

Abstract
The outcome of murine infection with Leishmania major is regulated by major histocompatibility complex class II–restricted T helper cells. Invariant chain-deficient (Ii −/−) mice have impaired ability to present major histocompatibility complex class II–restricted antigens, and reduced numbers of CD4+ T cells. Despite these deficits, C57BL/6 Ii −/− mice controlled L. major infection comparably to wild-type mice. As assessed by mRNA analysis and in vitro antigen restimulation for IFN-γ, Ii −/− mice had normal induction of Th1 subset differentiation even though antigen-dependent proliferation of their lymph node cells was substantially compromised. In addition, BALB/c Ii −/− mice exhibited a progressive course of infection and Th2 effector cell development that were comparable to that seen in wild-type BALB/c mice. We wished to determine whether this unexpected efficiency of T helper subset induction despite inefficient T cell stimulation could be modeled in vitro. In the presence of rIL-12 or rIL-4 naive parasite-specific transgenic T cells could mature into IFN-γ–or IL-4–secreting T helper cells, respectively, even when antigen presentation was suboptimal or antigen dose was submitogenic. These experiments demonstrate that activation of T helper cells to a threshold required for IL-2 production or proliferation is not required to achieve induction of disease-regulating T helper cell effector functions, and that pathogen-associated secondary activation signals may facilitate the full differentiation of T helper subsets during limiting presentation of antigenic peptides.
Experimental infection of inbred strains of mice with Leishmania major remains an exceptional model for analysis of CD4+ subset differentiation in vivo (1). Control of disease is dependent on class II–restricted Th type 1 (Th1) cells and their production of IFN-γ which is required to activate macrophages to restrain intracellular replication of the organism. Studies in T cell– (2–4) and IFN-γ–deficient (5) mice have confirmed the critical requirements for these elements in host immunity. MHC class II–deficient mice from a genetically resistant background are completely susceptible to infection (6, 7), while MHC class I–deficient mice from a genetically resistant background retain resistance to infection (8). In contrast to most strains of mice, BALB animals are unable to contain L. major due to the development of an aberrant Th type 2 (Th2) response during infection. The absence of class I does not impact Th2 development or susceptibility in BALB/c mice (9).
Leishmania replicates productively only in host macrophages within an endolysosomal-like compartment that contains MHC class II molecules, some of which are devoid of invariant chain (10, 11). Infection of macrophages in vitro is associated with diminished MHC class II–dependent presentation of exogenous antigens (12, 13). Although it is unclear whether this is due to degradation or inappropriate trafficking of MHC class II/peptide complexes from the parasitophorous vacuole (14), competent MHC class II molecules reach the cell surface as demonstrated by immunofluorescent, functional, and biochemical studies (12, 13, 15). Since invariant chain is involved in both targeting newly synthesized MHC class II molecules to peptide-generating compartments, and in protecting the peptide cleft during transit from the endoplasmic reticulum (16), we expected significant impairment in host immune responses to L. major using invariant chain–deficient (Ii −/−) mice. Unexpectedly, both Th1 and Th2 responses were maintained in mice on genetically resistant or susceptible backgrounds, respectively, emphasizing the capacity of the immune system to sustain T cell effector development even under conditions of suboptimal stimulation.
Materials and Methods
Mice.
Ii −/− mice (17), class II −/− mice (18), β2m −/− mice (19), BALB/c (Jackson Laboratory, Bar Harbor, ME), and C57BL/6 (Jackson Laboratory) mice were bred and maintained in the University of Chicago animal facilities. Double mutant Ii −/− β2m −/− mice were generated by interbreeding. Mice were screened by fluorescent cytometry using appropriate monoclonal antibodies for levels of MHC class I, class II, CD4+ and CD8+ T cells to confirm genotypes. Most mice used in these experiments were fourth generation C57BL/6, or fourth generation BALB/c. Mice on a 129 × C57 (L. major–resistant) background, but congenic for H-2d and H-2k, were used where designated.
TCR transgenic mice that express a clonotypic Vα8/Vβ4 TCR expanded early after infection by L. major (20), were established using standard methods, and are characterized elsewhere (Reiner, S., manuscript in preparation). T cells from these mice recognize an 18–amino acid peptide epitope restricted by I-Ad from an immunodominant antigen, Leishmanial receptor for activated protein kinase (LACK)1, that is expressed in both the promastigote and amastigote forms of the parasite (21). Thymic selection but not peripheral activation, of the transgene clonotype occurs on the mismatched H-2k background.
L. Major Infection.
Mice were inoculated in both hind footpads with 5 × 105 metacyclic promastigotes of L. major (WHOM/IR/-/173), maintained and purified as described (22). Designated mice received 2 mg of mAB XMG1.2 (neutralizing anti–IFN-γ, rat IgG1) intraperitoneally at the time of infection. Disease progression was monitored weekly by measuring the footpads with a metric caliper. After 6–8 wks, the popliteal lymph nodes draining the lesions were collected for the analysis of cell phenotypes, number, and cytokine production (below). Aliquots of the single-cell suspensions were analyzed for numbers of CD4+ and CD8+ T cells by fluorescent flow cytometry after staining with the appropriate mAb's. The footpads were washed in ethanol, rinsed in HBSS, and homogenized in 3 ml M-199 medium. The spleens were homogenized in 3 ml M-199 medium. Aliquots were serially diluted into flat-bottom 96-well microtiter plates, and the plates were sealed with parafilm and incubated at 26°C for 2 wks. Wells were examined for the presence of the motile promastigotes by inverted microscopy to determine the tissue parasite burdens.
Cytokine and Lymphoproliferation Analyses.
Popliteal lymph nodes draining the site of infection were collected 6–8 wks after infection. Single-cell suspensions were made and triplicate aliquots were distributed to 96-well round-bottom plates for determinations of cytokine production (106 cells/well) and proliferation (1.5 × 105 cells/well) in the presence of media or media plus 100 μg/ml soluble Leishmania antigen (SLA). For cytokine analysis, supernatants were collected after 48 h incubation and quantitated for IL-2, -4, or IFN-γ using ELISA (PharMingen, San Diego, CA). For proliferation analysis, wells were pulsed after 72 h with 1 μCi [3H]thymidine, and harvested 16 h later.
For cytokine mRNA analysis, total RNA was extracted from popliteal lymph nodes using RNAzol (Biotecx, Houston, TX), and reverse transcribed using random hexamer primers (Promega Corp., Madison, WI). A multiple cytokine–containing competitor construct was used for PCR-based semi-quantitation as described (23). Briefly, amounts of cDNA from the different samples were first normalized by adjusting for competition with the constitutively expressed hypoxanthine-guanine phosphoribosyltransferase (HPRT) gene product. The competitor PCR products were engineered as larger amplification products by insertion of an internal irrelevant DNA sequence, thus allowing their ready separation from the authentic transcripts by their differential mobility after electrophoresis in 2.5% agarose gels. These adjusted cDNA volumes were used in subsequent reactions with primers for IL-4 or IFN-γ which were also present in the competitor plasmid. The ratio of the competitor to the authentic cytokine transcripts was used to quantitate mRNA production in vivo.
Antigen Presentation Assays.
Bone marrow cells were flushed from the femurs of H-2d Ii +/− and −/− littermates, and adherent cells allowed to differentiate for 5–7 d in media containing 30% L929 cell supernatant. The adherent macrophages were harvested after incubation for 30 min in PBS with 2% glucose, washed, counted, and redistributed to wells containing fresh media, or media containing SLA, viable metacyclic promastigotes, or freshly thawed and washed amastigotes isolated from infected mice and stored in 10% DMSO/FCS at −70°C (Amastigotes retain infectivity when stored in this fashion.) Infection was monitored in concomitantly inoculated monolayers established on tissue-culture chambered slides which were fixed and stained with DifQuik (Dade Diagnostics, Aguada, P.R.). Infection of 5 × 104 macrophages with 2.5 × 105 parasites typically resulted in ∼50% of the cells infected with 1–3 parasites/macrophage. After 20 h, the antigen- or parasite-pulsed monolayers were washed extensively, and designated numbers of T cells from TCR transgenic mice (H-2k) lymph nodes were added to the wells. Supernatants were harvested after 48 h and assayed for IL-2 and IFN-γ production by ELISA. In designated experiments, splenocytes from Ii +/− or −/− littermates were irradiated and used as APCs. There were no measured alloreactive responses from the use of H-2k–derived TCR transgenic T cells with H-2d–derived APCs.
Results
Ii −/− Mice on a Resistant Background Control Infection with L major.
C57BL/6 Ii −/− and Ii +/− mice were challenged with L. major, and the course of infection monitored by measuring the size of the local lesions. Both groups of mice, in contrast to a concomitantly inoculated cohort of susceptible BALB/c mice, controlled infection and limited the size of the footpad lesion (Fig. 1 A). As in immunocompetent mice (24), control of the parasite was dependent on the production of IFN-γ, because treatment of mice with anti–IFN-γ neutralizing mAb abrogated the resistance phenotype. Delayed-type hypersensitivity responses to injected Leishmania antigens were also comparable in Ii −/− and +/− mice (data not shown). Mice on two additional MHC backgrounds (H-2d, and H-2k) with the Ii −/− genotype also maintained resistance to L. major (data not shown).
Figure 1.


Ii −/− mice on a resistant background control L. major infection. (A) Mice were inoculated in the hind footpads with promastigotes of L. major, and the course of disease followed by measuring the size of the footpads with time. A cohort of BALB/c mice was inoculated concurrently to assess virulence of the inoculum. Designated C57BL/6 Ii −/− mice were treated with neutralizing anti–IFN-γ antibody as noted. Data points are means and standard deviations of groups of animals, and are representative of nine separate experiments involving 30 Ii −/− mice. (B) Ii −/− mice on a resistant background were crossed to βm −/− to create double mutant animals and infected with promastigotes of L. major in the hind footpads. Cohorts of β2m −/− and Ii +/− animals, together with susceptible BALB/c mice, were inoculated at the same time. Data points are means and standard deviations and representative of results obtained with 6 Ii −/− β2m −/− mice.
The Ii −/− mice have an expanded population of CD8+ T cells that, in some instances, have been shown to exert immunologic activity in experimental leishmaniasis (25, 26). Since β2m −/− mice have been demonstrated to control primary L. major infection comparably to wild-type mice (8), these mice were bred into the Ii −/− background, and the experiment was repeated to rule out a contribution by the CD8+ T cell compartment. Again, Ii −/− mice controlled the local lesion as well as Ii +/− or +/+ mice in the presence or absence of β2m (Fig. 1 B).
To confirm that measurements of the local lesions correlated with tissue parasite burdens, dilutions of footpad and spleen were incubated in vitro and the numbers of recovered parasites were quantitated microscopically (Fig. 2). Although there was some variability among experiments, Ii −/− mice had comparable or even lower numbers of recovered organisms than Ii +/− mice in both tissues. Both Ii −/− and +/− mice, however, had significantly lower numbers of parasites than genetically susceptible BALB/c mice. The capacity of the Ii −/− mice to control L. major contrasted sharply with the inability of class II −/− mice to resist infection. These latter mice demonstrated unrestrained progression of the local lesion and large numbers of parasites in the footpad and spleen after 8 wk of infection (Fig. 2).
Figure 2.
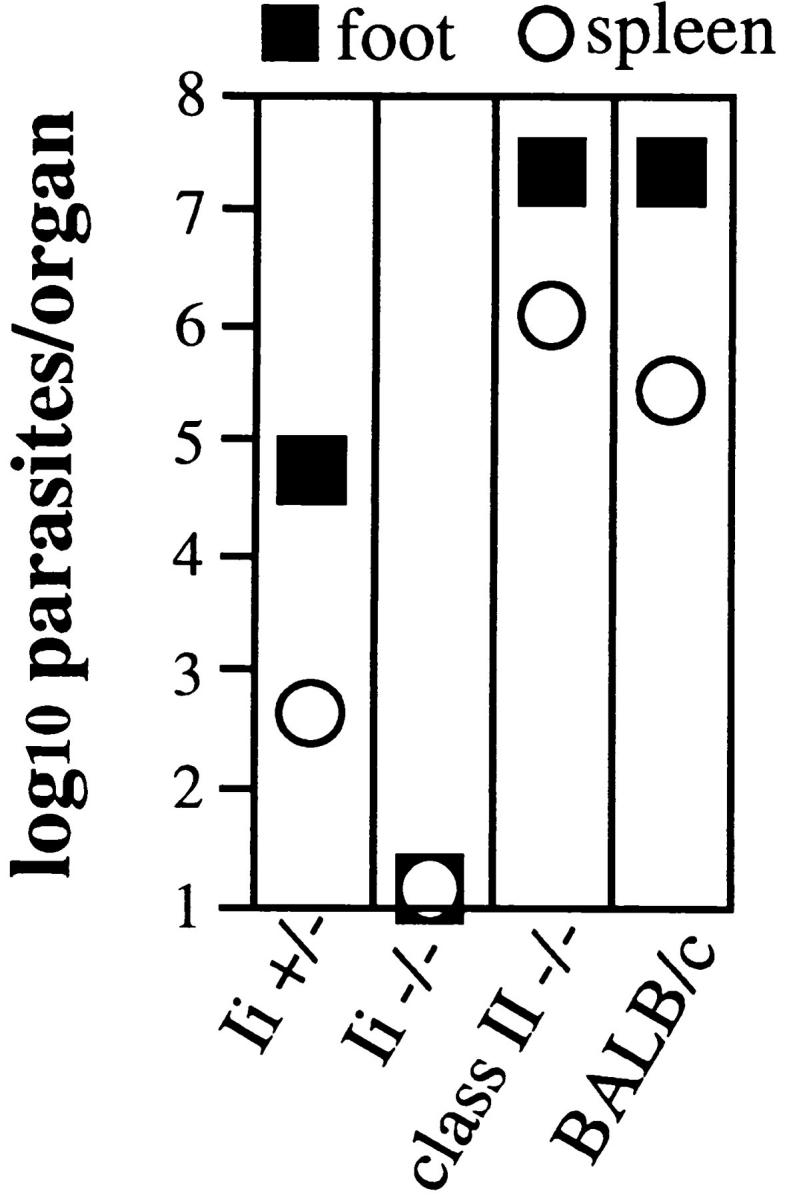
Resistance in Ii −/− mice represents control of parasite growth in tissues. Cohorts of C57BL/6 Ii −/− or Ii +/− mice, together with class II −/− and susceptible BALB/c mice, were infected with L. major and, after 8 wk, the popliteal lymph nodes and spleens were homogenized in equivalent volumes of media and serially diluted into growth media that supported the conversion of intracellular organisms to the extracellular promastigotes. Wells were examined 2 wk later for motile promastigotes using inverted microscopy.
Ii −/− Mice on a Resistant Background Generate Type I Immune Responses during Infection.
Popliteal lymph node cells draining the lesions of infected mice were restimulated in vitro with soluble parasite antigens to assess the qualitative cytokine response. IFN-γ was recovered from supernatants of stimulated cells from infected Ii −/− and β2m −/− Ii −/− mice at levels comparable to or greater than cells from Ii +/− mice (Fig. 3 A). Production of IFN-γ in vitro was reduced to less-than-detectable levels if anti–class II monoclonal antibody, but not an isotype control antibody, was included during the period of antigen stimulation (data not shown). IL-4, which was readily recovered by stimulation of cells from susceptible BALB/c mice, was not detected. Similar results were obtained analyzing the expression of IFN-γ and IL-4 transcripts in lymph node cells immediately after removal from infected animals (Fig. 3 B). Taken together, these data suggest that the deficiency in Ii expression does not impede the development of a class II– dependent type 1 immune response to L. major. Th1 effector development was comparable in Ii −/− mice on H-2b, H-2k, and H-2d backgrounds, despite the differences in efficient assembly of these different class II molecules in the absence of Ii (27).
Figure 3.


Ii −/− mice develop type 1 immune responses during infection with L. major. (A) Lymph node cells from the designated mice were collected 8 wk after infection with L. major, and incubated as described in the Materials and Methods, with or without soluble Leishmania antigens for 48 h. Supernatants were assayed for IFN-γ and IL-4 by ELISA. Points indicate quantities present in antigen-stimulated wells. All incubations in the absence of antigen resulted in cytokine recoveries <7 U/ml, except for one β2m −/− animal with an IFN-γ level of 19 U/ml, and the BALB/c with an IL-4 level of 100 U/ml. Data points represent means and standard deviations of triplicate determinations, and are representative of five separate experiments. (B) Total RNA from lymph node cells from infected Ii +/+ (+/+), −/− (−/−), or BALB/c (B/c) mice was reverse transcribed and subjected to PCR amplification in the presence of a competitor construct containing mutated cDNAs (upper bands) ∼75 bp larger than the authentic transcripts (lower bands). After standardizing the input cDNA for the amounts of the constitutively expressed HPRT gene product, the amounts of IFN-γ and IL-4 were determined using the requisite primers. These results are representative of three separate experiments.
T Cell Proliferation and IL-2 Production Induced by L. major Is Ii Dependent.
The unusual intracellular compartment occupied by Leishmania amastigotes raised the possibility that peptides from the organisms might normally access class II in an Ii-independent manner. However, we found that antigen-induced proliferation of lymph node cells taken from infected animals was significantly impaired in Ii −/−, as compared to I +/−, mice, demonstrating substantial Ii-dependence for optimal antigen presentation (Fig. 4). Assays using the same groups of cells showed comparable levels of antigen-induced IFN-γ, revealing dissociation of these two lymphocyte activities under the conditions used (Fig. 4).
Figure 4.

IFN-γ production is maintained in infected Ii −/− mice, despite suboptimal proliferation. Lymph node cells were prepared from Ii +/− or −/− mice infected 8 wk before with L. major, and incubated with or without soluble Leishmania antigens as described in the Materials and Methods. After 48 h, supernatants were collected and assayed for the presence of IFN-γ by ELISA (right). After 72 h, wells were pulsed with 1 μCi[3H]thymidine, and the cells harvested 16 h later to measure incorporation of radioactivity. Data points on the left represent results in the absence of antigen, and on the right, in the presence of antigen. Data represent three individual mice from each group, and are representative of three separate experiments. Values are means and standard deviations of triplicate determinations.
To further characterize the deficiency in presentation of parasite-derived antigens by Ii −/− cells, bone marrow– derived APCs were established from H-2d Ii −/− and +/− mice and used to measure activation of I-Ad–restricted TCR transgenic T cells that recognize an epitope from an immunodominant antigen of L. major designated LACK (21). After preincubating APCs for 20 h with the 18– amino acid peptide epitope, production of IL-2 in the supernatant (Fig. 5) was consistently enhanced using Ii −/−, as compared to Ii +/−, cells, in agreement with prior experiments (17, 28, 29). Processing and presentation of the LACK epitope from SLA, viable promastigotes, or intracellular amastigotes, however, revealed that only Ii +/− APCs could induce the transgenic T cells to release significant levels of IL-2. Thus, optimal activation of these transgenic T cells by processed antigen is Ii dependent.
Figure 5.

Optimal stimulation of LACK-reactive transgenic T cells is Ii dependent. Bone marrow–derived macrophages were established from Ii +/− or −/− littermates and incubated with varying amounts of the 18–amino acid peptide epitope of LACK (Peptide), soluble Leishmania antigens (SLA), live promastigotes (Promast.), or live amastigotes (Amast.) for 20 h as described in the Materials and Methods. The monolayers were washed and 105 TCR transgenic T cells added to 5 × 104 macrophages. After 48 h, the supernatants were assayed for the presence of IL-2 by ELISA. Data points represent means and standard deviations of triplicate determinations, and are representative of four separate experiments.
IFN-γ Production Can Be Induced by IL-12 at Suboptimal Antigen Thresholds.
The striking preservation of the IFN-γ response in vivo and in vitro suggested that costimulatory signals triggered by the organisms might compensate for the Ii −/− defect in antigen presentation. IL-12, induced by the intracellular forms of L. major (22), is required for optimal IFN-γ production and control of infection (30–32), and is required for Th1 effector cell development (33, 34). Stimulation of transgenic T cells (Fig. 6) with LACK peptide at levels >0.1 μM presented by normal Ii + APCs, allowed the ready recovery of IL-2, but not IFN-γ, from culture supernatants. Below this concentration of peptide, neither IL-2 nor proliferation was induced. Using suboptimal concentrations of the peptide, however, the addition of recombinant IL-12 (rIL-12) induced the dose-dependent production of IFN-γ by transgenic T cells not accompanied by IL-2 recovery in the supernatants (Fig. 6).
Figure 6.

IL-12 induces IFN-γ production from TCR transgenic T cells at suboptimal levels of stimulation. Splenocytes from Ii+ mice were incubated in either the absence (0) or presence of increasing amounts of the LACK peptide epitope, in the absence (open symbols) or presence of increasing amounts of rIL-12 (filled symbols), as designated on the lower axis. Each well contained 2 × 105 Ii + APCs and 2 × 105 TCR transgenic lymph node T cells. After 48 h, supernatants were collected and assayed for IL-2 and IFN-γ by ELISA. Data points represent means and standard deviations from duplicate determinations, and are representative of three separate experiments.
To confirm that rIL-12 could sustain IFN-γ production under conditions where the numbers of class II/peptide complexes might be limited, experiments were carried out using the same preparations of normal Ii + APCs in which either amounts of peptide, numbers of APCs, or numbers of accessible class II/peptide complexes were titrated. For the latter condition, dilutions of anti–class II monoclonal antibody were included in the cultures during the period of presentation to transgenic T cells. All of the experiments were performed in the presence of rIL-12. Supernatants were collected after 48 h and analyzed for the presence of IL-2 and IFN-γ (Fig. 7). Under each set of conditions, the amount of IL-2 recovered was directly related to the degree of class II–dependent stimulation. In contrast, under these same conditions, the amount of IFN-γ recovered was essentially stable. Thus, as assessed using these transgenic T cells, and as suggested by cells recovered from infected mice (Fig. 4), IL-12 preserves IFN-γ production under conditions suboptimal for inducing IL-2 or lymphoproliferation.
Figure 7.

IL-12 sustains T cell IFN-γ production at suboptimal levels of stimulation. Splenocytes from Ii+ mice were incubated with varying amounts of the LACK peptide epitope (Peptide; 5 × 105 APCs/well), inoculated into wells at varying numbers (APCs; 2.5 μM peptide), or inoculated into wells using a fixed number of cells and amount of peptide (α–class II; 5 × 105 APCs/well, 1.25 μM peptide). After washing, 2 × 105 TCR transgenic lymph node T cells were added to the wells in the presence of rIL-12 (2 ng/ml). In the right panel, anti-class II mAb M5/114 was titrated into the wells using the dilutions shown. Supernatants were collected after 48 h and assayed for IL-2 and IFN-γ using ELISA. Values represent means and standard deviations of triplicate determinations, and are representative of three separate experiments.
IL-12 Can Preserve IFN-γ Production by T Cells Primed on Ii −/− APCs.
To confirm that rIL-12 could similarly sustain IFN-γ production from T cells primed on Ii −/− APCs, Ii +/− or −/− APCs were incubated with varying concentrations of the recombinant LACK antigen and transgenic T cells in the presence of rIL-12. After 48 h, supernatants were analyzed for IL-2 and IFN-γ (Fig. 8 A). Consistent with the earlier results using normal APCs under conditions of low antigenic stimulation, T cells primed on Ii −/− APCs generated IFN-γ that was comparable to that generated by T cells primed on Ii +/− APCs, although only the latter generated IL-2 that could be recovered in supernatants. Recovery of IFN-γ was dependent on both antigen and rIL-12; in the absence of either, no cytokine could be recovered from transgenic T cells primed on Ii −/− APCs. This experiment additionally confirms that the LACK protein antigen, in contrast to the immunogenic LACK peptide (Fig. 5), requires Ii for optimal presentation.
Figure 8.


IL-12 can maintain IFN-γ production and implement Th1 development by T cells primed on Ii −/− APCs. (A) Irradiated spleen cells (105/ well) from Ii +− (filled symbols) or Ii −/− (open symbols) H-2d littermates were incubated with varying amounts of recombinant LACK Ag in the presence of 2 × 105 TCR transgenic lymph node cells and 2 ng/ml of rIL-12. After 48 h, supernatants were assayed for IL-2 and IFN-γ using ELISA. Values represent means and standard deviations from triplicate determinations, and are representative of four separate experiments. (B) B220+ cell–depleted TCR transgenic lymph node cells (2 × 105 cells/well) were stimulated in a 1 ml volume in 24-well plates with either Ii + or Ii −/− irradiated H-2d spleen cells (1 × 106 cells/well), plus recombinant LACK antigen (2 μg/ml), with (+) or without (−) rIL-12 (0.5 ng/ml) as designated on the x-axis. 6 d later, viable cells were purified and washed, and 2 × 105 T cells were restimulated with the same Ii-genotype APCs (1.2 × 106/well) used in the primary stimulation in the presence of antigen (2 μg/ml), but without rIL-12. After 48 h, supernatants were collected and assayed for IL-2 and IFN-γ by ELISA. Values represent means and standard deviations of triplicate determinations, and are representative of two separate experiments.
The development of Th1 effector cells is mediated by IL-12–dependent priming during the initial encounter of naive T cells with the appropriate peptide/MHC complex (33). To confirm that the IL-12-induced responses by T cells primed on Ii −/− APCs extended to secondary responses, transgenic T cells were primed using antigen-pulsed Ii +/− or −/− APCs in the presence or absence of rIL-12. After 6 d, viable cells were purified and redistributed to wells containing fresh antigen-pulsed Ii +/− or −/− APCs in the absence of IL-12. After 48 h, supernatants were collected and analyzed for IFN-γ (Fig. 8 B). As assessed under these conditions, transgenic T cells stimulated with Ii −/− APCs displayed even greater production of IFN-γ than T cells stimulated with Ii +/− APCs, and the production of IFN-γ was dependent on the presence of rIL-12 during the primary stimulation with LACK.
Ii −/− Mice on a Susceptible Background Generate Type 2 Immune Responses during Infection.
The finding that Th1 development was maintained in Ii −/− mice on a resistant background might be partially explained by effects of low antigen dose on Th subset development. Antigen dosage effects on subset differentiation have been demonstrated using TCR transgenic T cells in vitro (35, 36), and have been supported by observations in L. major infection. Thus, susceptible BALB/c mice have been rendered resistant by either administration of low numbers of parasites (37), or through interventions such as anti-CD4 monoclonal antibody (38) or sublethal irradiation (39) that decrease the numbers of responding T cells. The impaired antigen presentation and diminished CD4+ T cell numbers in Ii −/− mice might independently bias Th subset differentiation to the Th1 phenotype.
To test this prediction, Ii −/− mice were bred four generations onto the susceptible BALB/c background, and Ii −/− and +/− littermates were infected with L. major. As assessed by monitoring of the local footpad lesions, BALB/c Ii −/− suffered disease that was comparable to Ii +/− littermates, and both differed markedly from the course of infection in concurrently infected C57BL/6 Ii −/− or Ii +/+ mice (Fig. 9 A). Quantitation of parasites recovered from the footpads and spleen corroborated the lesion measurements (data not shown). Lymph node cells recovered from infected animals and incubated with SLA in vitro generated readily recovered IL-4 that was not obtained using lymph node cells from C57BL/6 mice (Fig. 9 B). Thus, Th2 cell differentiation in vivo was also maintained in the absence of invariant chain.
Figure 9.

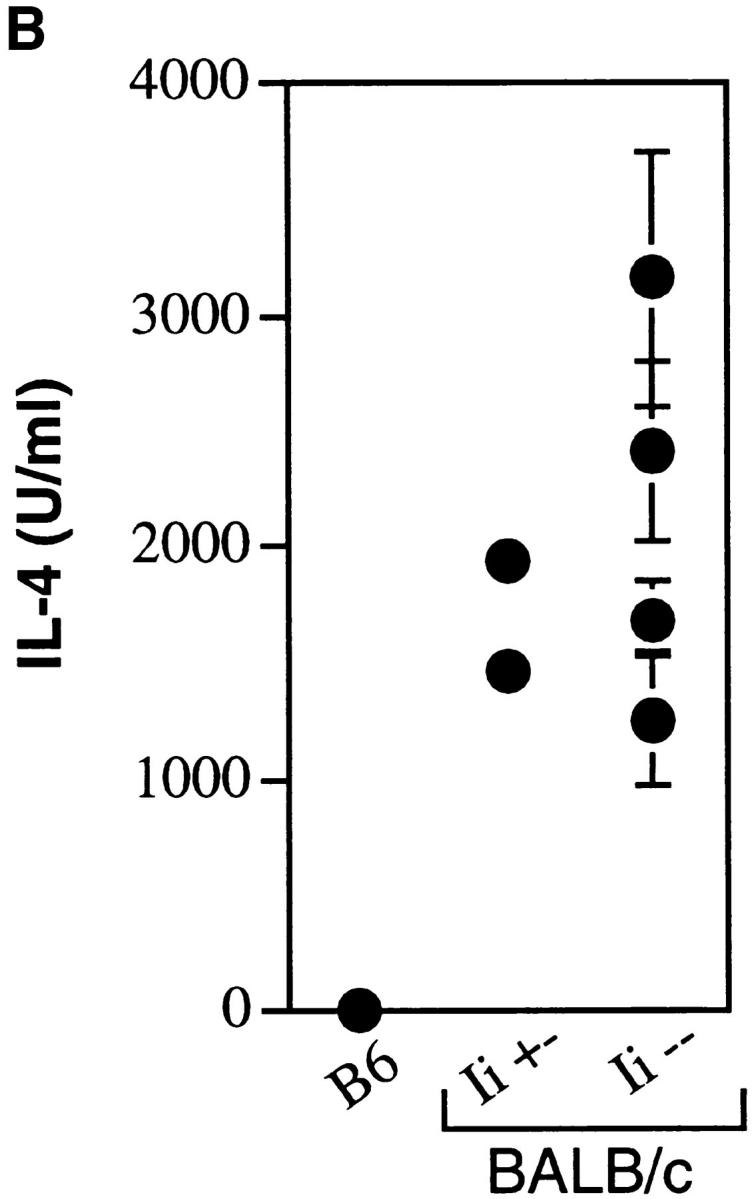
BALB/c Ii −/− mice are susceptible to L. major infection and generate type 2 immune responses. (A) Groups of fourth generation BALB/c Ii +/− or Ii −/− and C57BL/6 (B6) Ii + or Ii +/− mice were infected in the hind footpads with promastigotes of L. major, and the size of the footpad lesions measured using a metric caliper. Data represent means and standard deviations of footpad lesions in four separate experiments involving 12 BALB/c Ii −/− mice. (B) Lymph node cells were prepared from individual mice after 7 wk of infection and were cultured with and without soluble Leishmania antigens as described in the Materials and Methods. After 48 h, the supernatants were assayed for IL-4 by ELISA. Cells cultured without antigen yielded <20 U/ml IL-4. C57BL/6 (B6) mice are compared to BALB/c Ii +/− and −/− mice. Values represent means and standard deviations of triplicate determinations for individual animals, and are representative of three separate experiments.
To confirm that the threshold for Th2 development could be maintained in the absence of invariant chain, LACK-specific transgenic T cells were primed using antigen-pulsed, irradiated spleen cells from Ii + or Ii −/− mice in the presence or absence of rIL-4. After 6 d, viable cells were washed extensively, counted, and redistributed with freshly prepared, antigen-pulsed, irradiated spleen cells from the same Ii backgrounds, but in the absence of rIL-4. After 48 h, the supernatants were analyzed for IL-4 (Fig. 10). Recovery of IL-4 was substantially enhanced during the secondary stimulation when cells were primed using Ii −/− APCs, but only when rIL-4 was included during the primary incubation with LACK.
Figure 10.

IL-4 supports Th2 differentiation by T cells primed on Ii −/− APCs. CD4-selected TCR transgenic lymph node cells (2 × 105 cells/ well) were stimulated in a 1 ml volume in 24-well plates with either Ii + or Ii −/− irradiated H-2d spleen cells (1 × 106 cells/well), plus recombinant LACK antigen (0.5 μg/ml), with (+) or without (−) rIL-4 (5,000 U/ml) as designated on the x-axis. 6 d later, viable cells were purified and washed, and 2 × 105 T cells were restimulated with the same Ii-genotype APCs (1.2 × 106/well) used in the primary stimulation in the presence of antigen (0.5 μg/ml), but without rIL-4. After 48 h, supernatants were collected and assayed for IL-4 by ELISA. Values represent means and standard deviations of triplicate determinations.
Discussion
Infection with L. major is a well-characterized model in which differentiation of class II–restricted T cells into the two mature helper subsets is required for expression of the resistant and susceptible disease phenotype. Ii is required for stable expression of surface class II molecules and, as predicted, cells from Ii −/− mice have substantially lower amounts of surface class II that do not assume the compact conformation that characterizes stable peptide binding (17, 28, 29). The major immunologic consequences are twofold: a severely compromised ability to present processed antigens via the class II pathway, and a quantitatively and qualitatively altered CD4+ population due to aberrant selection by thymic epithelial cells unable to present self peptides in a normal manner (40, 41). Despite this drastic effect on the class II–dependent immune response, we could discern little consequence to the host in generating either Th1 or Th2 responses to L. major. How might we explain this unpredicted outcome?
One possibility is that effector T cells producing IFN-γ or IL-4 are responding to Ii-independent antigens, whereas proliferating (or nonproliferating, in the case of Ii −/− mice) T cells are responding to Ii-dependent antigens. Although it is not possible to dismiss this hypothesis completely, we think it unlikely for several reasons. First, we could demonstrate that an immunodominant L. major antigen, LACK, was presented optimally only by cells that contained Ii and, in studies of infected mice, expansion of LACK-reactive cytokine-producing T cells can be directly documented (20). Secondly, our in vitro studies demonstrated the possibility of achieving effector differentiation in the absence of discernible IL-2 production or proliferation. Dissociation of effector function from proliferation has been recently described among CTL clones that can be triggered to kill targets at doses of peptide which are several orders of magnitude below doses required for proliferation (42). Lastly, a recent study of antiviral immunity demonstrated that the same antigen which required Ii to elicit IL-2 production from a hybridoma could elicit specific T helper-dependent antibody from infected or immunized Ii −/− mice (43). We, therefore, consider it unlikely that distinct epitopes underlie proliferative and cytokine-effector responses.
The unique location of L. major within the class II pathway may also influence the ability of Ii −/− mice to generate strong anti-parasite immunity. Infection of macrophages does not limit surface expression of class II, and peptides eluted from surface class II exhibited comparable diversity to those from uninfected cells as well as parasite-specific complexes (15). Comparable experiments have not been done using infected Ii −/− macrophages, but analysis of the fine specificity of peptides presented by such cells has indicated that a varied range of epitopes might be presented by Ii −/−, as compared to Ii +/+ APCs (44). The route by which class II complexes exit the ER or cis-Golgi to transit to the surface in Ii −/− cells is not defined (17, 28, 29). Parasite-derived peptides could stabilize class II in the endoplasmic reticulum, as described for some endogenous class II–dependent antigen presentation pathways (45), or could load class II via a compartment that recycles from the cell membrane, as described for influenza hemagglutinin (46). The ability of the parasite to load class II with peptides or the potential recruitment of additional epitopes may contribute to an effective immune response in the absence of Ii. Our direct examination of an epitope known to be presented in vivo, however, demonstrated that parasitespecific class II complexes are functionally reduced in Ii −/− infected APCs (Fig. 5).
The effects of Ii deficiency on the T cell compartment must also be considered in understanding the remarkable preservation of subset effector differentiation. Peripheral CD4+ T cells are reduced to ∼25% of normal numbers in Ii −/− mice. CD4 −/− mice have even lower numbers of class II–restricted T cells in the periphery, yet readily control L. major infection (6). Acutely decreasing the number of CD4+ T cells can confer protection on otherwise susceptible BALB/c mice (38). Thus, it may not be surprising that Ii −/− mice have sufficient numbers of T cells to mount an adequate Th1 response. The reduction in the T cell compartment of Ii −/− mice is probably not an essential compensatory mechanism that allows for Th1 differentiation in C57BL/6 Ii −/− mice, however, since BALB/c Ii −/− mice readily develop Th2 responses despite similarly reduced numbers of CD4+ T cells. In addition to decreased numbers, CD4+ T cells from Ii −/− mice display an altered phenotype resembling prior activation (CD45RBlo, CD44hi, and L-selectinlo, with slightly diminished TCR expression [17] that is probably due to “incomplete” positive selection by abnormal class II/peptide complexes on thymic epithelial cells (41). The “activated” phenotype of CD4+ T cells in Ii −/− mice could contribute to a lower threshold of activation. Resting T cells activated in the presence of costimulatory ligands responded to lower antigen doses in a manner comparable to activated T cells (47). Even in the absence of Ii-deficient thymic education, however, transgenic T cells could be induced to secrete IFN-γ or IL-4 under conditions of limited peptide dose that was incapable of supporting proliferation or IL-2 production.
The finding that IFN-γ production by Th1 cells (and IL-4 production by Th2 cells) was comparable or greater from Ii −/− T cells, despite even fewer class II–restricted T cells in the lymph nodes, demonstrates striking preservation of T cell effector function in the setting of suboptimal antigen presentation. Titrating down the number of class II/peptide complexes on normal APCs in vitro to levels at which lymphoproliferation could not be sustained was accompanied by no diminution in IFN-γ production; similar findings could be demonstrated using Ii −/− APCs as compared to Ii +/− APCs, again consistent with retention of effector function at levels of class II/peptide that could not support IL-2 production. Just as the levels of cytokines produced in Ii −/− mice would sometimes exceed those seen in wild-type mice, activation of transgenic T cells in vitro often yielded slightly higher IFN-γ or IL-4 production when antigen was presented by Ii −/− APCs. These results suggest that weaker antigenic stimulation may favor, rather than limit, effector cytokine production at some ranges of antigen dose. The mechanism for this finding is unclear, but may also underly prior observations on the influence of peptide dose in T helper lineage commitment (35, 36).
For Th1 development, the preservation or augmentation of effector function was revealed only in the presence of rIL-12 and in a dose-dependent manner. The ability of IL-12 to augment IFN-γ production in response to antigen is well known, and was confirmed in IL-12 p40 −/− mice, in which IFN-γ production was severely impaired but IL-2 production and lymphoproliferation were unaffected (34). The studies reported here suggest that IL-12, at least for Th1 development, may be one of the signals implicated in “tuning” the threshold for activation by differing levels of TCR engagement (47). This activity is distinct from the costimulatory effect of IL-12 on committed Th1 cells, which includes a role in both proliferation and IFN-γ production (48). The ability of IL-4 to support robust Th2 development of parasite-specific T cells primed on Ii −/− cells in vitro suggests a similar role for IL-4 in affecting the threshold for Th2 effector cell function. The efficient development of Th1 and Th2 responses in Ii −/− mice, at least in this system, implies that the cellular sources for IL-12 and IL-4 in infected mice remain unperturbed by the loss of Ii.
It is intriguing to speculate that signals produced by cells of the innate immune system, such as IL-12, might be involved in lowering thresholds for distinct types of T cell effector responses to levels below those required for clonal expansion. This might enable maximal numbers of potentially protective naive T cells to be recruited at very low levels of peptide/MHC complex formation, thus limiting the spread of infection and allowing the sampling of multiple peptide/MHC complexes before clonal expansion and competition for growth factors and lymph node niches can occur. The production of inflammatory mediators by microbial pathogens has a major impact on T cell survival (49), so perhaps it should not be surprising that they greatly influence T cell effector functions at low thresholds for activation. In this manner, the immune system can focus attention on the few peptides presented in an inflammatory context, rather than the many peptides presented innocuously.
Footnotes
We gratefully acknowledge D. Mathis and C. Benoist for the generous gift of Ii −/− and class II −/− mice. We also thank R. Jaenisch for use of β2m −/− mice, N. Killeen for help with construction of TCR transgenic animals, N. Glaichenhaus for recombinant LACK antigen, and J. Miller for invaluable advice.
D.R. Brown is supported by the University of Chicago Medical Scientist Training Program and Immunology Training Grant (AI-07090), M.F. Naujokas is supported by an Arthritis Foundation Postdoctoral Fellowship, S.L. Reiner and R.M. Locksley are supported by the Burroughs Wellcome Fund and the National Institutes of Health (AI-01309 and AI-30663).
1 Abbreviations used in this paper: HPRT, hypoxanthine-guanine phosphoribosyl transferase; LACK, Leishmanial receptor for activated protein kinase C; SLA, soluble Leishmania antigen.
D.R. Brown and K. Swier contributed equally to this work.
References
- 1.Reiner, S.L., and R.M. Locksley. 1995. The regulation of immunity to Leishmania major. <JNL>Annu. Rev. Immunol. 13:151–177. [DOI] [PubMed]
- 2.Moll H, Scollay R, Mitchell GF. Resistance to cutaneous leishmaniasis in nude mice injected with L3T4+ T cells but not with Ly-2+ T cells. Immunol Cell Biol. 1988;66:57–63. doi: 10.1038/icb.1988.7. [DOI] [PubMed] [Google Scholar]
- 3.Holaday BJ, Sadick MD, Wang Z-E, Reiner SL, Heinzel FP, Parslow TG, Locksley RM. Reconstitution of Leishmaniaimmunity in severe combined immunodeficient mice using Th1- and Th2-like cell lines. J Immunol. 1991;147:1653–1658. [PubMed] [Google Scholar]
- 4.Varkila K, Chatelain R, Leal LMCC, Coffman RL. Reconstitution of C.B-17 scid mice with BALB/c T cells initiates a T helper type-1 response and renders them capable of healing Leishmania majorinfection. Eur J Immunol. 1993;23:262–268. doi: 10.1002/eji.1830230141. [DOI] [PubMed] [Google Scholar]
- 5.Wang, Z.-E., S.L. Reiner, S. Zheng, D.K. Dalton, and R.M. Locksley. 1994. CD4+ effector cells default to the Th2 pathway in interferon γ–deficient mice infected with Leishmania major. <JNL> J. Exp. Med. 179:1367–1371. [DOI] [PMC free article] [PubMed]
- 6.Locksley RM, Reiner SL, Hatam F, Littman DR, Killeen N. Helper T cells without CD4: control of leishmaniasis in CD4–deficient mice. Science (Wash DC) 1993;261:1448–1451. doi: 10.1126/science.8367726. [DOI] [PubMed] [Google Scholar]
- 7.Chakkalath HR, Theodos CM, Markowitz JS, Grusby MJ, Glimcher LH, Titus RG. Class II major histocompatibility complex–deficient mice initially control an infection with Leishmania majorbut succumb to the disease. J Infect Dis. 1995;171:1302–1308. doi: 10.1093/infdis/171.5.1302. [DOI] [PubMed] [Google Scholar]
- 8.Wang Z-E, Reiner SL, Hatam F, Heinzel FP, Bouvier J, Turck CW, Locksley RM. Targeted activation of CD8 cells and infection of β2-microglobulin–deficient mice fail to confirm a primary protective role for CD8 cells in experimental leishmaniasis. J Immunol. 1993;151:2077–2086. [PubMed] [Google Scholar]
- 9.Brown DR, Fowell DJ, Corry DB, Wynn TA, Moskowitz NH, Cheever AW, Locksley RM, Reiner SL. β2-microglobulin–dependent NK1.1+T cells are not essential for T helper cell 2 immune responses. J Exp Med. 1996;184:1295–1304. doi: 10.1084/jem.184.4.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Antoine, J.-C., C. Jouanne, T. Lang, E. Prina, C. de Chastellier, and C. Frehel. 1991. Localization of major histocompatibility complex class II molecules in phagolysosomes of murine macrophages infected with Leishmania amazonensis. <JNL>Infect. Immun. 59:764–775. [DOI] [PMC free article] [PubMed]
- 11.Russell DG, Xu S, Chakraborty P. Intracellular trafficking and the parasitophorous vacuole of Leishmania mexicana–infected macrophages. J Cell Sci. 1992;103:1193–1210. doi: 10.1242/jcs.103.4.1193. [DOI] [PubMed] [Google Scholar]
- 12.Fruth U, Solioz N, Louis JA. Leishmania majorinterferes with antigen presentation by infected macrophages. J Immunol. 1993;150:1857–1864. [PubMed] [Google Scholar]
- 13.Prina E, Jouanne C, Lao S, Szabo A, Guillet J-G, Antoine J-C. Antigen presentation capacity of murine macrophages infected with Leishmania amazonensisamastigotes. J Immunol. 1993;151:2050–2061. [PubMed] [Google Scholar]
- 14.Leao S, Lang T, Prina E, Hellio R, Antoine J-C. Intracellular Leishmania amazonensisamastigotes internalize and degrade class II molecules of their host cells. J Cell Sci. 1995;108:3219–3231. doi: 10.1242/jcs.108.10.3219. [DOI] [PubMed] [Google Scholar]
- 15.Campos-Neto A, Soong L, Cordova JL, Sant' D, Angelo, Skeiky YA, Ruddle NH, Reed SG, Jr, Janeway C, McMahon-Pratt D. Cloning and expression of a Leishmania donovanigene instructed by a peptide isolated from major histocompatibility complex class II molecules of infected macrophages. J Exp Med. 1995;182:1423–1433. doi: 10.1084/jem.182.5.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cresswell P. Invariant chain structure and class II function. Cell. 1996;84:505–507. doi: 10.1016/s0092-8674(00)81025-9. [DOI] [PubMed] [Google Scholar]
- 17.Viville S, Neefjes J, Lotteau V, Dierich A, Lemur M, Ploegh H, Benoist C, Mathis D. Mice lacking the MHC class II–associated invariant chain. Cell. 1993;72:635–648. doi: 10.1016/0092-8674(93)90081-z. [DOI] [PubMed] [Google Scholar]
- 18.Cosgrove D, Gray D, Dierich A, Kaufman J, Lemeur M, Benoist C, Mathis D. Mice lacking MHC class II molecules. Cell. 1991;66:1051–1066. doi: 10.1016/0092-8674(91)90448-8. [DOI] [PubMed] [Google Scholar]
- 19.Zijlstra M, Bix M, Simister NE, Loring JM, Raulet DH, Jaenisch R. Beta 2-microglobulin deficient mice lack CD4-8+ cytolytic T cells. Nature (Lond) 1990;344:742–746. doi: 10.1038/344742a0. [DOI] [PubMed] [Google Scholar]
- 20.Reiner S, Wang Z-E, Hatam F, Scott P, Locksley RM. Th1 and Th2 cell antigen receptors in experimental leishmaniasis. Science (Wash DC) 1993;259:1457–1460. doi: 10.1126/science.8451641. [DOI] [PubMed] [Google Scholar]
- 21.Mougneau E, Altare F, Wakil AE, Zheng S, Coppola T, Wang ZE, Waldmann R, Locksley RM, Glaichenhaus N. Expression cloning of a protective Leishmaniaantigen. Science (Wash DC) 1995;268:563–566. doi: 10.1126/science.7725103. [DOI] [PubMed] [Google Scholar]
- 22.Reiner SL, Zheng S, Wang Z, Stowring L, Locksley RM. Leishmania promastigotes evade interleukin 12 (IL-12) induction by macrophages and stimulate a broad range of cytokines from CD4+T cells during initiation of infection. J Exp Med. 1994;179:447–456. doi: 10.1084/jem.179.2.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reiner SL, Zheng S, Corry DB, Locksley RM. Constructing polycompetitor cDNAs for quantitative PCR. J Immunol Methods. 1993;165:37–46. doi: 10.1016/0022-1759(93)90104-f. [DOI] [PubMed] [Google Scholar]
- 24.Belosevic, M., D.S. Finbloom, P.H. Van der Meide, M.V. Slayter, and C.A. Nacy. 1989. Administration of monoclonal anti-IFN-γ antibodies in vivo abrogates natural resistance of C3H/HeN mice to infection with Leishmania major. <JNL>J. Immunol. 143:266–274. [PubMed]
- 25.Hill, J.O., M. Awwad, and R.J. North. 1989. Elimination of CD4+ suppressor T cells from susceptible BALB/c mice releases CD8+ T lymphocytes to mediate protective immunity against Leishmania. <JNL>J. Exp. Med. 169:1819–1827. [DOI] [PMC free article] [PubMed]
- 26.Muller I, Pedrazzini T, Kropf P, Louis J, Milon G. Establishment of resistance to Leishmania major infection in susceptible BALB/c mice requires parasite-specific CD8+T cells. Int Immunol. 1991;3:587–597. doi: 10.1093/intimm/3.6.587. [DOI] [PubMed] [Google Scholar]
- 27.Bikoff EK, Germain RN, Robertson EJ. Allelic differences affecting invariant chain dependency of MCH class II subunit assembly. Immunity. 1995;2:301–310. doi: 10.1016/1074-7613(95)90054-3. [DOI] [PubMed] [Google Scholar]
- 28.Bikoff EK, Huang L, Episkopou V, van Meerwijk J, Germain RN, Robertson EJ. Defective major histocompatibility complex class II assembly, transport, peptide acquisition, and CD4+T cell selection in mice lacking invariant chain expression. J Exp Med. 1993;177:1699–1712. doi: 10.1084/jem.177.6.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Elliott EA, Drake JR, Amigorena S, Elsemore J, Webster P, Mellman I, Flavell RA. The invariant chain is required for intracellular transport and function of major histocompatibility complex class II molecules. J Exp Med. 1994;179:681–694. doi: 10.1084/jem.179.2.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sypek JP, Chung CL, Mayor SEH, Subramanyam JM, Goldman SJ, Sieburth DS, Wolf SF, Schaub RG. Resolution of cutaneous leishmaniasis: interleukin 12 initiates a protective T helper type 1 immune response. J Exp Med. 1993;177:1797–1802. doi: 10.1084/jem.177.6.1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heinzel FP, Rerko RM, Ahmed F, Pearlman E. Endogenous IL-12 is required for control of Th2 cytokine responses capable of exacerbating leishmaniasis in normally resistant mice. J Immunol. 1995;155:730–739. [PubMed] [Google Scholar]
- 32.Scharton-Kersten T, Afonson LC, Wysocka M, Trinchieri G, Scott P. IL-12 is required for natural killer cell activation and subsequent T helper 1 cell development in experimental leishmaniasis. J Immunol. 1995;154:5320–5330. [PubMed] [Google Scholar]
- 33.Hsieh C-S, Macatonia SE, Tripp CS, Wolf SF, O'Garra A, Murphy KM. Development of Th1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Science (Wash DC) 1993;260:547–549. doi: 10.1126/science.8097338. [DOI] [PubMed] [Google Scholar]
- 34.Magram J, Connaughton SE, Warrier RR, Carvajal DM, Wu C, Ferrante J, Stewart C, Sarmiento U, Faherty DA, Gately MK. IL-12–deficient mice are defective in IFNg production and type 1 cytokine responses. Immunity. 1988;66:57–63. doi: 10.1016/s1074-7613(00)80413-6. [DOI] [PubMed] [Google Scholar]
- 35.Constant S, Pfeiffer C, Woodward A, Pasqualini T, Bottomly K. Extent of T cell receptor ligation can determine the functional differentiation of naive CD4+T cells. J Exp Med. 1995;182:1591–1596. doi: 10.1084/jem.182.5.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hosken NA, Shibuya K, Heath AW, Murphy KM, O'Garra A. The effect of antigen dose on CD4+T helper cell pheonotype development in a T cell receptor–αβ–transgenic model. J Exp Med. 1995;182:1579–1584. doi: 10.1084/jem.182.5.1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bretscher, P.A., G. Wei, J.N. Menon, and H. BielefeldtOhmann. 1992. Establishment of stable, cell-mediated immunity that makes “susceptible” mice resistant to Leishmania major. <JNL>Science (Wash. DC). 257:539–542. [DOI] [PubMed]
- 38.Titus RG, Ceredig R, Cerottini J-C, Louis JA. Therapeutic effect of anti-L3T4 monoclonal antibody GK1.5 on cutaneous leishmaniasis in genetically susceptible BALB/c mice. J Immunol. 1985;135:2108–2117. [PubMed] [Google Scholar]
- 39.Howard, J.G., C. Hale, and F.Y. Liew. 1981. Immunological regulation of experimental cutaneous leishmaniasis. IV. Prophylactic effect of sublethal irradiation as a result of abrogation of suppressor T cell generation in mice genetically susceptible to Leishmania major. <JNL>J. Exp. Med. 153:578-568. [DOI] [PMC free article] [PubMed]
- 40.Tourne S, Nakano N, Viville S, Benoist C, Mathis D. The influence of invariant chain on the positive selection of single T cell receptor specificities. Eur J Immunol. 1995;25:1851–1856. doi: 10.1002/eji.1830250709. [DOI] [PubMed] [Google Scholar]
- 41.Wong P, Rudensky AY. Phenotype and function of CD4+T cells in mice lacking invariant chain. J Immunol. 1996;156:2133–2142. [PubMed] [Google Scholar]
- 42.Valitutti S, Muller S, Dessing M, Lanzavecchia A. Different responses are elicited in cytotoxic T lymphocytes by different levels of T cell receptor occupancy. J Exp Med. 1996;183:1917–1921. doi: 10.1084/jem.183.4.1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Battegay M, Bachmann MF, Burhkart C, Viville S, Benoist C, Mathis D, Hengartner H, Zinkernagel RM. Antiviral immune responses of mice lacking MHC class II or its associated invariant chain. Cell Immunol. 1996;167:115–121. doi: 10.1006/cimm.1996.0014. [DOI] [PubMed] [Google Scholar]
- 44.Bodmer H, Viville S, Benoist C, Mathis D. Diversity of endogenous epitopes bound to class II molecules limited by invariant chain. Science (Wash DC) 1994;263:1284–1286. doi: 10.1126/science.7510069. [DOI] [PubMed] [Google Scholar]
- 45.Weiss S, Bogen B. Class II-restricted presentation of intracellular antigen. Cell. 1991;64:767–776. doi: 10.1016/0092-8674(91)90506-t. [DOI] [PubMed] [Google Scholar]
- 46.Pinet V, Vergelli M, Martin R, Bakke O, Long EO. Antigen presentation mediated by recycling of surface HLA-DR molecules. Nature (Lond) 1995;385:603–606. doi: 10.1038/375603a0. [DOI] [PubMed] [Google Scholar]
- 47.Viola A, Lanzavecchia A. T cell activation determined by T cell receptor number and tunable thresholds. Science (Wash DC) 1996;273:104–106. doi: 10.1126/science.273.5271.104. [DOI] [PubMed] [Google Scholar]
- 48.Murphy EE, Terres G, Macatonia SE, Hsieh CS, Mattson J, Lanier L, Wysocka M, Trinchieri G, Murphy K, O'Garra A. B7 and interleukin 12 cooperate for proliferation and interferon gamma production by mouse T helper clones that are unresponsive to B7 costimulation. J Exp Med. 1994;180:223–231. doi: 10.1084/jem.180.1.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vella AT, McCormack JE, Linsley PS, Kappler JW, Marrack P. Lipopolysaccharide interferes with the induction of peripheral T cell death. Immunity. 1995;2:261–270. doi: 10.1016/1074-7613(95)90050-0. [DOI] [PubMed] [Google Scholar]