

Abstract
Anaphylaxis is a life-threatening systemic allergic reaction with the potential for a recurrent or biphasic pattern. Despite an incidence of biphasic reaction between 5 and 20%, the molecular mechanism for the reaction is unknown. Using a murine model of penicillin V–induced systemic anaphylaxis, we show an autoregulatory cascade of biphasic anaphylactic reactions. Induction of anaphylaxis caused a rapid increase in circulating platelet-activating factor (PAF) levels. In turn, the elevated PAF contributes to the early phase of anaphylaxis as well as the subsequent activation of the nuclear factor (NF)-κB, a crucial transcription factor regulating the expression of many proinflammatory cytokines and immunoregulatory molecules. The induction of NF-κB activity is accompanied by TNF-α production, which, in turn, promotes late phase PAF synthesis. This secondary wave of PAF production leads eventually to the late phase of anaphylactic reactions. Mast cells do not appear to be required for development of the late phase anaphylaxis. Together, this work reveals the first mechanistic basis for biphasic anaphylactic reactions and provides possible therapeutic strategies for human anaphylaxis.
Keywords: early anaphylaxis, biphasic anaphylaxis, mast cell, penicillin V, TNF-α
Introduction
Anaphylaxis is a life-threatening systemic allergic reaction triggered by food antigens, medications or drugs, insect venom, and many other agents. Anaphylaxis has been shown to have the potential for a recurrent or biphasic pattern with an incidence between 5 and 20% (1–4). A few papers regarding biphasic reaction reveal that the recurrence can be severe enough to necessitate intubation and treatment with vasopressor agents (1, 4). These observations require that patients should be followed carefully after an apparent remission of an anaphylactic event.
The early anaphylactic reaction is thought to be caused by the release of platelet-activating factor (PAF),* histamine, serotonin, or many other chemical mediators from mast cells and basophils after antigen–antibody interactions (5). In a murine model of anaphylaxis, the major cause of fatal reaction is hypovolemic shock (6). PAF, a potent lipid messenger with diverse biological activities (7), is thought to be critical in the development of the hypovolemic shock. PAF antagonists can prevent fatal anaphylaxis by correcting PAF-induced hypovolemia (8, 9). However, the downstream pathways beyond the PAF-induced early reaction are unknown.
PAF is also known to play an important in vivo role in the initiation of inflammatory reactions through activation of nuclear factor (NF)-κB (10–12), a pivotal transcription factor responsible for regulating the expression of proinflammatory cytokines and many immunoregulatory molecules (13, 14). NF-κB is necessary for optimal expression of the proinflammatory cytokines TNF-α and IL-1, which promote further synthesis of PAF, thus completing a positive feedback system (15, 16). Therefore, it is intriguing to speculate that PAF-induced NF-κB–dependent release of inflammatory mediators, such as TNF-α and IL-1, may associate with development of the late phase of anaphylaxis. Using a murine model of penicillin V–induced systemic anaphylaxis (17, 18), we demonstrate in this work that the overall biphasic anaphylaxis is coincident with a biphasic increase of circulating PAF. The early release of PAF in response to the challenge injection induces a late or secondary synthesis of PAF through pathways involving NF-κB activation.
Materials and Methods
Animals.
Specific pathogen-free male C57BL/6 mice were obtained from the Korean Institute of Chemistry Technology, and were housed in a laminar flow cabinet and maintained on standard laboratory chow ad libitum. TNF-α knockout mice (B6: 129S-Tnftm1Gk1, TNF-α−/−), IL-1 receptor knockout mice (B6: 129S-Il1r1tm1Rom1, IL-1R−/−), and the original wild-type strain (B6129SF2/J) were obtained from the Jackson Laboratory. Female mast cell–deficient WBB6F1-W/W v (W/W v) and mast cell–sufficient control WBB6F1-+/+ mice were purchased from the Japan SLC Inc. Mice were 7–8-wk-old at the start of each experiment.
Induction of Active Systemic Anaphylaxis to Penicillin (Pen) V.
Mice were sensitized by i.p. injection of 500 μg Pen V–OVA conjugate plus 2 × 109 B. pertussis and 1.0 mg alum (17, 18). The challenge was performed 21 d later by giving i.v. injection of a sublethal dose (10–20 μg) of Pen V–BSA conjugate.
Reagents.
The PAF antagonist WEB 2170 was provided by Dr. C.K. Lee (Dankook University College of Medicine, Seoul, Korea) and CV 6209 was purchased from WAKO Chemical Co. 12.5 mg/kg WEB 2170 and 15.0 mg/kg CV 6209 were administered i.p. either 10 min before or 6 h after the challenge injection. 0.5 mg/kg N-acetyl-l-cysteine (NAC) and 120 mg/kg pyrrolidine dithiocarbamate (PDTC) were administered i.p. 2 h and 30 min before challenge, respectively. Control groups received the appropriate vehicles. Anti–TNF-α (MP6-XT22.11, rat IgG1; reference 19) and control rat IgG1, J4-1 (17), were injected i.p. 45 min after challenge.
Hematocrit Assay.
Anaphylaxis was quantified by measuring the increase in hematocrit value as described previously (20). Small blood samples were collected from the heart, which was cut open into a heparinized microhematocrit tube (Proper Manufacturing Co.). The tubes were centrifuged for 5 min using microhematocrit centrifuge (International Equipment Co.). Percent hematocrit change was expressed as [(value at postchallenge − value at prechallenge)/value at prechallenge] × 100.
Plasma PAF and Histamine Assay.
The postchallenge blood taken from the heart was mixed with a 0.1-vol 3.8% ice-chilled citrate solution and centrifuged immediately using an Eppendorf microcentrifuge. The plasma was stored at −20°C until use. Plasma levels of PAF and histamine were measured as described previously (18).
Gel Shift Assay.
The nuclear extracts were prepared from the lungs as described previously (10–12). To inhibit endogenous protease activity, 1 mM phenylmethylsulfonyl fluoride was added. As a probe for the gel retardation assay, oligonucleotides containing the Igκ-chain binding site (κB, 5′-CCGGTTAACAGAGGGGGCTTTCCGAG-3′) and cyclic AMP response element (CRE) sequence (5′-AGAGATTGCCTGACGTCAGAG AGCTAG-3) were synthesized. The two complementary strands were annealed and labeled with α-[32P]dCTP. Labeled oligonucleotides (10,000 cpm), 10 μg of nuclear extracts, and the binding buffer (10 mM Tris-HCl, pH 7.6, 500 mM KCl, 10 mM EDTA, 50% glycerol, 100 ng of poly (dI · dC), and 1 mM DTT) were incubated for 30 min at room temperature in a final volume of 20 μl. The reaction mixture was analyzed by electrophoresis on a 4% polyacrylamide gel in 0.5× TBE buffer. Specific binding of NF-κB was determined by competition with a 50-fold excess of cold κB or CRE oligonucleotide.
Reverse Transcriptase–PCR.
Total RNA was prepared at different time points from the lungs by a single extraction based on the guanidine isothiocyanate extraction method with the denaturing solution (Sol. D) consisting of 4 M guanidinium thiocyanate, 25 mM sodium citrate, pH 7.0, 0.5% sarcosyl, and 0.1 M 2-mercaptoethanol (21). Reverse transcription was performed using 1 μl total RNA in a 10-μl reaction mixture (Promega) containing oligo (dT)15 and avian myeloblastosis reverse transcriptase. 1 μl cDNA was amplified by PCR in a thermal cycler (denaturation for 30 s at 95°C, annealing for 30 s at 62°C, and elongation for 30 s at 72°C; PerkinElmer) using 25 cycles for each of three primers: TNF-α, IL-1β, or β-actin. The primers used in this analysis are as follows: TNF-α, 5′-CCTGTAGCCCACGTCGTAGC-3′ and 5′-TTGACCTCAGCGCTGATGTG-3′; IL-1β, 5′-GATACAAACTGATGAAGCTCGTCA-3′ and 5′-GAGATAGTGTTTGTCCACATCCTGA-3′; and β-actin, 5′-GGGTCAGAACTCCTATG-3′ and 5′-TTGACCTCAGCGCTGATGTG-3′.
Measurement of Pen V–Specific Serum Levels of IgE.
Pen V–specific serum IgE levels were determined by a passive cutaneous anaphylaxis reaction as described previously (18). In brief, serial dilutions of individual sera from mice immunized with Pen V–OVA were injected intracutaneously into the shaved backs of male Wistar rats (Japan SLC). After 24 h, 1 ml of 1% Evan's blue dye in PBS containing 4 mg Pen V–BSA was injected i.v. The rats were killed 30 min later, their skins were removed, and the blue spot with diameter >5 mm was regarded as a positive reaction.
Cytokine Assay.
Serum levels of TNF-α and IL-1β were determined using ELISA kits (Endogen) according to the manufacturer's instructions.
Results
Occurrence of Biphasic Anaphylactic Events Coincident with a Biphasic Increase in Circulating PAF Levels.
First, we examined whether a systemic biphasic reaction occurs in mice undergoing anaphylaxis. Anaphylaxis was monitored by measuring increases in hematocrit because hemoconcentration, which occurs due to extravasation of plasma, is the most prominent symptom in murine anaphylaxis (6). A biphasic increase in hematocrit (Ht) was observed in mice undergoing active systemic anaphylaxis (Fig. 1 a). The severe early hemoconcentration began within 2–3 min after challenge, peaked (40% increase) at 10–20 min, and subsequently returned to prechallenge values at 5–6 h. From these values, it could be calculated that 70–80% blood volume had disappeared from the circulation. A substantial second phase of hemoconcentration, albeit to a lesser degree relative to the duration and severity of the first, was evident 7–8 h after challenge. In this secondary phase of anaphylaxis, hematocrits increased by 20%, which represents a loss of ∼35–40% of the initial blood volume.
Figure 1.
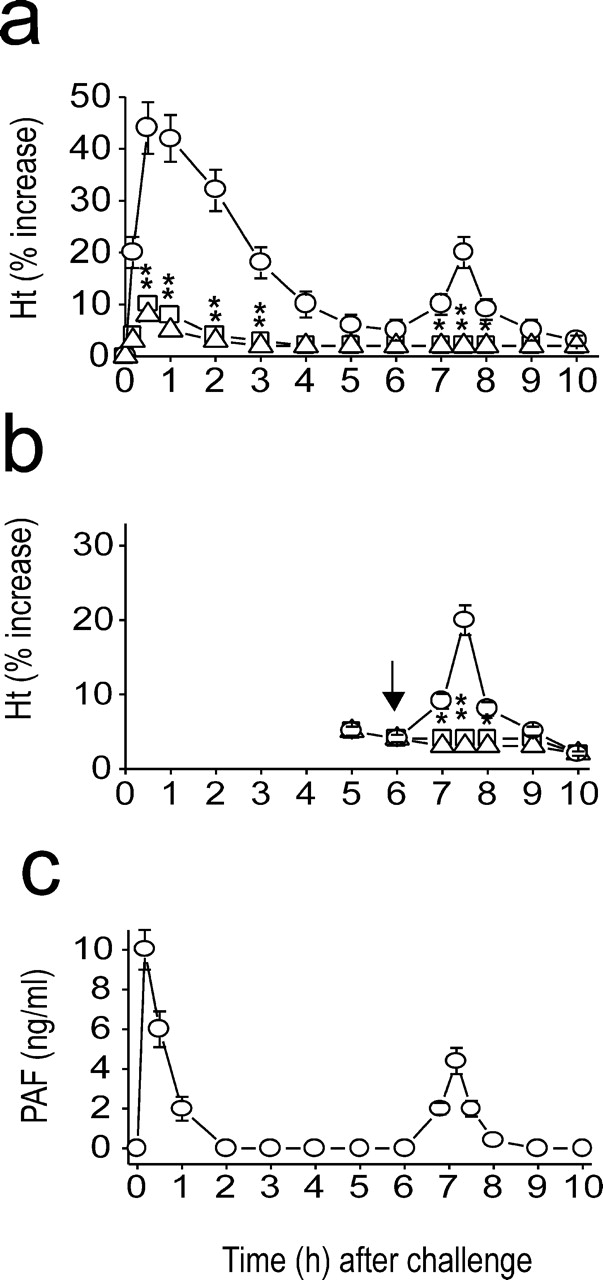
Induction of systemic anaphylaxis results in PAF-associated biphasic anaphylaxis. (a) Biphasic increase in hematocrit (Ht) values during anaphylaxis and its inhibition by the PAF antagonists WEB 2170 (□) and CV 6209 (▵) administered 10 min before challenge injection. ○, control group. (b) Inhibition of the late phase increase in hematocrit values by WEB 2170 (□) and CV 6209 (▵). They were administered 6 h after challenge injection. ○, control group. (c) Biphasic increase in plasma PAF levels during anaphylaxis. Results for all panels are expressed as the mean ± SEM of three to seven separate experiments (n = 4 for each time point). *, P < 0.05; **, P < 0.01 versus control. Statistical significance was determined by Mann-Whitney U test.
PAF is the crucial molecule responsible for murine anaphylactic reactions (8, 9, 18). To assess the association of PAF with the biphasic reaction, the PAF antagonists WEB 2170 and CV 6209 were used. Pretreatment with the antagonists before challenge abrogated the early as well as late phase of hemoconcentration (Fig. 1 a). The degree of inhibition of the early hemoconcentration reaction in the group receiving PAF antagonists was mirrored in their effect on other general anaphylactic symptoms, including sluggish gate, paresis, and prostration (unpublished data). Furthermore, PAF antagonists administered 6 h after challenge completely blocked the secondary phase of hemoconcentration (Fig. 1 b), which strongly suggests that PAF is also responsible for a second wave of anaphylactic events. The time course of plasma levels of PAF during anaphylaxis paralleled those of the biphasic anaphylactic reactions, with the major early peak appearing within 5–10 min followed by a small second PAF peak ∼7.0–7.5 h (Fig. 1 c). Collectively, these data suggest that the overall process of biphasic anaphylactic reactions is multiphasic with the initial release of PAF acting as an inducer for the next. In turn, this leads to systemic anaphylactic reaction that is also biphasic.
NF-κB Activity Is Required for the Secondary Increase in Plasma PAF and Hematocrit.
PAF is a potent inducer of NF-κB in vitro as well as in vivo (10–14). We measured NF-κB activity during anaphylaxis by a gel mobility shift assay. Induction of systemic anaphylaxis resulted in NF-κB activation in the lung (Fig. 2 a). NF-κB activity appeared within 30 min of the challenge. A similar pattern of NF-κB activation was also observed in the liver and spleen (unpublished data). Pretreatment of animals with PAF antagonists resulted in an almost complete inhibition of NF-κB activation (Fig. 2 a), confirming that PAF is responsible for the activation of NF-κB during anaphylaxis. To block NF-κB activation, we used the antioxidant, NAC and PDTC. NAC and PDTC also significantly inhibited NF-κB activation (Fig. 2 a). Complete blocking of NF-κB mobilization by adding the cold competitor, but not by adding of an irrelevant motif, CRE, indicated the specificity of NF-κB binding. In addition, NAC had no effect on CRE mobilization, further demonstrating the specificity of the inhibitors (Fig. 2 a). Next, we examined the possible association of NF-κB activity with the late anaphylactic reactions. Both NF-κB inhibitors significantly inhibited the second phase of increased plasma PAF levels (Fig. 2 b) and hematocrit (Fig. 2 c). However, the NF-κB inhibitors did not inhibit the first phase of increase in PAF release and hemoconcentration (unpublished data).
Figure 2.


PAF-induced activation of NF-κB during anaphylaxis and its association with the development of the late phase of anaphylaxis. (a) NF-κB activation during anaphylaxis and its inhibition by the pretreatment with PAF antagonists and NF-κB inhibitors. After the challenge, lungs were removed at the indicated time points and the time course of NF-κB activation was measured by gel shift assay with nuclear extracts (n = 3–5 for each time point). For gel shift assay of CRE mobilization, lungs were removed 1 h after challenge. A representative of four independent experiments is shown. (b and c) Inhibition of the second phase of increase in plasma PAF (b) and hematocrit value (c) by NF-κB inhibitors. Blood was collected 7.5 h after the challenge. Results for all panels are expressed as the mean ± SEM of three to seven separate experiments (n = 4 for each time point). *, P < 0.01 versus control; Mann-Whitney U test.
NF-κB–dependent TNF-α Production Leads to the Second Phase of Hemoconcentration through the Synthesis of PAF.
The activation of NF-κB results in the expression of proinflammatory cytokine genes, such as TNF-α and IL-1 (13, 14). Therefore, we investigated whether TNF-α and IL-1 are produced in an NF-κB–dependent manner during anaphylaxis. Serum TNF-α levels peaked 1–1.5 h after challenge (Fig. 3 a) and declined rapidly thereafter. In contrast, IL-1 was not detected in the circulation during the observation period. The administration of a PAF antagonist and an NF-κB inhibitor nearly completely inhibited the increase in serum TNF-α levels after the challenge (Fig. 3 b), indicating that production of TNF-α during anaphylaxis is a PAF-induced NF-κB–dependent process.
Figure 3.
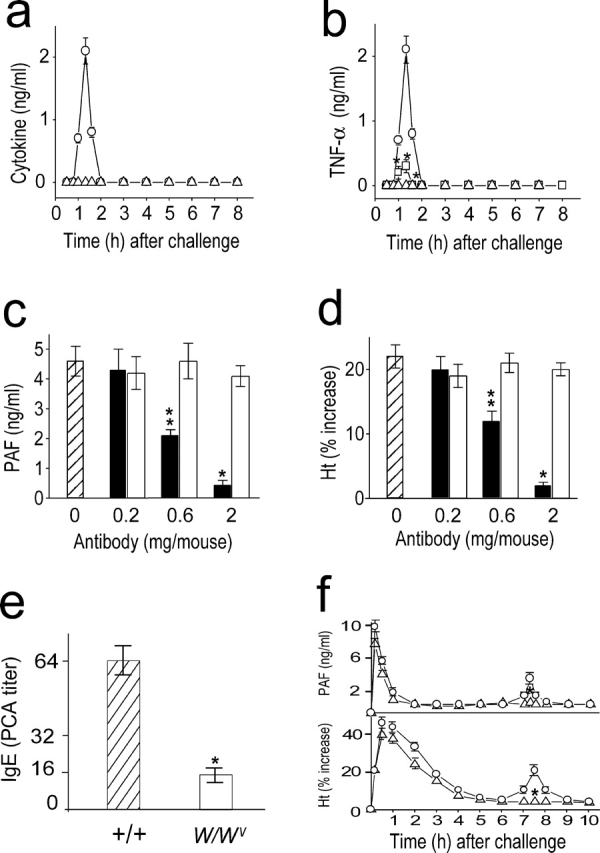
Involvement of TNF-α in the development of the late anaphylactic reactions. (a) Time course of serum levels of TNF-α (○) and IL-1 (▵) during anaphylaxis. (b) TNF-α inhibition by pretreatment with CV 6209 (▵) and PDTC (□). ○, control group. (c and d) Inhibition of the late phase increase in plasma PAF (c) and hematocrit value (d) by anti–TNF-α Ab (▪). □, control Ab. Abs were injected i.p. 45 min after challenge injection. Blood was collected 7.5 h after the challenge. (e) Serum levels of Pen V–specific IgE. Blood samples were taken 1 d before challenge. (f) No late phase increase in plasma PAF and hematocrit value in TNF-α2/2 mice (▵). ○, control littermates. Blood was collected 7.5 h after the challenge. Results for all panels are expressed as the mean ± SEM of three separate experiments (n = 4 for each time point). **, P < 0.05; *, P < 0.01 versus control; Mann-Whitney U test.
To assess the possibility that TNF-α produced after the early anaphylactic response is involved in the secondary increase in plasma PAF concentration and hemoconcentration, anti–TNF-α Ab-treated and TNF-α−/ − mice were challenged to induce anaphylaxis. Treatment with anti–TNF-α, MP6-XT22.11 (19), resulted in a dose-dependent inhibition of the secondary increase in plasma PAF (Fig. 3 c) and hemoconcentration (Fig. 3 d). At a concentration of 2 mg/mouse, anti–TNF-α almost completely inhibited the responses. We further examined the role for TNF-α in the development of the late phase of anaphylaxis using TNF-α−/ − mice. Given the fact that TNF-α–deficient mice exhibit abnormal antibody class switching (including IgE) due to a failure of germinal center formation (22), we first examined Pen V–specific IgE response in TNF-α–deficient mice. The Pen V–specific IgE level in TNF-α–deficient mice was ∼25% of control littermates (Fig. 3 e). Despite the reduced IgE response, a similar magnitude of the early phase of anaphylaxis developed in both control littermates and TNF-α−/ − mice (Fig. 3 f). In contrast, TNF-α−/ − mice exhibited little increase in the late phase plasma PAF concentration and hematocrit (Fig. 3 f). Given our previous information that a fatal Pen V–induced anaphylaxis occurs in mice of which Pen V–specific IgE level is reduced to 10% of control mice (17), the IgE level in TNF-α–deficient mice seems to be sufficient to induce anaphylaxis. Furthermore, when the mice were challenged with a lethal dose of antigen (100 μg Pen V–BSA), a similar mortality rate was observed in control littermates (9/10) and TNF-α–deficient mice (8/10; unpublished data). These data indicate that TNF-α is associated with the late phase of anaphylaxis through the synthesis of PAF.
Although IL-1 was not detected in the circulation during anaphylaxis by ELISA system, it is thought that even femtograms per milliliter of IL-1 can have pathophysiological activities (23, 24). Thus, to further clarify the role for IL-1 during anaphylaxis, we determined TNF-α and IL-1β mRNA levels in the lungs. A much weaker IL-1β mRNA expression was seen compared with the strong expression of TNF-α mRNA during anaphylaxis (Fig. 4 a). In contrast, LPS-induced IL-1β mRNA expression was comparable to that of TNF-α mRNA. Furthermore, similar levels of plasma PAF and hematocrit in the late phase were seen in IL-1R−/− mice (Fig. 4 b). These data indicate that IL-1 does not seem to play a significant role in Pen V–induced anaphylaxis.
Figure 4.


IL-1 is not an important mediator in developing a late phase anaphylactic reaction. (a) Comparison of mRNA expression between TNF-α and IL-1β during anaphylaxis. As a positive control, RNA was prepared at different time points from the lungs of 50-μg LPS-injected mice. (b) The late phase increase in plasma PAF and hematocrit value in IL-1R−/ − mice. Blood was collected 7.5 h after challenge injection. Results for all panels are expressed as the mean ± SEM of three separate experiments (n = 3 for each time point).
Mast Cells Are Not Involved in the Late Phase of Anaphylaxis.
To investigate a role for mast cells in the development of the late anaphylactic reactions, we determined the time course of plasma histamine levels after the challenge and examined whether the late anaphylactic reaction occurs in mast cell–deficient WBB6F1-W/W v (W/W v) mice. Levels of circulating histamine reached a peak within 10 min, but rapidly declined thereafter and remained within the basal level during the observation period (Fig. 5 a). The late anaphylactic reactions such as increases in plasma PAF concentration and hemoconcentration developed to a similar degree in both mast cell–sufficient control +/+ and W/W v mice (Fig. 5 b). From these data, mast cells do not appear to be required for development of the late anaphylactic reaction. Because mast cells are one source of both the cytokines TNF-α and IL-1 (25), we examined the circulating TNF-α levels during anaphylaxis in W/Wv mice. W/Wv mice were defective in producing TNF-α as W/Wv mice possess 20–30% of the wild-type level (Fig. 5 c). Circulating IL-1β was not detected in W/Wv mice during anaphylaxis (unpublished data). This reduced level of TNF-α in W/Wv mice seems to be due to the lower level of circulating PAF during anaphylaxis (18) and the absence of mast cell–derived TNF-α. Pretreatment with anti–TNF-α resulted in abrogation of the late increase in hematocrit in both wild-type and W/Wvmice (Fig. 5 d). Therefore, it is likely that TNF-α is also a key cytokine in the development of a late anaphylactic reaction in W/Wv, even with a reduced capacity to produce this cytokine.
Figure 5.

Mast cells are not involved in the late phase of anaphylaxis. (a) Time course of plasma histamine levels after challenge in C57BL/6 mice. (b) The late phase increase in plasma PAF and hematocrit value in W/Wv mice. Blood was collected 7.5 h after challenge injection. (c) Comparison of serum levels of TNF-α between +/+ and W/Wv mice during anaphylaxis. Blood was collected 80 min after challenge injection. (d) Inhibition of the second phase of increase in hematocrit value by anti–TNF-α Ab. 2 mg/mouse Ab was injected i.p. 45 min after challenge injection. Results for all panels are expressed as the mean ± SEM of three separate experiments (n = 3 for each time point). P < 0.01 versus control; Mann-Whitney U test.
Discussion
In a murine model of anaphylaxis, we were successful in demonstrating a biphasic systemic anaphylaxis and in describing an underlying molecular cascade. The early reaction is caused by the acute release of PAF, reaching a maximum 10 min after challenge. The first PAF wave eventually contributes to development of a late or secondary reaction by activating a sequential pathway involving NF-κB activation, TNF-α production, and a secondary phase of PAF synthesis. The secondary phase of PAF synthesis is responsible for eliciting the late phase reactions. Thus, overall, a biphasic reaction is coincident with a biphasic increase of circulating PAF.
PAF, produced by a variety of cells involved in inflammatory reactions, is a potent lipid messenger involved in a diverse inflammatory reactions (7) and cellular functions (26–28). Our studies have demonstrated that PAF acts as a potent inducer of NF-κB activation, leading to gene expression and protein synthesis of NF-kB–dependent cytokines in many pathological conditions, such as inflammation (10, 11), microbial infection (12), and angiogenesis (29). Therefore, PAF may be the most proximal mediator responsible for the pathogenesis of diseases in which NF-κB activity plays an important role.
We demonstrate for the first time an association of PAF-induced NF-κB activation with the late anaphylactic reactions. The late synthesis of PAF is in response to the presence of TNF-α, which was produced as a consequence of NF-κB activation. Regarding the molecules involved in the late phase of allergic reaction, TNF-α has been shown to participate in IgE-dependent cutaneous late phase reactions (30), which further supports the importance of TNF-α in the development of late phase reactions. NF-κB–dependent proinflammatory cytokines, such as TNF-α and IL-1, are known to stimulate the synthesis of PAF. In fact, many in vivo biological functions of TNF-α are mediated through the synthesis of PAF (31–33). Although the mechanism for TNF-α–induced PAF synthesis is not fully understood, it seems likely that TNF-α exerts its effect via a pathway involving phospholipase A2 (PLA2); given that TNF-α has been shown to enhance PLA2 activity (34, 35). An increase in PLA2 activity would lead to the biosynthesis of arachidonic acid metabolites, such as prostaglandins, leukotrienes, and PAF (36, 37). We also observed that the late phase increase in plasma PAF and hemoconcentration was completely inhibited by the PLA2 inhibitor, quinacrine (unpublished data), supporting the possibility that PLA2 activity is also an important component of the cascade leading to development of the late phase of anaphylaxis.
Mast cells have long been believed to be the central effector cells in the development of IgE-dependent anaphylaxis, although early immediate hypersensitivity reactions to protein antigens as well as hapten can be developed in W/W v mice (18, 38). The current paper showing an early single peak in plasma histamine level and similar late reactions in magnitude and kinetics in both +/+ and W/W v mice suggests that other inflammatory cells rather than mast cells participate in the late phase of anaphylaxis.
Currently, there is no evidence whether the same mechanisms are also in play in human anaphylaxis. However, based on the information that a late or secondary phase of human anaphylactic symptoms such as urticaria, rash, and hypotension develop after at least 5–6 h of lag period after the onset of anaphylaxis (4), it seems likely that a second wave of synthesis of chemical mediators also occurs in human anaphylaxis. Thus, it is plausible that a similar mechanism may operate in human anaphylaxis in terms of the involvement of NF-κB–induced proinflammatory cytokine expression resulting in the late synthesis of mediators such as PLA2-dependent arachidonic acid metabolites and PAF. If that is the case, selective inhibitors that target the various components of the pathway (for example, anti–TNF-α Ab or PLA2 inhibitor) might be of a great therapeutic value for controlling the late phase of human anaphylaxis.
Acknowledgments
We thank Dr. J.D. Hennebold for his critical review and reading of the manuscript.
This work was supported by the Medical Research Center project of the Korea Science and Engineering Foundation.
Footnotes
Abbreviations used in this paper: CRE, cyclic AMP response element; NAC, N-acetyl-l-cysteine; NF, nuclear factor; PAF, platelet-activating factor; PDTC, pyrrolidine dithiocarbamate; Pen, penicillin; PLA2, phospholipase A2.
References
- 1.Stark, B.J., and T.J. Sullivan. 1986. Biphasic and protracted anaphylaxis. J. Allergy Clin. Immunol. 78:76–83. [DOI] [PubMed] [Google Scholar]
- 2.Sampson, H.A., L. Mendelson, and J.P. Rosen. 1992. Fatal and near-fatal anaphylactic reactions to food in children and adolescents. N. Engl. J. Med. 327:380–384. [DOI] [PubMed] [Google Scholar]
- 3.Douglas, D.M., E. Sukenick, W.P. Andrade, and J.S. Brown. 1994. Biphasic systemic anaphylaxis: an inpatient and outpatient study. J. Allergy Clin. Immunol. 93:977–985. [DOI] [PubMed] [Google Scholar]
- 4.Lee, J.M., and D.S. Greenes. 2000. Biphasic anaphylactic reactions in pediatrics. Pediatrics. 106:762–766. [DOI] [PubMed] [Google Scholar]
- 5.Ishizaka, T., and K. Ishizaka. 1975. Biology of immunoglobulin E. Molecular basis of reaginic hypersensitivity. Prog. Allergy. 19:60–121. [PubMed] [Google Scholar]
- 6.Munoz, J., and R.K. Bergman. 1965. Mechanism of anaphylactic death in the mouse. Nature. 205:199–200. [DOI] [PubMed] [Google Scholar]
- 7.Braquet, P., L. Touqui, T.Y. Shen, and B.B. Vargaftig. 1987. Perspectives in platelet-activating factor research. Pharmacol. Rev. 39:97–145. [PubMed] [Google Scholar]
- 8.Vilain, B., V. Lagente, C. Touvay, S. Desquand, J. Randon, J. Lefort, P. Braquet, and Vargaftig. 1986. Pharmacological control of the in vivo passive anaphylactic shock by the PAF-acether antagonist compound BN 52021. Pharmacol. Res. Commun. 18(Suppl.):119–126. [DOI] [PubMed] [Google Scholar]
- 9.Hervert, J.M., L. Lespy., and J.P. Maffrand. 1991. Protective effect of SR27417, a novel PAF antagonist, on lethal anaphylactic and endotoxin-induced shock in mice. Eur. J. Pharmacol. 205:271–276. [DOI] [PubMed] [Google Scholar]
- 10.Im, S.Y., S.J. Han, H.M. Ko, J.H. Choi, S.B. Chun, D.G. Lee, T.Y. Ha, and H.K. Lee. 1997. Involvement of nuclear factor-κB in platelet-activating factor-mediated tumor necrosis factor-alpha expression. Eur. J. Immunol. 27:2800–2804. [DOI] [PubMed] [Google Scholar]
- 11.Han, S.J., J.H. Choi, H.M. Ko, H.W. Yang, I.W. Choi, H.K. Lee, O.H. Lee, and S.Y. Im. 1999. Glucocorticoids prevent NF-κB activation by inhibiting the early release of platelet-activating factor in response to lipopolysaccharide. Eur. J. Immunol. 29:1334–1341. [DOI] [PubMed] [Google Scholar]
- 12.Choi, J.H., H.M. Ko, J.W. Kim, H.K. Lee, S.S. Han, S.B. Chun, and S.Y. Im. 2001. Platelet-activating factor-induced early activation of NF-κB plays a crucial role for organ clearance of Candida albicans. J. Immunol. 166:5139–5144. [DOI] [PubMed] [Google Scholar]
- 13.Baeuerle, P.A., and D. Baltimore. 1988. Activation of DNA-binding activity in an apparently cytoplasmic precursors of the NF-κB transcription factor. Cell. 53:211–217. [DOI] [PubMed] [Google Scholar]
- 14.Kopp, E.B., and S. Ghosh. 1995. NF-κB and Rel proteins in innate immunity. Adv. Immunol. 58:1–27. [DOI] [PubMed] [Google Scholar]
- 15.Valone, F.H., and L.B. Epstein. 1988. Biphasic platelet-activating factor synthesis by human monocytes stimulated with IL-1β, tumor necrosis factor, or IFN-γ. J. Immunol. 141:3945–3950. [PubMed] [Google Scholar]
- 16.Bussolino, F., G. Camussi, and C. Baglioni. 1988. Synthesis and release of platelet-activating factor by human vascular endothelial cells treated with tumor necrosis factor or interleukin-1α. J. Biol. Chem. 263:11856–11861. [PubMed] [Google Scholar]
- 17.Park, J.S., I.H. Choi, D.G. Lee, S.S. Han, T.Y. Ha, H.H. Lee, W.H. Lee, Y.M. Park, and H.K. Lee. 1997. Anti-IL-4 monoclonal antibody prevents antibiotics-induced active fatal anaphylaxis. J. Immunol. 158:5002–5006. [PubMed] [Google Scholar]
- 18.Choi, I.H., Y.M. Shin, J.S. Park, M.S. Lee, E.H. Han, O.H. Chai, S.Y. Im, T.Y. Ha, and H.K. Lee. 1998. Immunoglobulin E–dependent active fatal anaphylaxis in mast cell–deficient mice. J. Exp. Med. 188:1587–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Suk, K., S. Kim, T.H. Kim, K.A. Kim, I. Chang, H. Yagita, M. Shong, and M.S. Lee. 2001. IFN-γ/TNF-α synergism as the final effector in autoimmune diabetes: a key role for STAT1/IFN regulatory factor-1 pathway in pancreatic beta cell death. J. Immunol. 166:4481–4489. [DOI] [PubMed] [Google Scholar]
- 20.Bowdre, J.H., M.D. Poole, and J.D. Oliver. 1981. Edema and hemoconcentration in mice experimentally infected with Vibrio vulnificus. Infect. Immun. 32:1193–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Han, S.J., H.M. Ko, J.H. Choi, K.H. Seo, H.S. Lee, E.K. Choi, I.W. Choi, H.K. Lee, and S.Y. Im. 2002. Molecular mechanisms for lipopolysaccharide-induced biphasic activation of nuclear factor-κB (NF-κB). J. Biol. Chem. 277:44715–44721. [DOI] [PubMed] [Google Scholar]
- 22.Pasparakis, M., L. Alexopoulou, V. Episkopou, and G. Kollias. 1996. Immune and inflammatory responses in TNF-α–deficient mice: a critical requirement for TNF-α in the formation of primary B cell follicles, follicular dendritic cell networks, and germinal centers, and in the maturation of the humoral immune response. J. Exp. Med. 184:1397–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dinarello, C.A. 1991. Interleukin-1 and interleukin-1 antagonism. Blood. 77:1627–1652. [PubMed] [Google Scholar]
- 24.Dinarello, C.A. 1996. Biologic basis for interleukin-1 in disease. Blood. 87:2095–2147. [PubMed] [Google Scholar]
- 25.Galli, S.J., Wershil, B.K., Gordon, J.R, and Martin, T.R. 1989. Mast cells: immunologically specific effectors and potential sources of multiple cytokines during IgE-dependent responses. Ciba. Found. Symp. 147:53–65; discussion 65–73. [DOI] [PubMed] [Google Scholar]
- 26.Buttke, T.M., and P.A. Sandstrom. 1995. Redox regulation of programmed cell death in lymphocytes. Free Radic. Res. 22:389–397. [DOI] [PubMed] [Google Scholar]
- 27.Fukuda, A.I., and K.F. Breuel. 1996. Effect of platelet activating factor on embryonic development and implantation in the mouse. Hum. Reprod. 11:2746–2749. [DOI] [PubMed] [Google Scholar]
- 28.Gay, J.C. 1993. Mechanism and regulation of neutrophil priming by platelet-activating factor. J. Cell. Physiol. 156:189–197. [DOI] [PubMed] [Google Scholar]
- 29.Ko, H.M., K.H. Seo, S.J. Han, K.Y. Ahn, I.H. Choi, G.Y. Koh, H.K. Lee, M.S. Ra, and S.Y. Im. 2002. NF-κB-dependency of platelet-activating factor-induced angiogenesis. Cancer Res. 62:1809–1814. [PubMed] [Google Scholar]
- 30.Wershil, B.K., Z.S. Wang, J.R. Gordon, and S.J. Galli. 1991. Recruitment of neutrophils during IgE-dependent cutaneous late phase reactions in the mouse is mast cell-dependent. Partial inhibition of the reaction with antiserum against tumor necrosis factor. J. Clin. Invest. 87:446–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Im, S.Y., H.M. Ko, J.W. Kim, H.K. Lee, T.Y. Ha, H.B. Lee, S.J. Oh, S. Bai, K.C. Chung, Y.B. Lee, et al. 1996. Augmentation of tumor metastasis by platelet-activating factor. Cancer Res. 56:2662–2665. [PubMed] [Google Scholar]
- 32.Montrucchio, G., E. Lupia, E. Battaglia, G. Passerini, F. Bussolino, G. Emanuelli, and G. Camussi. 1994. Tumor necrosis factor α–induced angiogenesis depends on in situ platelet-activating factor biosynthesis. J. Exp. Med. 180:377–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nourshargh, S., S.W. Larkin, A. Das, and T.J. Williams. 1995. Interleukin-1-induced leukocyte extravasation across rat mesenteric microvessels is mediated by platelet-activating factor. Blood. 85:2553–2558. [PubMed] [Google Scholar]
- 34.Hoeck, W.G., C.S. Ramesha, D.J. Chang, N. Fan, and R.A. Heller. 1993. Cytoplasmic phospholipase A2 activity and gene expression are stimulated by tumor necrosis factor: dexamethasone blocks the induced synthesis. Proc. Natl. Acad. Sci. USA. 90:4475–4479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leslie, C.C. 1997. Properties and regulation of cytosolic phospholipase A2. J. Biol. Chem. 272:16709–16712. [DOI] [PubMed] [Google Scholar]
- 36.Samuelsson, B., S.E. Dahlen, J.A. Lindgren, C.A. Rouzer, and C.N. Serhan. 1987. Leukotrienes and lipoxins: structures, biosynthesis, and biological effects. Science. 237:1171–1176. [DOI] [PubMed] [Google Scholar]
- 37.Irvine, R.F. 1982. How is the level of free arachidonic acid controlled in mammalian cells? Biochem. J. 204:3–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ha, T.Y., and N.D. Reed. 1987. Systemic anaphylaxis in mast cell-deficient mice of W/W v and SI/SI d genotypes. Exp. Cell Biol. 55:63–68. [DOI] [PubMed] [Google Scholar]