

Abstract
The transgenic (tg) expression of interleukin (IL)-4 under the control of a major histocompatibility complex (MHC) class I promoter leads to B cell hyperactivity in mice, characterized by increased B cell surface MHC class II and CD23 expression, elevated responsiveness of the B cells to polyclonal ex vivo stimulation, and increased immunoglobulin (Ig)G1 and IgE serum levels. Tg mice develop anemia, glomerulonephritis with complement and immune deposition in the glomeruli, and show increased production of autoantibodies. Treatment of IL-4 tg mice with anti-IL-4 neutralizing antibodies protected the mice from disease development, showing that IL-4 was responsible for the observed disorders. Deletion of superantigen responsive autoreactive T cells in the IL-4 tg mice was normal and treatment of mutant mice with deleting anti-CD4 antibodies failed to ablate the onset of autoimmune-like disease, suggesting that CD4+T cells were not the primary cause of the disorders. Furthermore, the deletion of B cells reacting against MHC class I molecules was also normal in the IL-4 tg mice. Therefore the most likely explanation for the increased production of autoantibodies and the autoimmunelike disorders is that IL-4 acts directly on autoreactive B cells by expanding them in a polyclonal manner. Taken together our results show that inappropriate multi-organ expression of IL-4 in vivo leads to autoimmune-type disease in mice.
Autoimmune diseases are caused by incomplete deletion and inappropriate peripheral activation of self-reactive T and/or B cells. Self-reactive T cells can cause autoimmune diseases either directly by interacting with self-target cells and/or organs, as seen in autoimmune diabetes, or indirectly by activating self-reactive B cells. The autoreactive B cells then in turn produce autoantibodies which can cause or contribute to autoimmune disorders such as rheumatoid arthritis, systemic lupus erythematosus (SLE)1, multiple sclerosis, or myasthenia gravis (1–3). Autoreactive T and B cells are present in normal animals and humans showing that factors other than self recognition per se are involved in the development of autoimmune disease (4–7). Even though human autoimmune disorders have been widely studied, and several and diverse animal models are available, it is known only in a few cases why autoreactive T or B cells become activated and manifest themselves. One important mechanism in the induction and maintenance of tolerance is the deletion of self-reactive T and B cells by apoptosis. Experiments with transgenic (tg) mice overexpressing the proto-oncogene bcl-2, which inhibits apoptosis, support this view. Some bcl-2 tg mouse strains develop glomerulonephritis and show inhibition of T and B cell apoptosis (8, 9). Another factor shown to be involved in apoptosis of T and B cells is fas. Mice with deficient expression of either fas or fas-ligand (lpr, lprcg, and gld mice) develop SLE-like symptoms (1, 2). Recent in vitro findings suggested that IL-4 can rescue apoptotic B cells from cell death (10–15). The expression of IL-4 in vivo could therefore lead to the survival and activation of autoreactive B cells and thus possibly contribute to the development of autoimmune disease. Whereas Th2 immune responses with highly localized but tightly regulated IL-4 production do not seem to cause autoimmune disease, it is possible that aberrant and continuous IL-4 secretion could lead to the development of autoimmune disorders either through polyclonal B cell activation, or by selectively rescuing apoptotic self-reactive B cells from cell death. To test this hypothesis we analyzed whether constitutive in vivo expression of IL-4 would lead to the production of autoantibodies and autoimmune disease. For this purpose we used tg mice expressing IL-4 under the control of a class I promoter leading to a low level of IL-4 production in virtually all cell types (16, 17). The use of this promoter ensures that autoreactive B cells undergoing negative selection are subject to the action of IL-4. In this report we show that IL-4 tg mice have elevated autoantibody titers and suffer from autoimmune-type disease.
Materials and Methods
Mice
The IL-4 tg mice were originally established from B6C3F1/ CrlBR mice and then bred with B6C3F1 mice (16). The mice used in this report were backbred eight generations with C3H mice. Littermates were used as wild-type controls. The CTLA-4 tg mice (18) were obtained from Peter Lane (Basel Institute of Immunology, Basel, Switzerland) and backbred eight generations on C57Bl/6 genetic background. To establish IL-4/CTLA-4 double tg mice, heterozygous IL-4 tg males were bred with heterozygous CTLA-4 tg females. The offspring were tested as previously described (16, 19) and non-tg or single-tg mice were used as control animals. The 3-83μ tg animals (H-2d and H-2k) were obtained from K. Buerki (Sandoz, Basel, Switzerland). IL-4/3-83μ doubletg mice were generated by breeding heterozygous IL-4 tg males with heterozygous 3-83μ tg females (H-2k). The offspring were tested using PCR technology. All animals were maintained under conventional conditions in an isolation facility.
Cell Preparations and FACS® Analysis
Spleens and lymph nodes were rubbed through a steel mesh and the cell debris excluded. Bone marrow cells were obtained by washing the cells out of the bones with a gauze needle mounted on a 1-ml syringe. The cells were washed and then microscopically counted in a thoma chamber. B cells from the spleen were obtained by depleting T cells through the addition of monoclonal anti-Thy-1 antibodies and subsequent complement lysis (16). B cells from the bone marrow were isolated by incubating 1 × 108 cells/ml with anti B220 coated magnetic beads (1:10 in BSS/1% BSA) for 15 min at 4°C, and then running the suspensions over a magnetic column and eluting the B220+ cells according to the manufacturer's instructions (Miltenyi Biotech, Bergisch Gladbach, Germany). The reactivity of cell populations with monoclonal antibodies was analyzed by immunofluorescence, using two- and threecolor analysis performed with a FACScan® (Becton Dickinson, Mountain View, CA). The FITC-labeled antibodies used were antiCD8 (Ly-2), CD23 (B3B4), and Thy-1.2 (53-2.1) (all purchased from PharMingen, San Diego, CA). The clone 13/4 reacting with IAkwas kindly provided by G. Hämmerling (Deutsches Krebsforschungs-Zentrum, Heidelberg, Germany). The FITC-labeled antibody reacting with the 3-83μ tg Ig receptor (mAb 54.1 from the rat) was a generous gift from K. Buerki. The purified anti-IL-4 receptor antibody (clone mIL-4R-M2) was obtained from Immunex (Seattle, WA). Phycoerythrin-labeled antibodies used were anti-CD4 (L3T4), CD8 (Ly-2) (both PharMingen), and B220 (RA3-692) (Serva, Heidelberg, Germany). Biotin labeled antibody used was antiαβTCR in conjunction with streptavidin cychrome (both from PharMingen). Specificity of antibody binding was controlled by staining with isotype-matched control antibodies.
Proliferation Assays
Splenic B cells.
2 × 105 T cell–depleted spleen cells/well were incubated in RPMI medium (Gibco BRL, Berlin, Germany) supplemented with 5% fetal calf serum and additives (RPMI+) in the presence of medium alone, anti-μF(ab′)2 antibodies (25 μg/ml; Jackson Immuno Research Labs. Inc., West Grove, PA), or LPS (10 mg/ml, Hoechst, Germany) for 40 h. 11B11 (anti IL-4 mAb, 10 μg/ml, kindly provided by Dr. G. LeGros [Malaghan Institute of Medical Research, Wellington, New Zealand]) was also added into parallel cultures at the onset of the experiments. To determine the amount of proliferation the cells were then pulsed with [3H]thymidine (0.25 mCi/well), 2 Ci (mmol) for 16 h. The incorporated radioactivity was measured on a β-plate (Pharmacia).
Bone Marrow–derived B Cells.
1 × 105bone marrow–derived B cells were incubated with 3 × 104 CD40 ligand transfected (20) or untransfected (both mitomycin-treated) L929 fibroblasts in the presence or absence of 11B11 (anti IL-4 mAb, 10 μg/ml) for 40 h. The proliferation was measured by pulsing the cells for the last 16 h with [3H]thymidine (0.25 mCi/well) and incorporated radioactivity measured as above.
Detection of Autoantibodies in the Serum
Anti-nuclear antigen (ANA) and anti-smooth muscle antigen test (ASMA). Serum from IL-4 tg and littermate control mice were serially diluted in PBS. The diluted serum was incubated with sections containing Hep2 cells (ANA) or rat gut and stomach (ASMA; both from Biolab, Germany) at rt for 1 h. After washing three times with PBS the sections were stained with FITC-conjugated rat anti–mouse Ig (Dianova, Hamburg, Germany). The binding of antibodies was visualized and scored using a fluorescent microscope. A serum was scored autoantibody positive when the titer was greater or equal to 1/30 (ANA, maximum detected titer 1/200) or 1/10 (ASMA, maximum detected titer 1/100).
Detection of Autoreactive Monoclonal Antibodies
Whole spleen from 4 IL-4 tg and 3 littermates (mice were 4-5 wk of age) were fused with P3X63.Ag8.653 myeloma cells with the help of polyethyleneglycol (Sigma Chem. Co., St. Louis, MO). Hybridomas were grown on 96-well flat bottom microtiter plates (Falcon, Oxnard, CA) in selection medium (RPMI+, 1× azaserine-hypoxanthine, 2.5% HECS; Sigma) for 1 wk and the supernatants tested in an ELISA assay for antibodies against endonuclear antigen (ENA-assay). In brief, 96-well microtiter plates were coated with calf thymus extract (Inova Diagnostics, San Diego, CA) in coating buffer (1:100 dilution in 0.05 M carbonate buffer, pH9.6), washed with ELISA buffer (PBS/0.05% Tween 20; Sigma), and then blocked with PBS/1% BSA for 1 h. 50 μl of each supernatant was incubated in doublets on the microtiter plates for 2 h at room temperature. Human serum containing autoantibodies against endonuclear antigens was used as a positive control; RPMI+ medium was used as a negative control. After washing, autoreactive antibody binding was detected with goat anti–mouse Ig or goat anti-human Ig coupled to alkaline phosphatase (Southern Biotech., UK) followed by substrate solution (1 mg/ml paranitrophenylphosphate, 0.02% NaN3, 0.08% MgCl2, 9.7% (vol/vol) diethanolamine; all Sigma). The color reaction was measured at OD405 after 30–50 min with an ELISA reader (Biorad, Munich, Germany). Supernatants were considered positive when the ratio of the absorbency between the supernatant and the negative control was equal to or greater than 2.0.
Treatment of Mice with Anti-CD4 and Anti-IL-4 Antibodies
3–4-d-old mice (6 litters) were treated either for 8 wk with anti-CD4 depleting antibodies (19 mice) or for 7 wk with antiIL-4 neutralizing antibodies (11B11) (16 mice). The mice were injected either with 1 mg of anti-CD4 once weekly or with 1 mg of 11B11 every 3 d, i.p. Less than 2% residual CD4 T cells could be detected after the anti-CD4 treatment. This was shown by triple staining using anti-CD4 PE, anti-CD8 FITC, and anti-αβTCR biotin/Strep-Chychrome. Individual IL-4 tg and wild-type mice were identified using PCR-technology (16), after sacrifice. The anti-CD4 antibodies (clone GK 1.5) and anti-IL-4 neutralizing antibodies (11B11) were generously provided by Dr. G. LeGros.
Hematology
Reticulocytes were identified by incubating fresh whole blood with 1% Brilliant cresyl blue in citrate saline at 37°C. Smears were prepared after 20 min and examined under oil immersion. The percentage of reticulocytes present in the blood was evaluated by counting 300 cells/smear. Hematocrit was measured by the microhematocrit method using heparinized capillary tubes. Bilirubin levels were quantified with a bilmeter (Mochida, Tokyo, Japan) using capillary tubes.
The coombs test was performed using standard technology and goat anti–mouse or rabbit anti–mouse total Ig (both from Dako Corp., Carpinteria, CA) with serial dilutions.
Immunohistology
Paraffin sections (2 μm) of kidney tissue obtained from normal and IL-4 tg mice were digested with bacterial protease XXIV (Sigma) for 10 min at 37°C before incubating the slides with antibodies for 1 h at room temperature. Frozen, acetone-fixed tissue sections were also stained with antibodies for 1 h at room temperature omitting the predigestion step. Antibodies used were: peroxidaseconjugated rabbit anti–mouse Ig (anti-total Ig; Dako Corp., Carpinteria, CA); peroxidase-conjugated sheep anti–mouse IgA (Serotec, Oxford, England), biotinylated rat anti–mouse IgM, and IgG2a (both PharMingen), biotinylated rat anti–mouse IgE (Ciba Geigy, Basel, Switzerland), peroxidase-conjugated sheep anti–mouse IgG1 (Serotec), FITC conjugated goat anti–mouse complement 3 (Nordic, Tilburg, NL). All antibodies were diluted in 1% BSA/Trisbuffer. Endogenous peroxidase activity was inhibited with 0.3% H2O2 in Tris-buffered saline for 10 min. Incubation of biotinylated antibodies was followed by application of peroxidase-conjugated streptavidin (BioGenex, San Ramon, CA) for 30 min. The substrate diaminobenzidine was added until the desired staining intensity occurred. After counterstaining with hematoxylin, the sections were dehydrated and mounted in DPX. For control experiments, biotinylated primary antibodies were omitted and peroxidase-conjugated antibodies were substituted with peroxidase conjugated rabbit anti–swine immunoglobulins (Dako). Blood smears were prepared, air-dried and fixed in acetone for 10 min. The same method as for the frozen sections was used to detect autoantibody binding to RBCs (see above).
Microscopy
Light Microscopy.
Tissues were fixed in 10% phosphate buffered formalin for 24 h, and embedded in paraffin wax. 2–3-μm sections were cut and stained with hematoxylin and eosin.
Transmission Electron Microscopy.
Renal cortex was fixed in half strength Karnovsky fixative for 1 h, transferred to cacodylate buffered osmium tetroxide, embedded in Epon 812, and ultrathin sections were cut. Grids were viewed in a Siemens (Munich, Germany,) 102 transmission electron microscope following staining with lead citrate and uranyl acetate.
Results
Pathological Disorders in IL-4 Tg Mice.
IL-4 tg mice of this particular line have a very high mortality rate (Fig. 1 A) and suffer from severe anemia as seen by the dramatic decrease in hematocrit (Fig. 1 B) and increase in reticulocyte numbers in the blood (Fig. 1 C). Furthermore, IL-4 tg mice showed extramedullary hematopoiesis in spleen and liver with elevated bilirubin levels in the blood (data not shown). These observations suggested that the mice develop an autoantibody-induced hemolytic anemia (AIHA). Staining of blood smears with rabbit anti–mouse Ig revealed that some of the tg mice with the most severe anemia had detectable levels of antibodies bound to their erythrocytes. However, a coombs test using erythrocytes from IL-4 tg mice and goat or rabbit anti–mouse total Ig failed to detect autoantibodies binding to the erythrocytes from the IL-4 tg mice (data not shown).
Figure 1.



Anemia in the IL-4 tg mice. (A) Survival rate of IL-4 tg mice. IL-4 tg (n = 31) and littermate control mice (n = 20) were monitored daily for 12 wk and the death of mice recorded. Shown is the survival rate of the mice in percentage. (B and C) Hematocrit counts and percentage of reticulocyte numbers in the blood of 4- and 12-wk-old IL-4 tg and control mice.
Kidneys of IL-4 tg mice showed progressive glomerular damage with complement and Ig deposition in glomeruli (Fig. 2) and developed progressive glomerular hypertrophy and glomerulosclerosis. The antibodies present in the kidneys of IL-4 tg mice were of the IgM, IgG1, IgG2a, and IgA but not IgE isotypes (data not shown). On electron microscopy the deposits were predominantly localized to subendothelial and mesangial areas (data not shown). Most, but not all, of the glomeruli in the kidneys of IL-4 tg mice showed signs of damage. IL-4 tg mice showing signs of wasting syndrome (n = 6) had an average 10-fold decrease in urine volume (measured over 24-h period) and a 6-fold increase in urinary protein excretion as compared to littermate control mice (Rüger, B., unpublished observation).
Figure 2.

Immune deposits in the kidneys of IL-4 tg mice. The kidneys of an IL-4 tg (A) and control mouse (B) were fixed in formalin and stained with peroxidase conjugated rabbit anti–mouse total Ig. Ig binding was visualized through the addition of diaminobenzidine. Shown is a representative example of immune deposition detected in IL-4 tg mice (n = 20). g, glomerulus.
Autoantibody Production in the IL-4 Tg Mice.
The association of autoimmune disease with elevated autoantibody titers lead us to analyze whether mutant mice had elevated levels of autoantibodies. For this purpose an ANA and ASMA test was used. Serum from IL-4 tg and control mice were incubated with sections of Hep 2 cells (ANA test) or rat gut (ASMA test). Bound autoantibodies were visualized by rat anti–mouse Ig-FITC under a fluorescence microscope. Fig. 3 shows a typical positive (A) and negative (B) ANA or ASMA staining. Antibody titers equal or greater to 1/30 (ANA) or 1/10 (ASMA) were scored positive. Fig. 4 summarizes the results of the ANA tests. The frequency of mice having detectable amounts of autoantibodies against nuclear as well as smooth muscle antigens (data not shown) were threefold higher in IL-4 tg mice than in littermate control mice both in younger and older mice. The average autoantibody titer of IL-4 tg mice scoring positive in both ANA and ASMA test was about two- to threefold higher than in littermate controls. Furthermore, there was no correlation between high total Ig titers and autoantibody production in IL-4 tg mice. In contrast, most of the control mice with the highest autoantibody titers also had the highest total Ig serum levels (data not shown).
Figure 3.

ANA and ASMA test. The sections containing Hep 2 cells (ANA) and rat gut (ASMA), were incubated with different concentrations of mouse serum (shown is the result using a 1/30 dilution). The binding of antibodies to the autoantigens was then detected by incubating the sections with rat anti–mouse Ig conjugated with FITC and analyzed under a fluorescence microscope. Shown is a typical positive ANA and ASMA staining using serum from a tg mouse (A), and a negative staining (B), using serum from a wt mouse.
Figure 4.

Percentage of IL-4 tg and littermate control mice with autoantibodies in their serum binding to nuclear antigens. The serum of the different mice were analyzed for the presence of autoantibodies against nuclear antigens in an ANA test. Shown are the results using a 1/30 serum dilution.
To assess whether autoantibody production in IL-4 tg mice was due to the presence of more autoantibody producing B cells, B cell hybridomas from pooled spleens of four IL-4 tg and three control mice (aged between 4–5 wk) were established and analyzed for autoantibody production using an anti-ENA test. Out of 1,064 tested IL-4 tg-hybridomas 9.1% scored positive for autoantibody production versus only 1.3% from 586 analyzed control-hybridomas. This was a sevenfold increase in the frequency of hybridomas producing autoantibodies against endonuclear antigens in the IL-4 tg mice as compared to littermate control mice.
Numbers of Ly1+ B cells which are often associated with the production of autoantibodies (21) were not elevated in the lymph nodes and peritoneal cavity of the IL-4 tg mice as compared to control mice. This suggests that Ly1+ B cells were not the major source of autoantibodies in the mutant mice (data not shown).
Constitutive Expression of IL-4 In Vivo Leads to Polyclonal B Cell Activation.
Autoantibody production is often associated with polyclonal B cell activation (22, 23). B cells from the spleen, lymph node and bone marrow of IL-4 tg mice express high levels of MHC class II and increased levels of CD23, indicating polyclonal in vivo activation (Fig. 5 A). To investigate whether the observed phenotype also correlates with a greater responsiveness to B cell activators, we stimulated B cells from lymph nodes with LPS and antiμF(ab′)2 antibodies. Fig. 5 B shows that B cells from IL-4 tg mice proliferated more strongly in response to LPS and anti-μF(ab′)2stimulationthan did B cells from littermate controls. Furthermore, splenic B cells from tg mice still showed proliferation to anti-μF(ab′)2at concentrations where normal B cells were no longer reactive (16). To assess whether IL-4 also had a costimulatory effect on B cells activated through the interaction of CD40 with CD40 ligand, we isolated B cells from the bone marrow and activated them with CD40 ligand–transfected L929 fibroblasts (20). Again, B cells from IL-4 tg mice responded more vigorously than did B cells from controls (Fig. 5 C). The addition of anti-IL-4 neutralizing antibodies to the B cell cultures (Fig. 5, B and C) from the IL-4 tg mice had no negative effect on the proliferation of the B cells. This shows that the B cells from the IL-4 tg mice have been activated in vivo and not by tg-derived IL-4 in vitro. Taken together these results, and previously published data (16), support the costimulatory effect of tg IL-4 expression on B cells in vivo leading to a constitutive activation of B cells in the IL-4 tg mice. Interestingly, this polyclonal B cell activation did not lead to a strong increase in total Ig levels in the serum of tg mice, where the average amount of total Ig was only about two- to threefold increased (data not shown). However, IgG1 and especially IgE levels were elevated (16).
Figure 5.
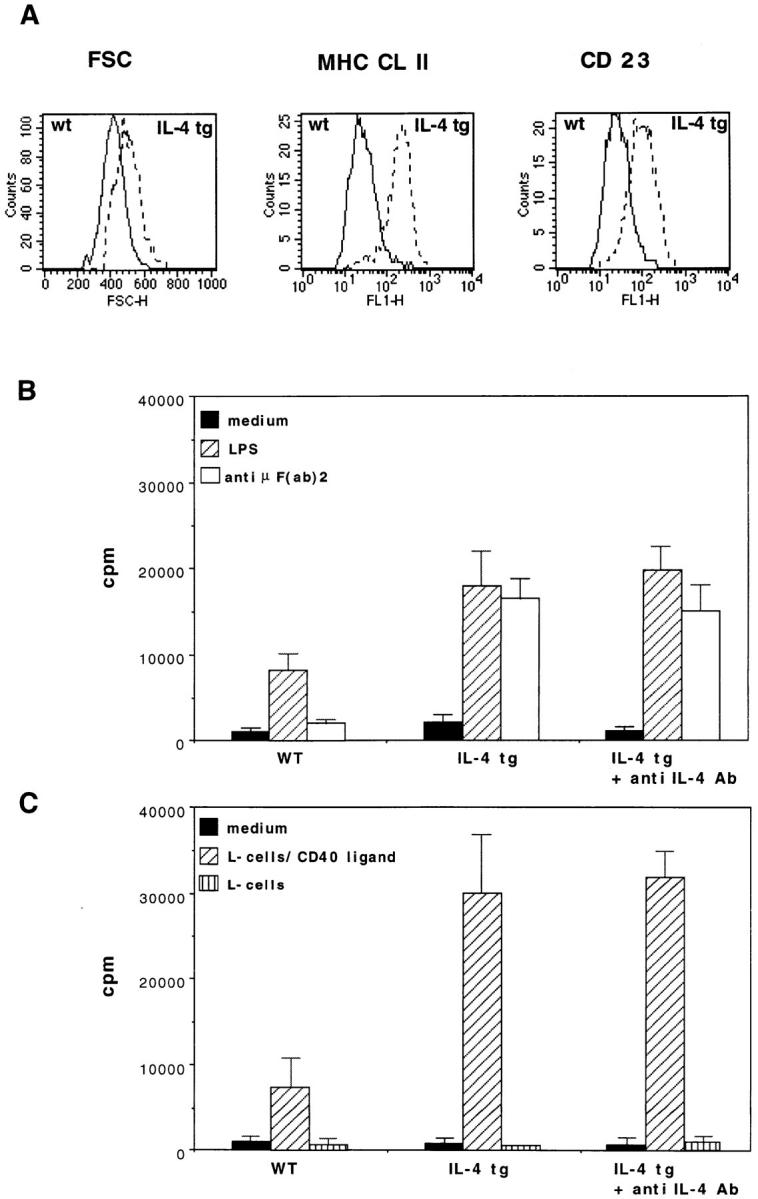
Hyperreactivity of B cells from the IL-4 tg mice. (A) Upregulation of surface MHC Cl II and CD23 expression and enlargement of tg B cells. Spleen cells were isolated and then stained with antibodies against B220, MHC CL II, and CD23. Experiments were repeated six times with similar results. (B) [3H]Thymidine uptake from B cells from the spleen of IL-4 tg and littermate control mice stimulated with LPS and anti-μ F(ab′)2 antibodies. 2 × 105B cells per well were incubated with medium, LPS (10 μg/ml) and anti-μ F(ab′)2 antibodies (10 μg/ml) in the absence or presence of 10 μg/ml anti-IL-4–neutralizing antibody (11B11) for 40 h, pulsed for the last 16 h of the culture period, and then harvested. Mean [3H]thymidine uptake of triplicates and standard deviations are shown representative of three separate experiments. (C) B cells from the bone marrow of IL-4 tg and control mice stimulated with CD40 ligand transfected L cells. 1 × 105B cells from the bone marrow of IL-4 tg and control mice were incubated with 3 × 104 CD40 ligand–transfected and –untransfected L929 fibroblasts (mitomycin D treated) in the absence or presence of 10 μg/ml anti-IL-4 neutralizing antibody (11B11). The proliferation assay was performed as described above.
Negative Selection of Superantigen-reactive T Cells and Selfreactive B Cells Is Normal in IL-4 Tg Mice.
Previously published data showed that the IL-4 tg mice used in this study have a perturbed T cell development in the thymus and the periphery (16, 17). To assess if these abnormalities lead to the generation of more autoreactive T cells, we analyzed whether the mutant mice deleted self-reactive T cells as efficiently as control mice. For this purpose the negative selection of T cells in thymus and lymph nodes of IL-4 tg mice was analyzed. The IL-4 tg mice used in this study were backbred for eight generations onto the C3H genetic background and therefore, expressed MHC class II molecules of the IE haplotype, and carried the murine mammary tumor viruses (MMTV) which, in association with IE (24, 25), are responsible for deleting Vβ3, 5 and 11+ T cells. Table 1 shows that the IL-4 tg mice delete T cells expressing the TCR elements Vβ3, 5 and 11 in the thymus and in the lymph nodes to a similar extent as did littermate controls. As expected, the control Vβ6 element was not affected by the deletion.
Table 1.
Negative Selection of CD3+ T Cells Expressing Vβ3, 5, and 11 in the Thymus and Lymph Nodes of Control and IL-4 tg Mice
| TCR Vβ elements | Thymus | Lymph nodes | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WT C57B1/6 (IA) | WT C3H (IE) | IL-4 tg C3H (IE) | WT C57B1/6 (IA) | WT C3H (IE) | IL-4 tg C3H (IE) | |||||||
| Vβ3 | 6.9 ± 1.4 | 1.8 ± 0.6 | 0.7 ± 0.3 | 1.9 ± 0.7 | 0.3 ± 0.1 | 0.5 ± 0.3 | ||||||
| Vβ5 | 7.6 ± 0.7 | 1.2 ± 0.7 | 1.7 ± 0.7 | 4.9 ± 1.1 | 1.5 ± 0.4 | 0.9 ± 0.4 | ||||||
| Vβ6 | 6.7 ± 1.8 | 7.9 ± 2.4 | 10.5 ± 3.1 | 7.1 ± 1.5 | 10.1 ± 2.5 | 12.3 ± 3.4 | ||||||
| Vβ11 | 5.2 ± 1.1 | 1.9 ± 0.4 | 1.1 ± 0.4 | 4.5 ± 1.0 | 2.1 ± 0.4 | 1.4 ± 0.3 | ||||||
Negative selection of CD3+ T cells expressing the TCR elements Vβ3, 5, and 11 in the thymus and lymph nodes of control and IL-4 transgenic mice. Thymocytes and lymph node cells from five IL-4 transgenic and five control mice (littermate and C57B1/6 mice), between the ages of 5-6 wk, were stained with monoclonal antibodies against CD3, Vβ3, 5, 6, and 11. Shown are the mean percentages and standard deviation of CD3+ cells in the thymus and lymph nodes expressing the different TCR-Vβ elements.
To address the question whether the deletion of self- reactive B cells is normal in the IL-4 tg mice we crossed the IL-4 tg mice (H-2k) with tg mice expressing genes for the variable region of an anti-H-2k antibody (3-83μ tg mice) (30). F1 offspring were typed by using PCR technology. The B cells in the bone marrow, lymph nodes, and spleen of the 3-83μ tg and 3-83μ × IL-4 double tg mice were double stained with anti-B220 and anti-3-83μ idiotype antibody (clone 54.1). FACS® analysis of the different lymphoid organs revealed that <1.0% of B220+ cells expressed the 3-83μidiotype in both the 3-83μ tg and 3-83μ × IL-4 double tg mice (Table. 2). Using the same staining protocol B220+/3-83μ+ double positive cells were readily detected in the bone marrow, lymph nodes and spleens of 3-83μ tg mice of a non deleting background (30 and Table 2). These results show that IL-4 tg mice delete self-reactive B cells to a similar extent as non tg mice.
Table 2.
IL-4 Transgenic Mice Delete Self-reactive B Cells Reacting to MHC Class I Molecules (H-2 k) as Efficiently as Control Animals
| Lymph nodes | Spleen | Bone marrow | ||||
|---|---|---|---|---|---|---|
| 3-83μtg | ||||||
| (H-2d) | 12.2 | 32.8 | 20.5 | |||
| 3-83μtg | ||||||
| (H-2k) | <1 | <1 | <1 | |||
| 3-83μ × IL-4tg | ||||||
| (H-2k) | <1 | <1 | <1 |
The cells in the bone marrow, lymph nodes and spleen of the 3-83μ transgenic (either of the H-2k or H-2d haplotype) and 3-83μ × IL-4 double transgenic mice (H-2k) were double stained with anti-B220 and anti-3-83μ idiotype antibodies (clone mAb 54.1). Shown are the percentages of B220+ cells expressing the anti H-2k idiotype in the lymphoid organs of the different groups of mice analyzed, representative for two separate experiments.
Treatment of IL-4 Tg Mice with Depleting Anti- CD4 and Neutralizing Anti-IL-4 (11B11) Antibodies and Analysis of Crosses between IL-4 Tg Mice and Tg Mice Expressing a Soluble CTLA-4 Human Ig Fusion Protein.
Even though negative selection of autoreactive T cells seems to have been normal in the IL-4 tg mice, it is possible that activated autoreactive B cells could interact with a small number of residual autoreactive T cells. These T cells could then further enhance the production of autoantibodies by providing help to autoreactive B cells, thus causing the autoimmune disorders. To address this question, we treated 11 tg and 8 control mice with anti-CD4 for 8 wk with a weekly dose of 1 mg of antibodies injected i.p. (starting at 3–4 d of age). Less than 2% residual CD4 T cells could be detected after the antibody treatment (data not shown). Fig. 6 A shows that 75% of the CD4 depleted IL-4 tg mice survived versus only 45% in the untreated age matched control mutant mice. However, the increased survival was only reflected by a marginal increase in hematocrit (0.33 ± 0.6 in treated mice versus 0.25 ± 0.7 in untreated mice). The treated control mice had similar hematocrit counts to untreated controls showing that the injection of anti-CD4 antibodies had no direct effect on hematocrit counts (data not shown). Interestingly, there was no difference between IL-4 tg mice treated with the anti-CD4 antibodies and non treated IL-4 tg mice in respect to extramedullary hematopoiesis in spleen and liver respectively. The IL-4 tg mice treated with antiCD4 antibodies also developed severe glomerulonephritis (Table 3 and data not shown).
Figure 6.
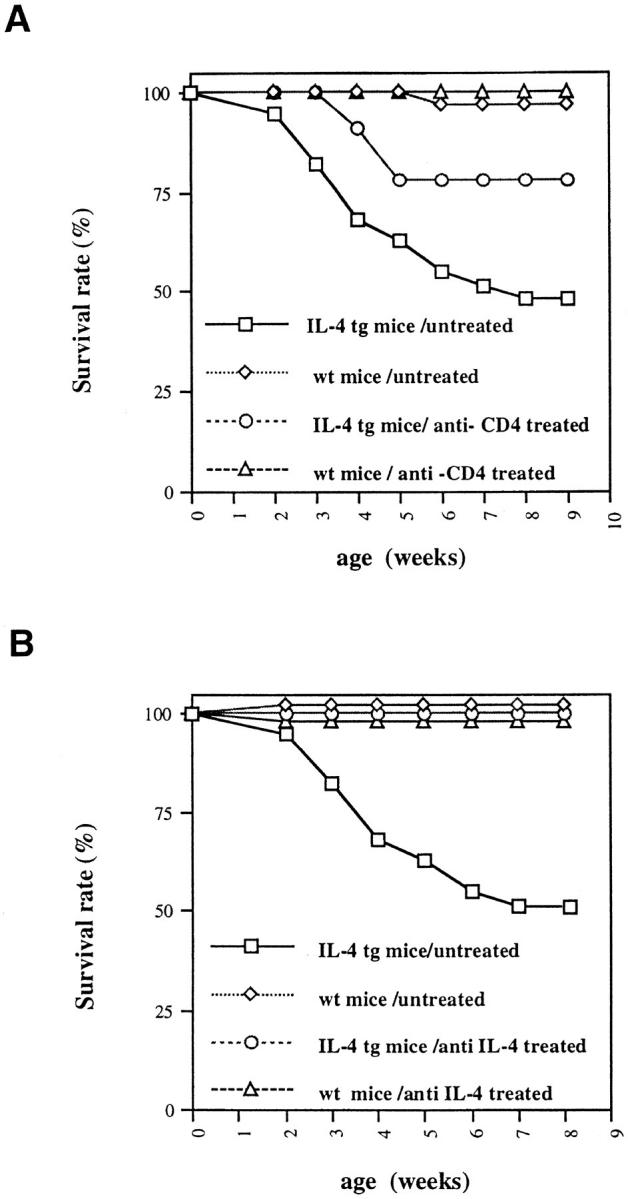
Survival rate of IL-4 tg mice after in vivo anti-CD4 (A) and anti-IL-4 neutralizing antibody (B) treatment. At 1 wk of age IL-4 tg and littermate control mice were injected with anti-CD4 antibodies (tg: n = 11; wt: n = 8) for 8 wk and anti-IL-4 neutralizing antibodies (11B11) for 7 wk (tg: n = 7; wt: n = 9). During this time the numbers of dead mice were recorded. Shown are the survival rates of treated versus untreated control and IL-4 tg mice.
Table 3.
Severity of Autoimmune Type Disorders Detected in Control and IL-4 tg Mice, Crossed with CTLA-4 tg Mice or Treated with Anti-IL-4 or Anti-CD4 Ab
| Anemia | Glomerulonephritis | Ig deposits in the kidneys | ||||
|---|---|---|---|---|---|---|
| Control mice | ||||||
| untreated | − | − | − | |||
| CTLA-4 tg mice | ||||||
| untreated | − | − | − | |||
| IL-4 tg mice | ||||||
| untreated | +++ | +++ | +++ | |||
| Control mice | ||||||
| anti-CD4 | ||||||
| treated | − | − | − | |||
| Control mice | ||||||
| anti-IL-4 | ||||||
| treated | − | − | − | |||
| Control mice × | ||||||
| CTLA-4 tg | ||||||
| crosses | − | − | − | |||
| IL-4 tg mice | ||||||
| anti-CD4 | ||||||
| treated | ++ | +++ | +++ | |||
| IL-4 tg | ||||||
| anti-IL-4 | ||||||
| treated | − | − | +/− | |||
| IL-4 tg × | ||||||
| CTLA-4 tg | ||||||
| crosses | +++ | +++ | +++ |
Summary of the autoimmune-type disorders detected in the IL-4 tg mice crossed with CTLA-4 tg mice or treated with anti-IL-4 or antiCD4 antibodies. The development of anemia, glomerulonephritis and Ig-depositions in the kidneys were determined for each individual mouse per group (for details of treatment and detection see Materials and Methods and Results). The severity of the disorder/group was scored as follows: undetectable (−), detectable (+), mild (++), severe (+++).
To further investigate the role of T cells in the development of the autoimmune-like disease in IL-4 tg mice, we crossed IL-4 tg mice with tg mice secreting a CTLA-4 human Ig fusion protein (CTLA-4 tg mice). The soluble CTLA-4 human Ig fusion protein binds to the B71 and B72 accessory molecules thus inhibiting T cell costimulation via CD28 (18). This leads to the inability of T cells to sufficiently activate B cells to produce antibodies due to the lack of in vivo cytokine secretion by T cells (19). Comparison of the F1 offspring from IL-4 tg × CTLA-4 tg crossings (both heterozygous for the transgene) revealed that 5–6-wk-old double tg mice were suffering from anemia as seen by the reduction of hematocrit counts (0.35 ± 0.02 in double IL-4/CTLA-4 tg mice [n = 4] versus 0.34 ± 0.07 [n = 6] in IL-4 tg mice and 0.48 ± 0.04 in single CTLA-4 tg mice [n = 5]). The double-tg mice also developed glomerulonephritis with Ig deposits in the glomeruli (data not shown).
To ascertain that the observed disorders were indeed due to the expression of IL-4, we treated seven IL-4 tg and nine littermate control mice with neutralizing anti-IL-4 antibodies. After 7 wk of antibody treatment, all of the mice were healthy and none had died (Fig. 6 B). Furthermore, anti-IL-4–treated tg mice did not develop an anemia as seen by the normal hematocrit levels (0.45 ± 0.03 versus 0.48 ± 0.04 in treated control mice) and the lack of extramedullary blood formation in the spleen and liver (data not shown). Furthermore, kidneys of anti-IL-4 treated tg mice showed no pathological changes by light microscopy with only minor Ig deposits in the glomeruli (data not shown). Table 3 summarizes the results obtained in IL-4 tg mice using the different experimental approaches.
Discussion
A striking feature of the IL-4 tg mice used in this study is that they have a high mortality rate and develop anemia and glomerulonephritis. The glomerulonephritis is characterized by proteinuria, and complement and Ig deposits in the glomeruli. The antibodies present in the glomeruli could either be directly binding to renal antigens or be part of immune complex depositions. Whether these Ig depositions in the glomeruli are responsible for nephritis remains to be determined, although together with the complement binding, this seems to be the most likely explanation. The kidney disease in the IL-4 tg animals was accompanied by severe anemia. This was apparent by the very low hematocrit counts, high reticulocyte numbers in the blood, and extramedullary hematopoiesis in liver and spleen. Mice with high reticulocyte and low hematocrit counts had elevated bilirubin levels in the serum, and in some cases, weak autoantibody binding to the erythrocytes could be detected (data not shown). This data suggest that the mice were suffering from an AIHA. However, a coombs test using erythrocytes from the IL-4 tg mice failed to detect any autoantibody binding. This suggests that anti-erythrocyte autoantibodies were possibly not the cause for the hemolytic anemia observed in the IL-4 tg mice. In humans, progressive renal failure can lead to deficiency of erythropoietin production, which may also cause anemia (26). Therefore it is possible that the glomerulonephritis in the IL-4 tg mice was contributing to the anemia. However, it is very unlikely that renal failure was responsible for the initial development of anemia in these mice, since both extramedullary blood formation and increased production of reticulocytes is dependent upon the action of erythropoietin. Taken together the anemia detected in the IL-4 tg mice has many of the features typical for an AIHA. However, we were not able to readily detect autoantibodies binding to the erythrocytes suggesting that the hemolytic anemia might be due to other mechanisms. The treatment of the IL-4 tg mice with anti-IL-4 antibodies completely abolished the development of the observed anemia strongly suggesting that the anemia was due to either a direct or indirect immunological mechanism mediated by IL-4.
It is not clear what is causing the high mortality of the IL-4 tg mice. Severe anemia and glomerulonephritis in IL-4 tg mice correlated with the onset of the wasting syndrome, with no detectable signs of bacterial or viral infections (by histology and blood cultures). Taken together, these results strongly indicate that IL-4 tg mice die as a direct result of an autoimmune-type disorder, although it is not possible to completely rule out other causes of death.
The findings that IL-4 tg mice had higher autoantibody levels and more detectable autoantibody producing B cells in the spleen, together with the deposition of antibodies in the kidney suggests that the increase in autoantibody levels could be causing autoimmune disease leading to the observed glomerulonephritis and anemia. In the murine animal models for SLE, anemia and glomerulonephritis are usually mutually exclusive (1, 27). This is not true for human disease where individuals can suffer from both anemia and glomerulonephritis (28). Because the IL-4 tg mice develop both anemia and a glomerulonephritis it is possible that these mice might be a very useful novel animal model for human SLE disease.
What is the reason for this increase in self-reactive B cells? One possibility is that the tg expression of IL-4 leads to the generation of more self-reactive Th cells, which then in turn activate and expand the small number of normally present self-reactive B cells. The negative selection of superantigen reactive T cells in the thymus and the periphery was normal in the IL-4 tg mice. Therefore it is unlikely that an increase in autoreactive T cells was responsible for the generation of more self-reactive B cells.
Another possibility for the increase in autoantibody levels is that the constitutive expression of IL-4 leads to polyclonal B cell activation. Polyclonal B cell activation has been shown to lead to the production of autoantibodies and autoimmune disease (22, 23). Our experiments clearly show that the B cells in the IL-4 tg mice are hyperreactive, as seen by the high level of MHC class II expression and the greater reactivity to in vitro stimulation via antiμF(ab′)2antibodies orCD40 ligand transfected L cells. The addition of 10 μg/ml 11B11 (anti-IL-4 neutralizing antibodies) had no effect on the in vitro proliferation of the B cells from the IL-4 tg mice (Fig. 5) or B cells from the control animals (data not shown). This shows that the enhanced in vitro reactivity to B cell stimulators was due to prior in vivo activation and not in vitro activation by tgderived IL-4. Furthermore, IgM and especially IgG1 and IgE serum levels were greatly enhanced (16). Therefore, polyclonal B cell activation could be a simple explanation for the increase in autoantibody levels.
A further possibility which could explain how IL-4 leads to increased development of autoreactive B cells in IL-4 tg mice is that IL-4 selectively rescues autoreactive B cells from cell death. This effect was described in vitro. It was reported that cross-linking sIgM on immature bone marrow–derived B cells resulted in apoptotic cell death, and that the sIgM-induced apoptosis could be blocked by the addition of IL-4 (12). IL-4 also rescued mature B cells, stimulated via plastic-immobilized anti-μ or anti-δ antibodies from apoptosis, when it was added to the cultures in conjunction with anti-CD40 antibodies (13). Furthermore, immature B cells stimulated with anti-μ, anti-IgD, and CD40 ligand–transfected fibroblasts, only proliferated when IL-4 was added to the culture (10). These in vitro assays mimic the in vivo activation of potentially autoreactive B cells through noncognate pathways in the presence of IL-4. The expression of tg IL-4 in vivo could, as shown in vitro, selectively rescue autoreactive B cells from apoptosis thus causing the increase in autoantibody levels and the autoimmune-type disorders. To address this issue we crossed IL-4 tg mice with tg mice expressing genes for the variable region of an anti-H-2k antibody (3-83μ tg mice [30]). The resulting F1 single 3-83μ tg mice and double tg mice, expressing both IL-4 and the 3-83μ transgene, express H-2k MHC class I molecules and should, therefore, delete the anti-H-2k receptor bearing B cells. Our results showed that virtually no anti-H-2k Ig receptor bearing B cells could be detected in the bone marrow, lymph nodes, and spleen of both the single 3-83μ tg mice and 3-83μ × IL-4 tg mice. These results show that negative selection of self-reactive B cells is normal in the IL-4 tg mice and using superantigen induced T cell depletion as an indicator for normal negative selection of autoreactive T cells suggest that polyclonal B cell activation is the most likely explanation for the increase in autoantibody levels in the IL-4 tg mice.
To evaluate the role of CD4+ T cells in the development of the pathological disorders detected in IL-4 tg mice, we treated the mice with deleting anti-CD4 antibodies for 8 wk. The increased survival (75% versus 45% in untreated mice), suggests that CD4+T cells were contributing to disease exacerbation. However, anti-CD4–treated IL-4 tg mice still developed severe anemia and glomerulonephritis suggesting that CD4+T cellswere not responsible for disease development. To rule out the possibility that low numbers of residual CD4+ T cells (2%) present in the anti-CD4 treated mice had an effect on the development of autoimmune-like disorders, we crossed IL-4 tg with tg mice expressing a soluble CTLA-4 human Ig fusion protein (CTLA-4 tg mice [18]). The soluble CTLA-4 binds to the B71 and B72 accessory molecules thus inhibiting T cell costimulation via CD28, leading to the inability of T cells to give help to B cells (19). The double-tg mice expressing both IL-4 and soluble CTLA-4 had a similar death rate, hematocrit counts and kidney damage, as did the mice only expressing IL-4 (Table 3 and data not shown). Together with the data from the anti-CD4 treatment this argues that CD4+ T cell interaction with B cells was not necessary for the generation of the disorders detected in the IL-4 tg mice.
The treatment of the IL-4 tg mice with neutralizing anti-IL-4 antibodies (11B11) clearly showed that development of the pathological disorders were due to expression of IL-4. Surprisingly, it has not been previously reported that other IL-4 tg mice models, where B cells and/or T cells expressed tg IL-4 (31–33), develop autoimmune-like disease. The reason for this discrepancy between our model and the other tg models is not clear, but probably reflects the different expression patterns of IL-4 in the different mouse lines. However, although there is no evidence suggesting that other IL-4 tg mice models develop autoimmune-like disease, two recent reports demonstrated that the development of autoimmunity was associated with the production of IL-4. In a murine model of graft versus host disease, anti-IL4 antibody treatment prevented development of disease (34). Furthermore, autoimmune disorders developing after neonatal thymectomy have also been reported to be associated with IL-4 production and a humoral immune response (35). These two reports demonstrate involvement of in vivo IL-4 production and development of autoimmune disease, and support our findings that the inappropriate expression of IL-4 can under some circumstances lead to autoimmune-like disorders. However, in these two models disease development was dependent on CD4+ T cells. This does not seem to be the case in our model. We therefore suggest that the presence of IL-4 alone (in sufficient amounts and at the appropriate place) might be able to substitute for self-reactive CD4+ T cells secreting IL-4, leading to increased levels of autoantibodies and the development of autoimmune disease.
In conclusion, our results demonstrate that the constitutive in vivo expression of IL-4 leads to the activation and/ or expansion of autoreactive B cells and development of autoimmune-type disease. From our data we cannot conclude whether autoantibodies were indeed causing the detected anemia and glomerulonephritis; however, it seems the most likely explanation. It has been proposed that recombinant IL-4 could be used for treatment of Th1 mediated inflammatory autoimmune diseases in humans (36). Immune deviation mediated by IL-4 might prove very useful for treatment of some of these disorders, but in view of our results, the possible effects of IL-4 treatment on humans should be further scrutinized.
Footnotes
The authors wish to thank Dr. G. LeGros, Dr. F. Ronchese, and Dr. Paige Lacy (Malaghan Institute of Medical Research, Wellington, New Zealand) for providing reagents and their critical discussions.
This work was supported by the Deutsche Forschungsgemeinschaft (SFB 165), the Bundesministerium für Forschung und Technologie (AIDS-Scholarship), Germany, and Fonds der Chemischen Industrie.
1 Abbreviations used in this paper: AIHA, autoantibody-induced hemolytic anemia; ANA, anti-nuclear antigen; ASMA, anti-smooth muscle antigen; ENA, endonuclear antigen; MMTV, murine mammary tumor viruses; SLE, systemic lupus erythematosus; tg, transgenic.
References
- 1.Singer GG, Carrera AC, Marshak RA, Martinez C, Abbas AK. Apoptosis, Fas and systemic autoimmunity: the MRL-lpr/lpr model. Curr Opin Immunol. 1994;6:913–920. doi: 10.1016/0952-7915(94)90013-2. [DOI] [PubMed] [Google Scholar]
- 2.Theofilopoulos AN. The basis of autoimmunity: Part II. Genetic predisposition. Immunol Today. 1995;16:150–159. doi: 10.1016/0167-5699(95)80133-2. [DOI] [PubMed] [Google Scholar]
- 3.Theofilopoulos AN. The basis of autoimmunity: Part I. Mechanisms of aberrant self-recognition. Immunol Today. 1995;16:90–98. doi: 10.1016/0167-5699(95)80095-6. [DOI] [PubMed] [Google Scholar]
- 4.Arnold B, Schonrich G, Hammerling GJ. Multiple levels of peripheral tolerance. Immunol Today. 1993;14:12–14. doi: 10.1016/0167-5699(93)90317-E. [DOI] [PubMed] [Google Scholar]
- 5.Avrameas S. Natural autoantibodies: from ‘horror autotoxicus' to ‘gnothi seauton'. Immunol Today. 1991;12:154–159. doi: 10.1016/S0167-5699(05)80045-3. [DOI] [PubMed] [Google Scholar]
- 6.Hämmerling GJ, Schonrich G, Ferber I, Arnold B. Peripheral tolerance as a multi-step mechanism. Immunol Rev. 1993;133:93–104. doi: 10.1111/j.1600-065x.1993.tb01511.x. [DOI] [PubMed] [Google Scholar]
- 7.Pircher H, Buerki K, Lang R, Hengartner H, Zinkernagel RM. Tolerance induction in double specific T-cell receptor transgenic mice varies with antigen. Nature (Lond) 1989;342:559–561. doi: 10.1038/342559a0. [DOI] [PubMed] [Google Scholar]
- 8.Strasser A, Harris AW, Cory S. bcl-2 transgene inhibits T cell death and perturbs thymic self-censorship. Cell. 1991;67:889–899. doi: 10.1016/0092-8674(91)90362-3. [DOI] [PubMed] [Google Scholar]
- 9.Strasser A, Whittingham S, Vaux DL, Bath ML, Adams JM, Cory S, Harris AW. Enforced bcl-2 expression in B-lymphoid cells prolongs antibody responses and elicits autoimmune disease. Proc Natl Acad Sci USA. 1991;88:8661–8665. doi: 10.1073/pnas.88.19.8661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brines RD, Klaus GG. Polyclonal activation of immature B cells by preactivated T cells: the role of IL-4 and CD40 ligand. Int Immunol. 1993;5:1445–1450. doi: 10.1093/intimm/5.11.1445. [DOI] [PubMed] [Google Scholar]
- 11.Illera VA, Perandones CE, Stunz LL, Mower DJ, Ashman RF. Apoptosis in splenic B lymphocytes. Regulation by protein kinase C and IL-4. J Immunol. 1993;151:2965–2973. [PubMed] [Google Scholar]
- 12.Norvell A, Mandik L, Monroe JG. Engagement of the antigen-receptor on immature murine B lymphocytes results in death by apoptosis. J Immunol. 1995;154:4404–4413. [PubMed] [Google Scholar]
- 13.Parry SL, Holman MJ, Hasbold J, Klaus GG. Plastic-immobilized anti-mu or anti-delta antibodies induce apoptosis in mature murine B lymphocytes. Eur J Immunol. 1994;24:974–979. doi: 10.1002/eji.1830240429. [DOI] [PubMed] [Google Scholar]
- 14.Parry SL, Hasbold J, Holman M, Klaus GG. Hypercross-linking surface IgM or IgD receptors on mature B cells induces apoptosis that is reversed by costimulation with IL-4 and anti-CD40. J Immunol. 1994;152:2821–2829. [PubMed] [Google Scholar]
- 15.Ohashi PS, Pircher H, Buerki K, Zinkernagel RM, Hengartner H. Distinct sequence of negative or positive selection implied by thymocyte T-cell receptor densities. Nature (Lond) 1990;346:861–863. doi: 10.1038/346861a0. [DOI] [PubMed] [Google Scholar]
- 16.Erb KJ, Holtschke T, Muth K, Horak I, Schimpl A. T cell subset distribution and B cell hyperreactivity in mice expressing interleukin-4 under the control of major histocompatibility complex class I regulatory sequences. Eur J Immunol. 1994;24:1143–1147. doi: 10.1002/eji.1830240520. [DOI] [PubMed] [Google Scholar]
- 17.Erb KJ, Hanke T, Schimpl A, Hunig T, Stingl G, Elbe A. Impaired survival of T cell receptor V gamma 3+ cells in interleukin-4 transgenic mice. Eur J Immunol. 1995;25:1442–1445. doi: 10.1002/eji.1830250546. [DOI] [PubMed] [Google Scholar]
- 18.Lane P, Gerhard W, Hubele S, Lanzavecchia A, McConnell F. Expression and functional properties of mouse B7/BB1 using a fusion protein between mouse CTLA4 and human gamma 1. Immunology. 1993;80:56–61. [PMC free article] [PubMed] [Google Scholar]
- 19.Ronchese F, Hausmann B, Hubele S, Lane P. Mice transgenic for a soluble form of murine CTLA-4 show enhanced expansion of antigen-specific CD4+ T cells and defective antibody production in vivo. J Exp Med. 1994;179:809–817. doi: 10.1084/jem.179.3.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wohlleben G, Gray D, Schimpl A. In vitro immunisation of naive mouse B cells: establishment of IgM secreting hybridomas specific for soluble protein or hapten from B cells cultured on CD40 ligand transfected mouse fibroblasts. Int Immunol. 1996;8:343–349. doi: 10.1093/intimm/8.3.343. [DOI] [PubMed] [Google Scholar]
- 21.Hardy RR, Hayakawa K. Development and physiology of Ly-1 B and its human homolog, Leu-1 B. Immunol Rev. 1986;93:53–79. doi: 10.1111/j.1600-065x.1986.tb01502.x. [DOI] [PubMed] [Google Scholar]
- 22.Klinman DM, Steinberg AD. Systemic autoimmune disease arises from polyclonal B cell activation. J Exp Med. 1987;165:1755–1760. doi: 10.1084/jem.165.6.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Steinberg AD, Krieg AM, Gourley MF, Klinman DM. Theoretical and experimental approaches to generalized autoimmunity. Immunol Rev. 1990;118:129–163. doi: 10.1111/j.1600-065x.1990.tb00815.x. [DOI] [PubMed] [Google Scholar]
- 24.Acha OH, MacDonald HR. Superantigens of mouse mammary tumor virus. Annu Rev Immunol. 1995;13:459–486. doi: 10.1146/annurev.iy.13.040195.002331. [DOI] [PubMed] [Google Scholar]
- 25.Hodes RJ, Abe R. T cell recognition of Mlslike superantigens: analysis of TCR requirements, superantigenic ligands, and signal transduction. Semin Immunol. 1992;4:319–327. [PubMed] [Google Scholar]
- 26.Eschbach, J.W., and J.W. Adamson. 1991. The pathophysiology and treatment of the anemia of chronic renal failure. In Current Nephrology. H.C. Gonick, editor. Moshy Year Book Press, NY. 259–291.
- 27.Eastcott JW, Schwartz RS, Datta SK. Genetic analysis of the inheritance of B cell hyperactivity in relation to the development of autoantibodies and glomerulonephritis in NZB × SWR crosses. J Immunol. 1983;131:2232–2239. [PubMed] [Google Scholar]
- 28.Boumpas DT, Austin H, III, Fessler BJ, Balow JE, Klippel JH, Lockshin MD. Systemic lupus erythematosus: emerging concepts. Part 1: renal, neuropsychiatric, cardiovascular, pulmonary, and hematologic disease. Ann Int Med. 1995;122:940–950. doi: 10.7326/0003-4819-122-12-199506150-00009. [DOI] [PubMed] [Google Scholar]
- 29.Nemazee D, Buerki K. Clonal deletion of autoreactive B lymphocytes in bone marrow chimeras. Proc Natl Acad Sci USA. 1989;86:8039–8043. doi: 10.1073/pnas.86.20.8039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nemazee DA, Buerki K. Clonal deletion of B lymphocytes in a transgenic mouse bearing anti-MHC class I antibody genes. Nature (Lond) 1989;337:562–566. doi: 10.1038/337562a0. [DOI] [PubMed] [Google Scholar]
- 31.Muller W, Kuhn R, Rajewsky K. Major histocompatibility complex class II hyperexpression on B cells in interleukin 4-transgenic mice does not lead to B cell proliferation and hypergammaglobulinemia. Eur J Immunol. 1991;21:921–925. doi: 10.1002/eji.1830210410. [DOI] [PubMed] [Google Scholar]
- 32.Tepper RI, Levinson DA, Stanger BZ, Campos TJ, Abbas AK, Leder P. IL-4 induces allergic-like inflammatory disease and alters T cell development in transgenic mice. Cell. 1990;62:457–467. doi: 10.1016/0092-8674(90)90011-3. [DOI] [PubMed] [Google Scholar]
- 33.Lewis DB, Yu CC, Forbush KA, Carpenter J, Sato TA, Grossman A, Liggitt DH, Perlmutter RM. Interleukin 4 expressed in situ selectively alters thymocyte development. J Exp Med. 1991;173:89–100. doi: 10.1084/jem.173.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ushiyama C, Hirano T, Miyajima H, Okumura K, Ovary Z, Hashimoto H. Anti-IL-4 antibody prevents graft-versus-host disease in mice after bone marrow transplantation - the IgE allotype is an important marker of graft-versus-host disease. J Immunol. 1995;154:2687–2696. [PubMed] [Google Scholar]
- 35.Bonomo A, Kehn PJ, Payer E, Rizzo L, Cheever AW, Shevach EM. Pathogenesis of post-thymectomy autoimmunity—role of syngeneic mlr-reactive T cells. J Immunol. 1995;154:6602–6611. [PubMed] [Google Scholar]
- 36.Rocken M, Racke M, Shevach EM. IL-4- induced immune deviation as antigen-specific therapy for inflammatory autoimmune disease. Immunol Today. 1996;17:225–231. doi: 10.1016/0167-5699(96)80556-1. [DOI] [PubMed] [Google Scholar]