen induces immunological tolerance mediated by functionally disabled CD4+ T cells. J. Immunol. 163:2592–2600. [PubMed] [Google Scholar]
- 37.De Smedt, T., B. Pajak, E. Muraille, L. Lespagnard, E. Heinen, P. De Baetselier, J. Urbain, O. Leo, and M. Moser. 1996. Regulation of dendritic cell numbers and maturation by lipopolysaccharide in vivo. J. Exp. Med. 184:1413–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sparwasser, T., E.S. Koch, R.M. Vabulas, K. Heeg, G.B. Lipford, J.W. Ellwart, and H. Wagner. 1998. Bacterial DNA and immunostimulatory CpG oligonucleotides trigger maturation and activation of murine dendritic cells. Eur. J. Immunol. 28:2045–2054. [DOI] [PubMed] [Google Scholar]
- 39.Fujii, S., K. Shimizu, C. Smith, L. Bonifaz, and R.M. Steinman. 2003. Activation of natural killer T cells by α-galactosylceramide rapidly induces the full maturation of dendritic cells in vivo and thereby acts as an adjuvant for combined CD4 and CD8 T cell immunity to a co-administered protein. J. Exp. Med. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Metlay, J.P., M.D. Witmer-Pack, R. Agger, M.T. Crowley, D. Lawless, and R.M. Steinman. 1990. The distinct leukocyte integrins of mouse spleen dendritic cells as identified with new hamster monoclonal antibodies. J. Exp. Med. 171:1753–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Crowley, M.T., K. Inaba, M.D. Witmer-Pack, S. Gezelter, and R.M. Steinman. 1990. Use of the fluorescence activated cell sorter to enrich dendritic cells from mouse spleen. J. Immunol. Methods. 133:55–66. [DOI] [PubMed] [Google Scholar]