

Abstract
Programmed death-1 (PD-1) receptor, an inhibitory costimulatory molecule found on activated T cells, has been demonstrated to play a role in the regulation of immune responses and peripheral tolerance. We investigated the role of this pathway in the development of autoimmune diabetes. PD-1 or PD-L1 but not PD-L2 blockade rapidly precipitated diabetes in prediabetic female nonobese diabetic (NOD) mice regardless of age (from 1 to 10-wk-old), although it was most pronounced in the older mice. By contrast, cytotoxic T lymphocyte–associated antigen 4 (CTLA-4) blockade induced disease only in neonates. Male NOD mice also developed diabetes after PD-1–PD-L1 pathway blockade, but NOR mice, congenic to NOD but resistant to the development of diabetes, did not. Insulitis scores were significantly higher and frequency of interferon γ–producing GAD-reactive splenocytes was increased after PD-1–PD-L1 pathway blockade compared with controls. Interestingly, PD-L1 but not PD-L2 was found to be expressed on inflamed islets of NOD mice. These data demonstrate a central role for PD-1–PD-L1 interaction in the regulation of induction and progression of autoimmune diabetes in the NOD mouse and provide the rationale to develop new therapies to target this costimulatory pathway in this disease.
Keywords: diabetes mellitus, insulin-dependent; mice, inbred NOD; autoimmunity; self-tolerance; programmed cell death protein 1
Introduction
Termination of an immune response is achieved through a number of different mechanisms (1) including regulatory costimulatory molecules (2). Overall, the balance between the positive and negative signaling costimulatory pathways dictates the fate of individual T cells and the immune response. The inhibitory costimulatory molecule programmed death-1 (PD-1)*and its ligands, PD-L1 and PD-L2, have been shown to play an important role in regulating T cell activation and peripheral tolerance (3–6). PD-1 deficiency leads to lupus-like syndrome with glomerulonephritis and arthritis or fatal dilated cardiomyopathy, depending on the genetic background of the animal (7, 8). We investigated the role of this novel inhibitory costimulatory pathway in regulating the autoimmune response in nonobese diabetic (NOD) mice.
The NOD mouse has proved to be a valuable model for studying autoimmune diabetes. Indeed, the recent report by Herold et al. (9) on the efficacy of nonmitogenic anti-CD3 mAb in humans with type 1diabetes was a direct translation of the original studies reported in the NOD mouse almost a decade earlier (10). In NOD mice, spontaneous insulitis, the hallmark pathologic lesion, evolves through several characteristic stages that begin with peri-insulitis and end with invading and destructive insulitis and overt diabetes. Peri-insulitis is first observed at 3–4 wk of age, invading insulitis at 8–10 wk, and destructive insulitis appears just before the onset of clinical diabetes, with the earliest cases at 10–12 wk. By 20 wk of age, 70–80% of females become diabetic. By contrast, in male NOD mice, diabetes is typically delayed with only a 40–50% incidence by 30 wk of age (11). Thus, before islet cell damage, a period of nondestructive inflammation is observed with a heavy cellular infiltrate surrounding the individual islets beginning at least 7 wk before the onset of overt diabetes (12). Interestingly, insulitis does not uniformly progress to disease. For example, in male mice insulitis is as prevalent as in females, but fewer males go on to develop overt diabetes (11). The mechanisms regulating the initiation and progression of insulitis to overt diabetes are unclear. However, a role for negative T cell signaling pathways through inhibitory receptors has been demonstrated in the initiation of disease in neonatal animals. Using the BDC2.5/NOD TCR transgenic model, in which diabetes develops in 15–30% of animals at 5–6 mo of age, blocking cytotoxic T lymphocyte–associated antigen 4 (CTLA-4) induced disease by 3 wk of age in almost all of the transgenic animals. Interestingly, late (after 17 d of age) blockade did not induce disease (13). Similarly, early (at 2–3 wk of age) anti–B7-1 mAb treatment of female NOD mice increased the incidence of diabetes and CTLA-4Ig or anti–B7-2 mAb treatment reduced the incidence of diabetes, whereas late (at 10 wk of age) treatment with any of these reagents had no effect on disease progression (14), suggesting that CD28/B7/CTLA-4 costimulatory pathway plays an important role in regulating disease induction but not progression. By contrast, our data demonstrate that PD-1–PD-L1 pathway is involved in the regulation of both initiation and progression of autoimmune diabetes in NOD mice.
Materials and Methods
Mice.
Female and male NOD, female NOR, BALB/c mice of various ages, and female NOD.SCID mice were obtained from The Jackson Laboratory and cared for in accordance with institutional guidelines.
Antibodies and Treatment Protocol.
The anti–mouse PD-1 mAb (J43, hamster IgG) has been described (4). The anti–mouse PD-L1 mAb (MIH6, rat IgG2a) and the anti–mouse PD-L2 mAb (TY25, rat IgG2a) were generated as recently described (15). PD-1–transfected BHK cells (baby hamster kidney cell line; reference 4) were pretreated with control hamster IgG (BD Biosciences) or the anti–PD-1 mAb (J43) and then stained with PD-L1-Ig (16) or PD-L2-Ig (17) followed by PE-labeled goat anti–human IgG (Caltag). Flow cytometry analysis showed that the anti–PD-1 mAb inhibited the binding of both PD-L1-Ig and PD-L2-Ig to PD-1 transfectants (Fig. 1 A). PD-L1-Ig and PD-L2-Ig were also preincubated with control rat IgG2a (BD Biosciences), anti–PD-L1 mAb (MIH6), or anti–PD-L2 mAb (TY25), and then used for staining of the PD-1/BHK cells. Flow cytometry analysis showed that anti–PD-L1 mAb inhibited the binding of PD-L1-Ig and anti-PD-L2 inhibited the binding of PD-L2-Ig specifically (Fig. 1 B). Fab fragments of these mAbs showed similar results (not depicted). These results indicate the blocking properties of the mAbs used in this study. Anti–CTLA-4 (4F10) mAb-producing hybridoma was provided by J. Bluestone (University of California San Francisco, San Francisco, CA). All mAbs were manufactured and purified by BioExpress Inc. Hamster IgG (ICN Pharmaceuticals Inc.) and rat IgG (Sigma-Aldrich) served as controls. All mAbs were given intraperitoneally: 500 μg on day 0, followed by 250 μg on days 2, 4, 6, 8, and 10.
Figure 1.
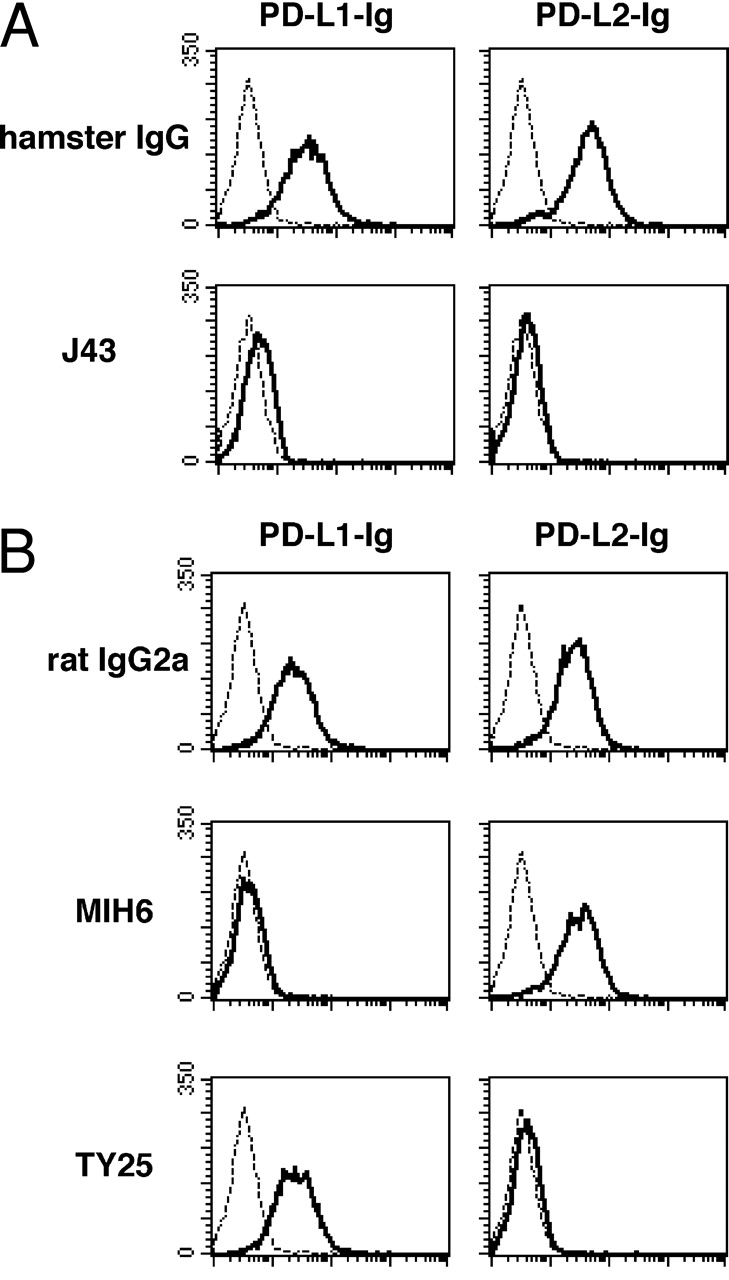
Blocking properties of mAbs against PD-1, PD-L1, and PD-L2. (A) Anti–PD-1 (J43) inhibits binding of both PD-L1-Ig and PD-L2-Ig (bold lines) to PD-1 transfectants. Dashed lines indicate staining with control human IgG. (B) Anti–PD-L1 (MIH6) inhibits PD-L1-Ig binding and anti–PD-L2 (TY25) inhibits PD-L2-Ig binding to PD-1 transfectants (bold lines). Dashed lines indicate staining with control human IgG.
Monitoring for Diabetes.
Clinical diabetes was defined as a random blood glucose reading of >250 mg/dL for three consecutive days. Blood glucose was measured by Accu-Chek Advantage glucometers (Roche Diagnostics). 4- and 10-wk-old mice were monitored daily by measuring blood glucose for the first 3 wk followed by three times a week until the mice were killed. The neonatal mice were monitored initially for urine glucose on alternate days until 3 wk of age, followed by blood glucose measurement according to the above schedule.
Histopathology.
Mice in the 4- and 10-wk-old group were killed on days 11–13 after initiation of treatment for histology specimens and harvesting of spleens for ELISPOT. Pancreas sections were stained with hematoxylin and eosin. Immunoperoxidase staining of frozen sections of pancreata for PD-1, PD-L1, and PD-L2 was performed using the appropriate purified mAbs and isotype controls using the avidin-biotin technique (Vector Laboratories). The sections were counter stained with hematoxylin. Insulitis scoring was performed as described by Yoon et al. (18) by examining at least 20 islets per mouse and graded as follows: grade 0, normal islets; grade 1, mononuclear infiltration, largely in the periphery, in <25% of the islet; grade 2, 25–50% of islet showing mononuclear infiltration; grade 3, >50% of islet showing mononuclear infiltration; and grade 4, small, retracted islet with few mononuclear cells.
Micro-Insulin Autoantibody Assay.
This assay was performed by G. Isenbarth's laboratory (Barbara Davis Center, Denver, CO), according to previously published methods (19).
ELISPOT.
Splenocytes from treated and control mice were obtained as single cell suspensions. The ELISPOT assay was adapted to measure IFN-γ–secreting cells. ELISASpot plates (Cellular Technology Limited) were coated with capture mAb against IFN-γ (BD Biosciences) in PBS and left overnight at 4°C. The plates were blocked with PBS-BSA 1% for 1 h and then washed with PBS. 106 splenocytes were added to each well in 100 μl complete RPMI medium containing 10% fetal calf serum (Sigma-Aldrich), 2 mM l-glutamine, 100 U/ml penicillin/streptomycin (BioWhittaker), and 50 mM 2-mercaptoethanol (Sigma-Aldrich). Control wells contained responder splenocytes plus medium alone. Test cells were added with GAD antigen (Diamyd Inc.) at 10 μg/ml. After 48 h, the plates were washed, biotinylated detection mAbs (BD Biosciences) were added, and the plates were left for an additional overnight incubation at 4°C. After further washing, horseradish peroxidase–conjugated avidin (DakoCytomation) was added for 2 h at room temperature. Color was developed with AEC (Sigma-Aldrich). The resulting spots were counted on a computer-assisted ELISASpot Image Analyzer (Cellular Technology Limited). The frequencies were then expressed as cytokine-producing cells per million splenocytes.
Flow Cytometry.
Single cell splenocyte suspensions were obtained and two color flow cytometry was performed. Cells were left unstimulated or activated with 10 μg/ml plate-bound anti-CD3 mAb (BD Biosciences) for 72 h, after which 106 cells were stained with anti–CD3-FITC (BD Biosciences) and either anti–PD-1–PE or anti–PD-L1–PE (eBioscience). Hamster IgG–PE or Rat Ig–PE (BD Biosciences) served as controls. Cells were analyzed on a FACSCalibur™ (Becton Dickinson) and the percentages of CD3+ or CD3− cells expressing PD-1 or PD-L1 were quantified using CELLQuest™ software (Becton Dickinson).
Results and Discussion
To explore the involvement of PD-1 and its ligands in the development of diabetes, we administered blocking mAbs against PD-1, PD-L1, and PD-L2 into prediabetic female NOD mice of various ages. In 10-wk-old female NOD mice, PD-L1 blockade by anti–PD-L1 mAb precipitated diabetes in 82.4% of the animals 6 d after initiation of therapy with nearly a quarter developing diabetes after a single injection of the antibody. Anti–PD-1 mAb had a similar effect (76.5% of mice developed diabetes). In contrast, only one mouse in the anti–PD-L2 mAb-treated group developed diabetes (not statistically significant over controls) and none in the anti–CTLA-4 mAb-treated group for the duration of follow up. The incidence of diabetes in our colony of unmanipulated NOD female mice is 20% at 10 wk of age, and >70% by 20 wk of age. However, in keeping with previous reports, control IgG–treated animals had slightly delayed onset of overt diabetes (20), demonstrating that the accelerated onset in our treated cohort was a true biological effect related to PD-1–PD-L1 blockade (Fig. 2 A). In 4-wk-old female NOD mice, anti–PD-L1 mAb treatment induced diabetes in 86.2% of mice by 20 d after initiation of treatment with the first cases appearing around day 10. Anti–PD-1 mAb had a less pronounced effect with over a third (41.4%) developing diabetes by day 15, and none of the animals developed diabetes in the anti–PD-L2 mAb-treated group. Again, anti–CTLA-4 mAb and control IgG did not result in diabetes for the duration of follow up (Fig. 2 B). By contrast, in 1-wk-old female NOD mice, anti–CTLA-4 mAb treatment was able to induce diabetes in 54.5% of mice by 28 d after initiation of treatment with the first cases appearing around day 21. PD-1–PD-L1 blockade was able to induce diabetes in 47.6% of mice in the anti–PD-1 group and 57.1% in the anti–PD-L1 group (not statistically significant vs. CTLA-4) and this effect was not seen until day 28, more than 1 wk after the first cases seen with anti–CTLA-4 antibody treatment (Fig. 2 C).
Figure 2.
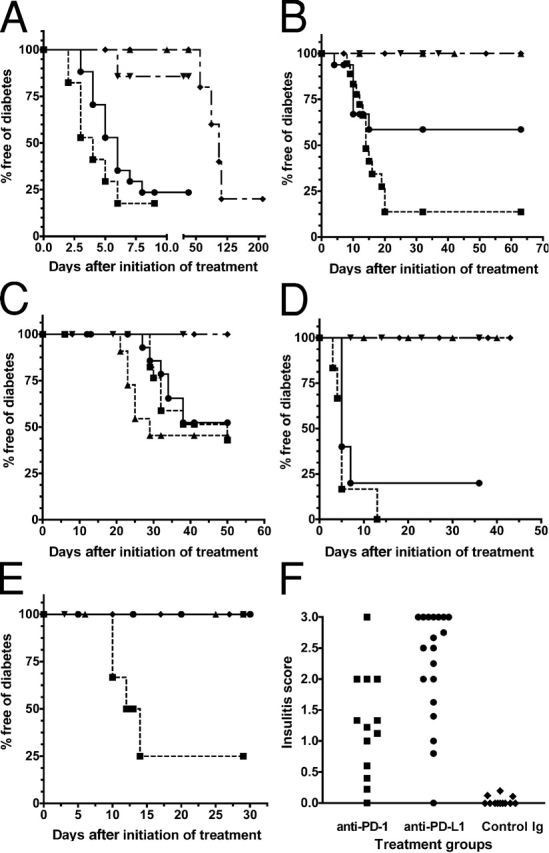
PD-1–PD-L1 blockade accelerates autoimmune diabetes in NOD mice. All p-values are for comparisons with respective controls. (A) Diabetes-free survival after PD-1–PD-L1 blockade in 10-wk-old female NOD mice: anti–PD-1 (•; n = 17; P < 0.0001), anti–PD-L1 (▪; n = 17; P < 0.0001), anti–PD-L2 (▾; n = 7; P = 0.1904), anti–CTLA-4 (▴; n = 6; P = NS), and control antibody (♦; n = 12) treatment. (B) Diabetes-free survival after PD-1–PD-L1 blockade in 4-wk-old female NOD mice: anti–PD-1 (•; n = 15; P < 0.0184), anti–PD-L1 (▪; n = 18; P < 0.0001), anti–PD-L2 (▾; n = 7; P = NS), anti–CTLA-4 (▴; n = 10; P = NS), and control antibody (♦; n = 12) treatment. (C) Diabetes-free survival after PD-1–PD-L1 blockade in 1-wk-old female NOD mice: anti–PD-1 (•; n = 18; P = 0.0071), anti–PD-L1 (▪; n = 18; P = 0.0018), anti–PD-L2 (▾; n = 11; P = NS), anti–CTLA-4 (▴; n = 11; P = 0.0016), and control antibody (♦; n = 10) treatment. (D) Diabetes-free survival after PD-1–PD-L1 blockade in 10-wk-old male NOD mice: anti–PD-1 (•; n = 5; P = 0.0126), anti–PD-L1 (▪; n = 5; P = 0.0126), and control antibody (♦; n = 5) treatment. In 10-wk-old female NOR mice: anti–PD-1 (▾; n = 5; P = NS) and anti–PD-L1 (▴; n = 5; P = NS) treatment. (E) Diabetes-free survival after PD-1–PD-L1 blockade in 4-wk-old male NOD mice: anti–PD-1 (•; n = 5; P = NS), anti–PD-L1 (▪; n = 6; P = 0.038), control Ig (♦; n = 5) treatment. In 4-wk-old female NOR mice: anti–PD-1 (▾; n = 5; P = NS) and anti–PD-L1 (▴; n = 6; P = NS) treatment. (F) Insulitis scores in 4–5-wk-old female NOD mice treated with anti–PD-1 (▪; mean score 1.249 ± 0.2349; P < 0.001), anti–PD-L1 (•; mean score 2.275 ± 0.1987; P < 0.001), and control IgG (♦; mean score 0.03634 ± 0.01987).
At 10 wk of age, in male NOD mice that rarely develop overt diabetes, PD-1–PD-L1 blockade had a similar effect to that seen in female NOD mice of the same age, with an 80% incidence of diabetes after anti–PD-1 and 100% after anti–PD-L1 mAb treatment. At 4 wk of age, anti–PD-1 mAb treatment had no effect whereas anti–PD-L1 mAb induced diabetes in 75% of mice by day 14 after initiation of treatment. Neither NOR mice (Fig. 2, D and E) nor BALB/c mice (not depicted) developed diabetes after PD-1–PD-L1 blockade, indicating that the effect observed in NOD mice is not due to nonspecific islet cell toxicity related to mAb treatment.
We then examined the pathology of pancreata of young (4–5-wk-old) diabetic mice from the experimental group in which treatment was started at 1 wk of age for evidence of insulitis to compare the effect of PD-1 or PD-L1 blockade with control mice (which normally exhibit minimal insulitis at this age) and with that of CTLA-4 blockade (the only experimental age group where CTLA-4 blockade induced disease). Pathologically, there was marked destructive insulitis in the young diabetic animals after either CTLA-4, PD-1, or PD-L1 blockade, whereas the control animals had minimal inflammation of the islets at that time (Fig. 3, A–D). Insulitis scores were significantly higher in the PD-1 (mean score 1.249 ± 0.2349) and PD-L1 (mean score 2.275 ± 0.1987) blockade groups compared with the control group (mean score 0.03634 ± 0.01987, P < 0.001 for both anti–PD-1 and anti–PD-L1 vs. controls; Fig. 2 F).
Figure 3.
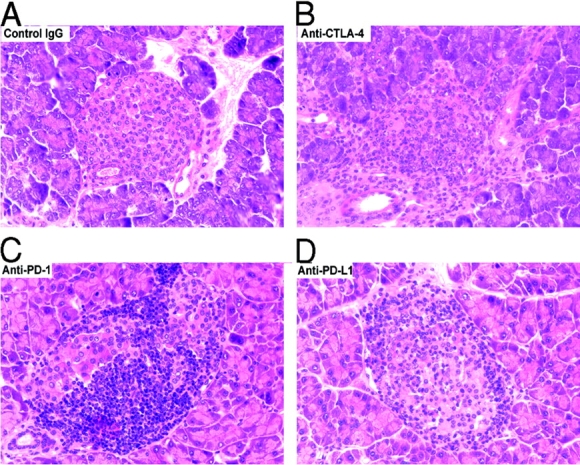
PD-1–PD-L1 and CTLA-4 blockade accelerates insulitis in prediabetic female NOD mice. (A) Islets from control IgG–treated mouse at 5 wk of age. (B–D) Inflamed islets from anti–CTLA-4, anti–PD-1, or anti–PD-L1 mAb-treated mice, respectively, at 5 wk of age (all stained with hematoxylin and eosin; ×400).
To assess whether PD-1–PD-L1 blockade exerted some of its diabetogenic effect by its action on B cells, we measured insulin autoantibody levels in 4–5-wk-old mice treated with PD-1–PD-L1 blockade, but found no correlation between insulin autoantibody levels and development of diabetes. Specifically, some animals developed diabetes with no antibodies and some developed antibodies but did not develop diabetes (not depicted).
To assess the effect of PD-1–PD-L1 blockade on islet-reactive T cells, we transplanted diabetic female NOD mice (at least 2 wk after the last mAb treatment) with islet isografts from female NOD.SCID mice. These isografts reestablish euglycaemia in the recipients until a time that they are destroyed by the islet-specific autoreactive T cells (21). In the mice that had developed spontaneous diabetes, islet isografts were rejected within 21 d (median survival was 15.5 d). By contrast, in the mice rendered diabetic after PD-1–PD-L1 blockade, islet isografts suffered accelerated rejection with five of six recipients losing their grafts within 3 d. These data suggest that after PD-1–PD-L1 blockade, autoaggressive T cells might be expanded in number or maintained in a heightened state of activation, capable of full effector function leading to accelerated destruction of islet isografts.
To address these two possibilities, we quantified the frequencies of IFN-γ–producing GAD-reactive T cells and assessed the state of activation of T cells in mice receiving anti–PD-1 mAb, anti–PD-L1 mAb, or control IgG. Using ELISPOT analysis we found increased frequency of IFN-γ–producing GAD-reactive splenocytes in 4- or 10-wk-old mice from anti–PD-1 or anti–PD-L1 mAb-treated group, compared with age-matched control IgG–treated mice (Fig. 4). Flow cytometry analysis showed that the percentages of CD4+ CD25+ and CD4 or CD8 T cells expressing a CD62Llow CD44high memory/effector phenotype were similar in the anti–PD-L1, anti–PD-1, and control groups (not depicted). Therefore, taken together, it appears that PD-1–PD-L1 blockade resulted in expansion of activated GAD-reactive cells. This is consistent with the results of Salama et al. (22), who used antigen-specific transgenic T cells and demonstrated a critical role of PD-1 in regulating autoimmune encephalomyelitis in the C57BL/6 mouse model. Interestingly, blockade of PD-L1 resulted in greater insulitis, overt diabetes, and higher frequencies of GAD-reactive T cells than PD-1 blockade. These data are in keeping with a recent report indicating that PD-L1 may mediate apoptosis of activated T cells through ligation of a receptor distinct from PD-1 (16).
Figure 4.

Increased frequency of IFN-γ–producing GAD-specific splenocytes in NOD mice treated with anti–PD- or anti–PD-L1 mAb. ELISPOT responses are expressed as number of IFN-γ spots per million splenocytes from anti–PD-1 mAb, anti–PD-L1 mAb, and control IgG–treated 4-wk-old female NOD mice.
MHC class I–restricted CD8+ T cells play an important role in the initiation and mediation of autoimmune β cell destruction (23). CD8+ T cells are more sensitive to the PD-1–PD-L1 inhibitory pathway and costimulation through CD28 can overcome PD-1–mediated inhibition of CD4+ but not CD8+ T cell responses through IL-2 production (24). This might be of significance in diabetes where in the context of inflamed islets, autoreactive CD8+ T cells would be inactivated after encounter with MHC class I on parenchymal cells coexpressing PD-L1 (5, 25). PD-L1 expression has also been demonstrated in a variety of human cancers and tumor cell lines (16). Iwai et al. (26), in a tumor immunity model, recently demonstrated that transgenic expression of PD-L1 in tumor cells rendered them less susceptible to the specific T cell antigen receptor–mediated lysis by cytotoxic T cells. We examined the expression of PD-1, PD-L1, and PD-L2 in NOD islets in unmanipulated animals with established insulitis. No staining was seen with isotype control or anti–PD-L2 mAb. PD-1 expression was seen only on infiltrating cells in the inflamed islets, whereas PD-L1 was limited to inflamed islets from unmanipulated 10-wk-old female NOD mice (Fig. 5 A). Uninflamed islets from younger NOD mice and those from BALB/c and C57Bl/6 mice lacked PD-L1 expression. Moreover, direct staining with anti–rat antibody of inflamed islets from a mouse that developed diabetes after anti–PD-L1 mAb treatment (Fig. 5 B) confirmed the localization of PD-L1 in the inflamed islets. These findings and the fact that a significant number of 10-wk-old female NOD mice developed diabetes after a single injection of the mAbs suggests that this pathway may inhibit the function of islet-reactive T cells already present at the site of inflammation. However, in younger mice, in whom islet expression of PD-L1 was not seen, blockade of this pathway also precipitated diabetes, albeit more gradually, and increased GAD-reactive T cell frequencies were found in the spleens of anti–PD-1– and anti–PD-L1–treated mice. Thus, the PD-1 pathway may regulate effector T cells within the target tissue and within the peripheral lymphoid organs. Moreover, in the experimental autoimmune encephalomyelitis (EAE) model, the findings that PD-L2 blockade augments disease despite minimal PD-L2 expression in the brain of animals with EAE (22) provides further evidence for lymphoid compartment regulation by this pathway.
Figure 5.
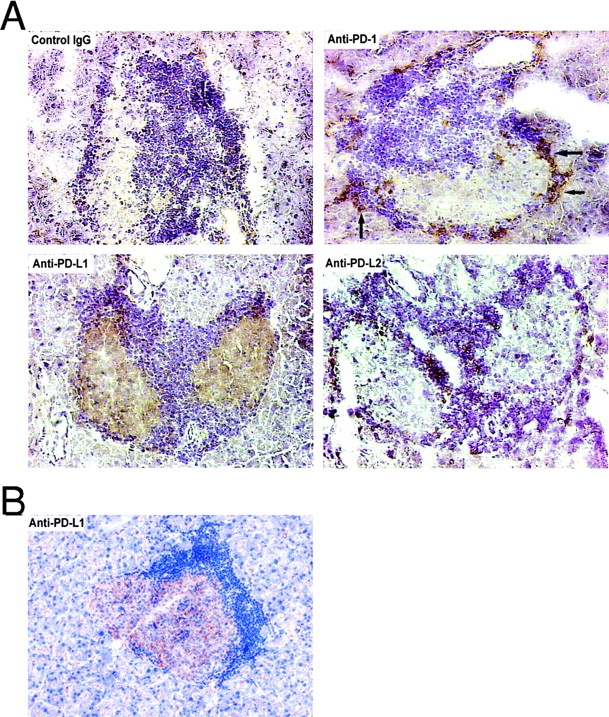
(A) PD-L1 expression in inflamed islets of NOD mice and PD-1 expression on infiltrating cells. Inflamed islets from 10-wk-old unmanipulated prediabetic female NOD mice showing negative staining with isotype control Ig, positive PD-1 staining of infiltrating cells (arrows), positive PD-L1 staining of islets, and negative PD-L2 staining (×400). (B) Injected anti–PD-L1 mAb binds to islets. Inflamed islet, from a 10-wk-old NOD mouse that developed diabetes after injection of anti–PD-L1, showing positive direct staining with anti–rat antibody (×400).
In the case of NOD mice, this inhibitory pathway ultimately fails resulting in diabetes as the mice age. This may in part relate to decreased levels of expression of PD-L1 or PD-1 in aging mice. Indeed, we found that anti-CD3–activated splenocytes from female NOD mice had decreasing percentages of cells expressing PD-L1 (Fig. 6, A–D) and decreasing mean fluorescence intensity of PD-L1 expression (not depicted) on both CD3+ and CD3− cells with increasing age, compared with age-matched control BALB/c mice. We found no change in PD-1 expression with age (not depicted).
Figure 6.



PD-L1 expression is down-regulated on stimulated splenocytes of older NOD mice. Expression of PD-L1 on CD3+ (A and B) or CD3− (C and D) splenocytes of female NOD (A and C) or BALB/c (B and D) mice of various ages before and after stimulation with immobilized anti-CD3 mAb for 72 h. *, P < 0.01 and **, P < 0.05 versus stimulated CD3+ cells from 9-wk-old female NOD mice.
Given the autoimmune phenotypes of PD-1–deficient mice and other in vitro data (7, 8), it appears that the PD-1 pathway is inhibitory. Our data demonstrate an important role for this pathway in the regulation of autoimmune diabetes in NOD mice, where PD-1–PD-L1 interaction inhibits the activation, expansion, and effector function of islet-reactive T cells. However, there are data suggesting that PD-L1 (B7-H1) costimulates T cell proliferation and IL-10 secretion (27), whereas PD-L2 (B7-DC) may activate naive T cells in vitro and promote Th1 cytokine production (17). Further, cross-linking of PD-L2 by IgM antibodies has been shown to activate the immune functions of dendritic cells and potentiate T cell responses (28). Therefore, there is also a remote possibility that the antibodies used in our experiments are stimulatory in vivo. However, this seems unlikely because in vivo administration of the anti–PD-1 Fab fragments augments immune responses to the same extent as the whole antibody (unpublished data). These data and the disparate effects of anti–PD-L1 and anti–PD-L2 mAbs in NOD diabetes and EAE models (22) argue against the antibodies acting agonistically in vivo.
This is the first study to establish, in the NOD mouse, that there is negative physiologic regulation of autoimmune diabetes by CTLA-4 only in the early stages of life compared with the PD-1–PD-L1 pathway, which appears to be regulating autoreactive T cells throughout the life span of the animal. This may suggest differing modes of action of these two pathways. The CTLA-4 inhibitory pathway appears to only inhibit activation of naive T lymphocytes but the PD-1 pathway appears to inhibit both activation of naive T cells and effector function of activated autoreactive cells. Further, there is PD-L1 expression in the inflamed islets of Langerhans, suggesting a novel mechanism of down-regulating lymphocyte function at the site of inflammation by parenchymal cells. Therefore, although both CTLA-4 and PD-1 are important in regulating the initiation of the autoimmune response, PD-1 appears to be critical for progression of autoimmune diabetes. These data provide a rationale for therapeutic manipulation of the PD-1 pathway in the treatment of autoimmune disease, as has recently been suggested in a transplantation model (29).
Acknowledgments
This study was supported by National Institutes of Health grant P01 AI-41521 and a funding grant from the Juvenile Diabetes Research Foundation Center at Harvard Medical School to M.H. Sayegh. T. Chitnis is funded by the Nancy Davis Center Without Walls.
Footnotes
Abbreviations used in this paper: CTLA-4, cytotoxic T lymphocyte–associated antigen 4; EAE, experimental autoimmune encephalomyelitis; NOD, nonobese diabetic; PD-1, programmed death-1.
References
- 1.Van Parijs, L., and A.K. Abbas. 1998. Homeostasis and self-tolerance in the immune system: turning lymphocytes off. Science. 280:243–248. [DOI] [PubMed] [Google Scholar]
- 2.Carreno, B.M., and M. Collins. 2002. The B7 family of ligands and its receptors: new pathways for costimulation and inhibition of immune responses. Annu. Rev. Immunol. 20:29–53. [DOI] [PubMed] [Google Scholar]
- 3.Ishida, Y., Y. Agata, K. Shibahara, and T. Honjo. 1992. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 11:3887–3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Agata, Y., A. Kawasaki, H. Nishimura, Y. Ishida, T. Tsubata, H. Yagita, and T. Honjo. 1996. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int. Immunol. 8:765–772. [DOI] [PubMed] [Google Scholar]
- 5.Freeman, G.J., A.J. Long, Y. Iwai, K. Bourque, T. Chernova, H. Nishimura, L.J. Fitz, N. Malenkovich, T. Okazaki, M.C. Byrne, et al. 2000. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 192:1027–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Latchman, Y., C.R. Wood, T. Chernova, D. Chaudhary, M. Borde, I. Chernova, Y. Iwai, A.J. Long, J.A. Brown, R. Nunes, et al. 2001. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2:261–268. [DOI] [PubMed] [Google Scholar]
- 7.Nishimura, H., M. Nose, H. Hiai, N. Minato, and T. Honjo. 1999. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 11:141–151. [DOI] [PubMed] [Google Scholar]
- 8.Nishimura, H., T. Okazaki, Y. Tanaka, K. Nakatani, M. Hara, A. Matsumori, S. Sasayama, A. Mizoguchi, H. Hiai, N. Minato, et al. 2001. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science. 291:319–322. [DOI] [PubMed] [Google Scholar]
- 9.Herold, K.C., W. Hagopian, J.A. Auger, E. Poumian-Ruiz, L. Taylor, D. Donaldson, S.E. Gitelman, D.M. Harlan, D. Xu, R.A. Zivin, et al. 2002. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N. Engl. J. Med. 346:1692–1698. [DOI] [PubMed] [Google Scholar]
- 10.Chatenoud, L., E. Thervet, J. Primo, and J.F. Bach. 1994. Anti-CD3 antibody induces long-term remission of overt autoimmunity in nonobese diabetic mice. Proc. Natl. Acad. Sci. USA. 91:123–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leiter, E. 1998. NOD mice and related strains: origins, husbandry, and biology introduction. NOD Mice and Related Strains: Research Applications in Diabetes, AIDS, Cancer and Other Diseases. E. Leiter and M. Atkinson, editors. R.G. Landes Company, Austin, Texas. 1–26.
- 12.Miyazaki, A., T. Hanafusa, K. Yamada, J. Miyagawa, H. Fujino-Kurihara, H. Nakajima, K. Nonaka, and S. Tarui. 1985. Predominance of T lymphocytes in pancreatic islets and spleen of pre-diabetic non-obese diabetic (NOD) mice: a longitudinal study. Clin. Exp. Immunol. 60:622–630. [PMC free article] [PubMed] [Google Scholar]
- 13.Luhder, F., P. Hoglund, J.P. Allison, C. Benoist, and D. Mathis. 1998. Cytotoxic T lymphocyte–associated antigen 4 (CTLA-4) regulates the unfolding of autoimmune diabetes. J. Exp. Med. 187:427–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lenschow, D.J., S.C. Ho, H. Sattar, L. Rhee, G. Gray, N. Nabavi, K.C. Herold, and J.A. Bluestone. 1995. Differential effects of anti–B7-1 and anti–B7-2 monoclonal antibody treatment on the development of diabetes in the nonobese diabetic mouse. J. Exp. Med. 181:1145–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamazaki, T., H. Akiba, H. Iwai, H. Matsuda, M. Aoki, Y. Tanno, T. Shin, H. Tsuchiya, D.M. Pardoll, K. Okumura, et al. 2002. Expression of programmed death 1 ligands by murine T cells and APC. J. Immunol. 169:5538–5545. [DOI] [PubMed] [Google Scholar]
- 16.Dong, H., S.E. Strome, D.R. Salomao, H. Tamura, F. Hirano, D.B. Flies, P.C. Roche, J. Lu, G. Zhu, K. Tamada, et al. 2002. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat. Med. 8:793–800. [DOI] [PubMed] [Google Scholar]
- 17.Tseng, S.Y., M. Otsuji, K. Gorski, X. Huang, J.E. Slansky, S.I. Pai, A. Shalabi, T. Shin, D.M. Pardoll, and H. Tsuchiya. 2001. B7-DC, a new dendritic cell molecule with potent costimulatory properties for T cells. J. Exp. Med. 193:839–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yoon, J.W., C.S. Yoon, H.W. Lim, Q.Q. Huang, Y. Kang, K.H. Pyun, K. Hirasawa, R.S. Sherwin, and H.S. Jun. 1999. Control of autoimmune diabetes in NOD mice by GAD expression or suppression in beta cells. Science. 284:1183–1187. [DOI] [PubMed] [Google Scholar]
- 19.Yu, L., D.T. Robles, N. Abiru, P. Kaur, M. Rewers, K. Kelemen, and G.S. Eisenbarth. 2000. Early expression of antiinsulin autoantibodies of humans and the NOD mouse: evidence for early determination of subsequent diabetes. Proc. Natl. Acad. Sci. USA. 97:1701–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Todd, I., C. Davenport, J.H. Topping, and P.J. Wood. 1998. IgG2a antibodies non-specifically delay the onset of diabetes in NOD mice. Autoimmunity. 27:209–211. [DOI] [PubMed] [Google Scholar]
- 21.Crawford, M., D. Daniel, D. Wegmann, H. Yang, and R.G. Gill. 1997. Autoimmune islet damage mediated by insulin-specific T cells. Transplant. Proc. 29:758–759. [DOI] [PubMed] [Google Scholar]
- 22.Salama, A.D., T. Chitnis, J. Imitola, H. Akiba, F. Tushima, M. Azuma, H. Yagita, M.H. Sayegh, and S.J. Khoury. 2003. Critical role of the programmed death-1 (PD-1) pathway in regulation of experimental autoimmune encephalomyelitis. J. Exp. Med. 198:71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wong, F.S., I. Visintin, L. Wen, R.A. Flavell, and C.A. Janeway, Jr. 1996. CD8 T cell clones from young nonobese diabetic (NOD) islets can transfer rapid onset of diabetes in NOD mice in the absence of CD4 cells. J. Exp. Med. 183:67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carter, L., L.A. Fouser, J. Jussif, L. Fitz, B. Deng, C.R. Wood, M. Collins, T. Honjo, G.J. Freeman, and B.M. Carreno. 2002. PD-1:PD-L inhibitory pathway affects both CD4(+) and CD8(+) T cells and is overcome by IL-2. Eur. J. Immunol. 32:634–643. [DOI] [PubMed] [Google Scholar]
- 25.Eppihimer, M.J., J. Gunn, G.J. Freeman, E.A. Greenfield, T. Chernova, J. Erickson, and J.P. Leonard. 2002. Expression and regulation of the PD-L1 immunoinhibitory molecule on microvascular endothelial cells. Microcirculation. 9:133–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iwai, Y., M. Ishida, Y. Tanaka, T. Okazaki, T. Honjo, and N. Minato. 2002. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA. 99:12293–12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dong, H., G. Zhu, K. Tamada, and L. Chen. 1999. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat. Med. 5:1365–1369. [DOI] [PubMed] [Google Scholar]
- 28.Nguyen, L.T., S. Radhakrishnan, B. Ciric, K. Tamada, T. Shin, D.M. Pardoll, L. Chen, M. Rodriguez, and L.R. Pease. 2002. Cross-linking the B7 family molecule B7-DC directly activates immune functions of dendritic cells. J. Exp. Med. 196:1393–1398. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 29.Ozkaynak, E., L. Wang, A. Goodearl, K. McDonald, S. Qin, T. O'Keefe, T. Duong, T. Smith, J.C. Gutierrez-Ramos, J.B. Rottman, et al. 2002. Programmed death-1 targeting can promote allograft survival. J. Immunol. 169:6546–6553. [DOI] [PubMed] [Google Scholar]