

Abstract
Interferon regulatory factor-1 (IRF-1) is a transcription factor that regulates interferon-induced genes and type I interferons. Recently, studies of IRF-1-deficient mice have revealed that IRF-1 regulates the induction of molecules that play important roles in inflammation, such as inducible nitric oxide synthase (iNOS) and interleukin-1β-converting enzyme (ICE). To study the role of IRF-1 in autoimmunity, we investigated type II collagen-induced arthritis (CIA), and experimental allergic encephalomyelitis (EAE), in mice lacking IRF-1. The incidence and severity of CIA were significantly decreased in IRF-1−/− mice compared with IRF-1+/− mice, as was the production of interferon (IFN)-γ in lymph node cells. Both IRF-1+/− and IRF-1−/− mice exhibited mild and transient disease after adoptive transfer of a type II collagen (CII)-specific T cell line together with sera from arthritic mice, but the IRF-1−/− mice were less severely affected than the IRF-1+/− mice. In addition, the incidence of EAE in IRF-1−/− mice was decreased as compared with IRF-1+/− mice. Reverse transcription polymerase chain reaction showed that IRF-1 mRNA was constitutively expressed in the spinal cords of IRF-1+/− mice, and was upregulated in mice with clinical EAE. Expression of iNOS was also detected in inflamed spinal cords. These results suggest that IRF-1 plays a key role in promoting inflammation and autoimmunity in CIA and EAE animal models.
Interferon regulatory factor 1 (IRF-1)1 was originally identified as a transcriptional activator of type I IFN (1, 2). It binds to IRF-E sequence elements in DNA, which overlap the IFN-stimulated response element (ISRE) (3–5). Recently, studies of IRF-1-deficient mice constructed with gene targeting (6) have revealed that IRF-1 has additional important functions, including that of tumor suppression (7, 8). Kamijo et al. (9) and others (10, 11) have reported that the induction of inducible nitric oxide synthase (iNOS) in macrophages is dependent on IRF-1.
Nitric oxide (NO) has numerous known functions in a variety of tissues, including activities against tumors and microorganisms (12). The transient high level of production of NO by iNOS in macrophages and other cells has been shown to be an important factor in host defense against bacteria, inflammatory responses, and endotoxin shock (13, 14). Furthermore, a role for NO in murine and rat autoimmune disease models has been suggested (15–20). The finding that IRF-1 controls the induction of iNOS suggested the possibility that iNOS induction and subsequent NO secretion might be modified by manipulating the function or induction of IRF-1. It has recently been established that IL-1β convertase (ICE) is also regulated by IRF-1 (21). ICE is important in IL-1 secretion, as well as in cell death induction (22, 23). These studies prompted us to investigate whether IRF-1 is involved in the pathogenesis of inflammation and autoimmune disease.
In this study, we used IRF-1-deficient mice to investigate the role of IRF-1 in collagen-induced arthritis (CIA), and in experimental allergic encephalomyelitis (EAE). Our results indicate that IRF-1−/− mice exhibit significantly decreased incidence and severity of both diseases, particularly CIA. Reverse transcription studies (RT-PCR) showed that IRF-1 mRNA was constitutively expressed in the spinal cord and that its expression was further enhanced after the onset of EAE. IRF-1 mRNA expression also correlated with the expression of iNOS found in inflamed spinal cords of IRF-1+/− mice with clinical EAE.
Materials and Methods
Mice.
To generate mice with an MHC haplotype susceptible to CIA or EAE, IRF-1−/− mice (5) were back-crossed into DBA/1 (H-2q) or PL/J (H-2u) mice and the fourth generation (DBA/1), or the second and the third generation (PL/J), of mice were used for experiments. Mice were typed by PCR or Southern blot (5) during back-crossing. Homozygous (IRF-1−/−) mice were obtained by intercrossing IRF-1+/− mice. IRF-1+/− and IRF-1−/− mice were mated and progeny littermates were analyzed for CD8+ T cell populations in peripheral blood using flow cytometry (5). CD8+ T cell populations were as follows: IRF-1+/− DBA/1, 15.1–35.0% (of CD3+ cells); IRF-1−/− DBA/1, 3.1– 10.2%; IRF-1+/− PL/J, 19.5–34.6%; IRF-1−/− PL/J: 3.8–11.2%. In all experiments, only IRF-1+/− littermates were used as controls. All mice were 8–16 wk of age at the time of immunization and were maintained at the Ontario Cancer Institute Animal Facility. Animals were cared for in accordance with the guidelines of the Canadian Medical Research Council.
Induction of CIA.
Mice were immunized intradermally at the base of the tail with 150 μg bovine collagen type II (CII) (Elastin Products, Owensville, MO) emulsified with an equal volume of CFA, containing 200 μg of H37RA Mycobacterium tuberculosis (Difco, Detroit, MI). Mice were boosted by intradermal injection with 150 μg bovine CII in IFA on day 21. Arthritis development was monitored by inspection three times per week and inflammation of the four paws was graded from 0 to 3 as follows: grade 0, paws with no swelling; grade 1, paws with swelling of finger joints or mild swelling of ankle or wrist joints; grade 2, paws with severe inflammation of the entire paw; grade 3, paws with deformity or ankylosis. Each paw was graded and the four scores added so that the maximum score per mouse was 12. The arthritis index was calculated by dividing the total score of the experimental mice by the total number of arthritic mice. Parameters used to assess arthritis in each group were disease incidence, arthritis index, and the number of arthritic paws.
Measurement of Serum Anti-CII Antibody Levels.
The level of serum antibodies to CII was measured by ELISA. Native bovine CII was dissolved in 0.1 M acetic acid at 1 mg/ml and diluted with 0.1 M sodium bicarbonate at 10 μg/ml. Microtiter plates (Maxisorp, Nunc, Denmark) were coated with 100 μl of CII antigen solution and incubated overnight at 4°C. After washing with PBS containing 0.05% Tween 20, nonspecific binding was blocked with PBS containing 1% bovine serum albumin for 1 h at room temperature. After two washes, serum samples were added in serial dilutions and incubated for 1 h at 37°C. After four additional washes, peroxidase-conjugated goat anti–mouse IgG (Tago, Camarillo, CA) was added and incubated for 1 h at 37°C. Antibody binding was visualized using orthophenylenediamine (Sigma Chemical Co., St. Louis, MO) dissolved in citrate buffer (pH 5) containing 0.012% H2O2; color was developed for 5–10 min. The optical density at 450 nm was measured using a microplate reader (Molecular Devices, Sunnyvale, CA). A standard serum composed of a mixture of sera from arthritic mice was added to each plate in serial dilutions to establish a standard curve. The standard serum was defined as 100 U and antibody titers of serum samples were calculated from this standard curve by Softmax software (Molecular Devices).
Histological Examination.
To evaluate the histology of joints, mice were killed at 7–8 wk after immunization. Inflamed toes or random toes (when macroscopic inflammation was not observed) were excised, fixed in 10% buffered formalin, decalcified in EDTA, embedded in paraffin, sectioned, and stained with hematoxylin and eosin.
IFN-γ Production of Lymph Node Cells.
IRF-1+/− and IRF-1−/− DBA/1 mice were killed 14 or 21 d after initial immunization. Lymph nodes (inguinal, paraaortic, axillary, and popliteal) were removed, and cells were resuspended in DMEM supplemented with 5 × 10−5 M 2-mercaptoethanol, 20 mM Hepes, and 4% autologous mouse serum. Cells were seeded at 4 × 106/plate in 96well flat-bottomed microtiter plates (Nunc) and stimulated with denatured bovine CII at 200 μg/ml or with purified anti-CD3 (2C11; PharMingen, San Diego, CA) at 5 μg/ml. For denaturation, CII was dissolved in 0.1 M acetic acid, dialyzed against DMEM for 48 h, and heat-denatured for 15 min at 56°C. Plated cells were incubated at 37°C in 5% CO2 for 48 h, after which culture supernatants were collected. IFN-γ was measured using an ELISA kit (Genzyme, Cambridge, MA) according to the manufacturer's instructions.
Proinflammatory Cytokine Production of Peritoneal Macrophages.
Peritoneal macrophages from IRF-1+/− and IRF-1−/− DBA/1 mice were obtained 3 d after injection of thioglycolate. Cells were resuspended in DMEM with 10% FCS at 2 × 106/ml and seeded in 24-well plates (1 ml/well). After 2-h incubation at 37°C, nonadherent cells were removed by gentle washing with PBS. The total peritoneal cell population and nonadherent cell population were checked by surface staining of cells with anti-CD11b and anti-B220 antibodies (PharMingen) to ensure that cell populations from IRF-1+/− and IRF-1−/− DBA/1 mice were comparable. Adherent macrophages were stimulated with 1 μg/ml LPS (Sigma), or 0.1 μg/ml LPS plus 100 U/ml IFN-γ (Genzyme), for 3 h and supernatants were collected for measurement of TNF-α. Cells were then washed with DMEM, stimulated with 30 μM of nigericin (Sigma) at 37°C for 30 min, and supernatants were collected to measure IL-1α and IL-1β. Cytokine concentrations were determined with ELISA kits (Genzyme). IL-1β could not be detected in the supernatant before nigericin addition. Viability of cells as judged by the trypan blue exclusion test was more than 95% after treatment with nigericin.
Adoptive Transfer of Arthritis Using a CII-specific T Cell Line and Sera from Arthritic Mice.
A CII-specific T cell line was established from CD4-deficient mice (24). These cells respond to denatured CII (dCII) and native CII (nCII) in MHC-restricted (H-2q) fashion, and the majority of cells have a phenotype of TCRαβ+ CD4− CD8− (24). Sera from arthritic DBA/1 mice were pooled, precipitated with saturated ammonium sulfate (50% vol/vol), resuspended in PBS, and dialyzed against PBS for 2 d. Final volume was 40–50% of the original sera. Anti-CII antibody level was measured as described above. On day 0, mice were injected intravenously with 0.3 ml of concentrated sera containing 4–4.5 × 107 cells that had been stimulated with dCII for 3 d. Mice received a second injection of sera (0.2 ml) on day 2. Mice were examined and scored for signs of arthritis daily. Because almost all mice developed only mild disease (grade 1), the severity of arthritis was measured by counting the number of fingers and toes that developed inflammation.
Induction of EAE.
Mice were immunized intradermally at the base of the tail with 200 μg of rabbit myelin basic protein (Sigma) emulsified with CFA on day 0. In addition, 200 ng of pertussis toxin (List Biochemical Research, Campbell, CA) was injected intravenously on days 0 and 2. Immunized mice were examined daily and scored as previously described (25).
RT-PCR.
Total RNA was prepared from spinal cord and joint tissue. Before removing spinal cords, mice were anesthetized and perfused with 5 ml of PBS to reduce contamination with peripheral leukocytes. Joint tissue was prepared from soft tissues around the ankle joint. Total RNA was extracted with TRI-ZOL Reagent (GIBCO BRL, Gaithersburg, MD). cDNA was obtained by reverse transcription of 2 μg of total RNA using oligodT and a RT-PCR kit (Stratagene, La Jolla, CA). The manufacturer's protocol was followed, using 10% of the reaction sample as the template for PCR. Primers used were as follows: IRF-1, GGTCAGAGACCCAAACTATGGTGC and TCTGAGTGGCATATGCAGATGGAC; β-actin, ACCCACACTGTGCCCATCTA and CGGAACCGCTCATTGCC; iNOS, CCCTTCCGAAGTTTCTGGCAGCAGCGGC and GGCTGTCAGAGCCTCGTGGCTTTGG (26). Reaction conditions for amplification with PCR were the following: 94°C for 40 s; 56°C for 1 min; 72°C for 2 min. The detection of IRF-1 and β-actin mRNAs required 24–30 PCR cycles. IRF-1 signals in the normal spinal cord and normal joint tissues could usually be detected after 26 cycles of amplification, while detection of iNOS mRNA required 36 cycles. PCR products were separated on 2% agarose gels and visualized with ethidium bromide.
Statistics.
Statistical significance of differences in incidence of CIA or EAE, number of arthritic paws, and number of arthritic fingers and toes were determined using χ2 analysis. Student's t test was used to compare the arthritis index in CIA, severity in EAE, and cytokine production in T cells and macrophages.
Results
Clinical Course of CIA in IRF-1−/− DBA/1 Mice.
IRF-1−/− and IRF-1+/− littermates were immunized with CII and observed for signs of arthritis for up to 10 wk after immunization. Disease incidence and the maximal arthritis index were assessed at the end of this period. The incidence of arthritis calculated from four independent experiments is shown in Fig. 1 A. As shown in Table 1, the incidence in IRF-1−/− mice (37.9%, 11 out of 29 mice) was significantly decreased as compared with IRF-1+/− mice (82.6%, 19 out of 23 mice; P <0.005). There was also a delay in the development of disease in IRF-1−/− mice, because the median day of onset among diseased individuals was day 34 in IRF-1+/− mice, and day 44 in IRF-1−/− mice. Furthermore, the arthritis index (calculated only from arthritic mice) was significantly decreased in IRF-1−/− mice (2.7 ± 0.6) compared with IRF-1+/− mice (6.9 ± 0.7; P <0.01) throughout the course of disease (Fig. 1 B). Histological examination of the phalangeal joints of toes of 4 out of 5 IFR-1+/− mice showed typical arthritis, characterized by dense cellular infiltration and bone erosion (Fig. 2 A). In contrast, most joints of IRF-1−/− mice (6 out of 7 mice) showed either mild infiltration of cells or no sign of inflammation (Fig. 2 B). These data imply that the incidence and the severity of CIA is decreased in IRF-1−/− mice; however, there was no difference in anti-CII IgG antibody level between the two groups of mice at 5 wk after immunization (IRF-1+/−, 60 ± 10 U; IRF-1−/−, 52 ± 12 U).
Figure 1.
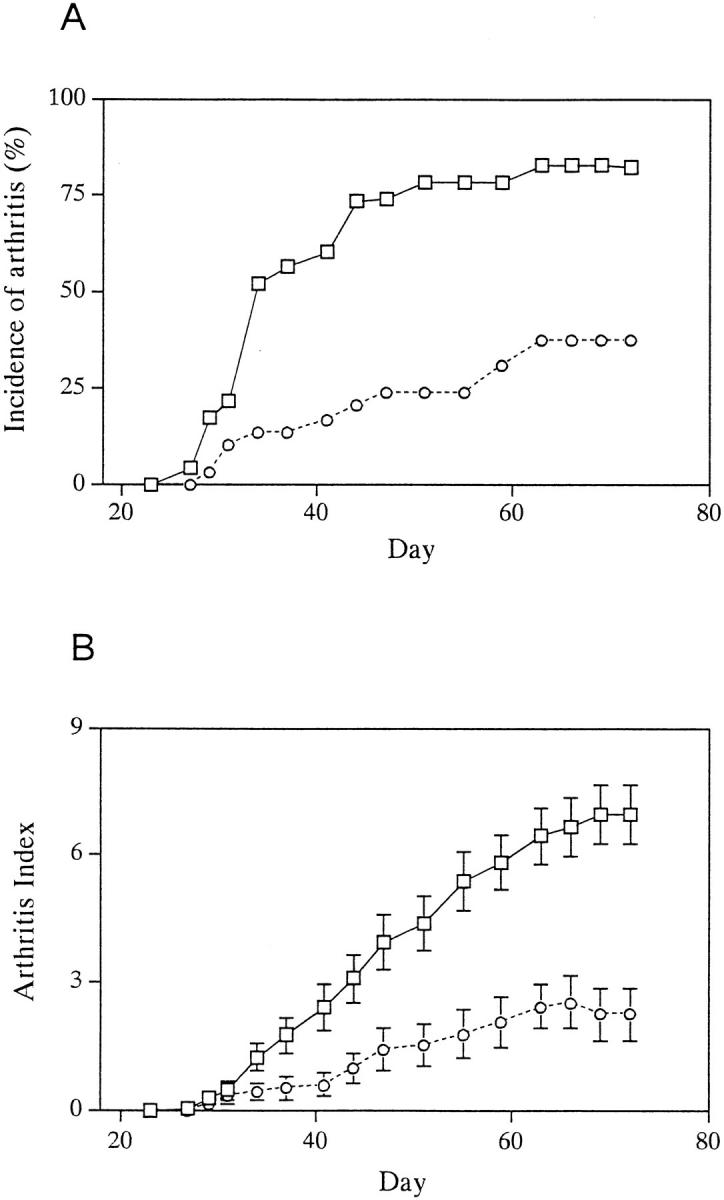
Development of CIA in IRF-1+/− and IRF-1−/− DBA/1 mice. IRF-1+/− (squares, n = 23) and IRF-1−/− (circles, n = 29) mice were immunized with bovine CII and signs of arthritis were monitored as described in Materials and Methods. A summary of four experiments is shown. (A) Incidence of arthritis in IRF-1−/− mice was decreased as compared with that of control IRF-1+/− mice (P <0.005). (B) Arthritis index, calculated from arthritic mice only, was also decreased in IRF-1−/− mice after day 42 (P <0.05). Mean arthritis index ± SEM is shown.
Table 1.
CIA in IRF-1+ /− and IRF-1− /− DBA/1 Mice
| Mouse genotype | Incidence of arthritis | Arthritis index* | Number of arthritic paws‡ | Anti-CII IgG antibody (U) | ||||
|---|---|---|---|---|---|---|---|---|
| IRF-1+/− | 19/23 (82.6%) | 6.9 ± 0.7 | 56/92 (60.9%) | 60 ± 10 | ||||
| IRF-1−/− | 11/29 (37.9%)§ | 2.7 ± 0.6‖ | 20/116 (17.2%)§ | 52 ± 12 |
Mean ± SEM of the maximal scores of arthritic mice is shown.
Number of paws with arthritis per total paws of immunized mice.
P <0.005 (χ2 analysis).
P <0.01 (Student's t test).
Figure 2.

Histological examination of the joints from IRF1+/− and IRF-1−/− DBA/1 mice. Cellular infiltrate in the synovium and erosion of cartilage and bone were observed in joints from control IRF-1+/− DBA/1 mice (A). In contrast, limited infiltration of cells was detected in IRF-1−/− DBA/1 mice (B). Sections were excised from CII-immunized mice at 7–8 wk after immunization and stained with hematoxylin and eosin. (×50).
IFN-γ Production of LN Cells from IRF-1+ /− and IRF-1− /− DBA/1 Mice.
To analyze the antigen-specific T cell response in IRF-1+/− and IRF-1−/− DBA/1 mice, IFN-γ production by LN cells in response to dCII was examined. Mice were killed at 14 or 21 d after immunization, and LN cells were purified and stimulated with dCII or anti-CD3 antibodies. Anti-CD3 stimulation induced large amounts of IFN-γ secretion from LN cells in both groups of mice. However, as shown in Fig. 3, LN cells from IRF-1−/− mice secreted a reduced amount of IFN-γ in response to dCII as compared with IRF-1+/− mice on day 14. On day 21, IFN-γ production in response to dCII was decreased in IRF-1+/− mice, while it remained at low levels in IRF-1−/− mice. These results suggest that IFN-γ production by LN cells in response to dCII is defective in IRF-1−/− mice.
Figure 3.

IFN-γ production of LN cells from CII-immunized IRF1+/− and IRF-1−/− DBA/1 mice. LN cells were prepared on day 14 or day 21 after immunization and stimulated with dCII or anti-CD3. IFN-γ production on day 14 was decreased by fourfold in IRF-1−/− mice (P <0.05). Mean IFN-γ concentration ± SD from four (day 14) and two (day 21) experiments is shown.
Proinflammatory Cytokine Production of Peritoneal Macrophages from IRF-1− /− DBA/1 Mice.
Macrophages produce proinflammatory cytokines, such as IL-1 or TNF-α, which have been shown to be important factors in perpetuating arthritis (27–29). Recent studies have shown that IRF-1 regulates the induction of ICE in splenocytes (21). To test the ability of macrophages from IRF-1−/− mice to secrete these cytokines, peritoneal macrophages from IRF-1+/− and IRF-1−/− mice were stimulated with LPS (1 μg/ml), or with LPS (0.1 μg/ml) plus IFN-γ (100 U), and secreted cytokine levels were measured by ELISA. There were no differences in the amounts of secreted IL-1α, IL-1β, or TNF-α detected after stimulation with LPS, or LPS plus IFN-γ, between macrophages of IRF-1+/− and IRF-1−/− mice (Table 2).
Table 2.
Proinflammatory Cytokine Production of Peritoneal Macrophages from IRF-+ /− and IRF-1− /− DBA/1 Mice*
| Stimulant/mouse genotype | IL-1α (ng/ml) | IL-1β (pg/ml) | TNF-α (ng/ml) | |||
|---|---|---|---|---|---|---|
| LPS (1 μg/ml) | ||||||
| IRF-1+/− | 0.56 ± 0.06‡ | 71 ± 53 | 5.4 ± 0.5 | |||
| IRF-1−/− | 0.71 ± 0.19 | 75 ± 21 | 4.4 ± 1.5 | |||
| LPS (0.1 μg/ml) + IFN-γ (100 U) | ||||||
| IRF-1+/− | 0.22 ± 0.01 | 56 ± 22 | 10.9 ± 0.8 | |||
| IRF-1−/− | 0.23 ± 0.05 | 42 ± 23 | 8.4 ± 0.8 |
Peritoneal macrophages were stimulated with LPS, or LPS plus IFN-γ, for 3 h. IL-1 secretion was further induced by stimulating cells with nigericin as described in Materials and Methods. A representative result from three independent experiments is shown. Each experiment included 2–3 mice per group.
Mean ± SD is shown.
Adoptive Transfer of Arthritis in IRF-1+ /− and IRF-1− /− DBA/1 Mice by a CII-specific T Cell Line and Sera from Arthritic Mice.
Because IRF-1 appeared to be involved in both the induction and the the effector phases of CIA, the effector phase of CIA in IRF-1−/− mice was examined by adoptive transfer. Adoptive transfer of disease has been used in the EAE model and in nonobese diabetes (NOD) mice, but there is no established regimen for CIA. We evaluated a method for adoptive transfer of CIA in which a CII-specific T cell line (established as described in Materials and Methods), together with sera from arthritic mice, is injected into naive DBA/1 mice. The helper T cells and antiCII antibodies required for the initiation of arthritis are injected, while the macrophages and other effector cells are derived from the recipient. Recipient mice developed mild and transient arthritis, which was generally grade 1 in severity (per paw). After day 10–14, these mice either developed stable arthritis, or underwent remission. As shown in Fig. 4 A, the arthritis index in IRF-1−/− mice decreased as compared with that in control IRF-1+/− mice after day 7. The number of arthritic fingers and toes was also lower, confirming the decreased severity of disease in IRF-1−/− mice (Fig. 4 B). These results suggest that IRF-1−/− mice develop less severe arthritis than their IRF-1+/− counterparts, even in the presence of equal numbers of CII-specific T cells and anti-CII antibodies.
Figure 4.

Adoptive transfer of arthritis in IRF-1+/− and IRF-1−/− DBA/1 mice. A CII-reactive cell line together with sera from arthritic mice was injected into naive mice as described in Materials and Methods. Arthritis index (A), and number of fingers and toes with arthritis (B), were evaluated. Arthritis index and number of arthritic fingers and toes were decreased in IRF-1−/− mice (circles) after day 7 (P <0.05 and P <0.01, respectively) as compared with those in IRF-1+/− mice (squares). In this experiment, all IRF-1+/− and IRF-1−/− mice developed arthritis. A representative result from a total of three experiments is shown.
EAE in IRF-1− /− PL/J Mice.
We investigated a second antigen–induced autoimmune disease model, EAE, in IRF-1−/− mice. Mice were injected with myelin basic protein in CFA and pertussis toxin, and observed for signs of disease. As shown in Table 3, the incidence of EAE was significantly decreased in IRF-1−/− mice (P <0.005). However, 3 out of 4 IRF-1−/− mice that developed the disease showed moderate to significant severity (maximal scores were 1, 2.5, 3.5, and 3.5). The mean maximal score calculated from diseased mice only was similar for IRF-1+/− and IRF-1−/− mice (IRF-1+/−, 3.3 ± 1.0; IRF-1−/−, 2.6 ± 1.2; Table 3).
Table 3.
EAE in IRF-1+ /− and IRF-1− /− PL/J Mice
| Mouse genotype | Incidence of disease | Severity* (total mice) | Severity‡ (diseased mice) | Day of onset | ||||
|---|---|---|---|---|---|---|---|---|
| IRF-1+/− | 12/15 (80.0%) | 2.6 ± 0.4 | 3.3 ± 0.3 | 25.8 ± 2.1 | ||||
| IRF-1−/− | 4/13 (30.8%)§ | 0.8 ± 0.3‖ | 2.6 ± 0.5 | 30.3 ± 1.4 |
Mean ± SEM of the maximal scores of total mice is shown.
Mean ± SEM of the maximal scores of diseased mice is shown.
P <0.005 (χ2 analysis),
P <0.005 (Student's t test).
Expression of IRF-1 and iNOS mRNA in Spinal Cord and Joint Tissue.
IRF-1 plays an important role in the induction of iNOS (9–11), and the resulting NO production in macrophages and other cells is considered to be crucial in promoting inflammation (12). To examine the expression of IRF-1 and iNOS in inflamed tissues, RT-PCR of mRNA from spinal cord (EAE) and joint tissue (CIA) of IRF-1+/− mice with or without clinical disease was carried out. IRF-1 mRNA was detected both in normal spinal cord (Fig. 5, lanes 2 and 3), and in cells from spinal cords with clinical EAE (lane 4, score 3.5; lane 5, score 4). In fact, IFR-1 expression was enhanced in the latter, suggesting an upregulation of IRF-1 mRNA in infiltrating cells and/or resident cells in EAE spinal cord. iNOS mRNA expression was also detected in spinal cords of IRF-1+/− mice with EAE (Fig. 5, lanes 4 and 5). In contrast, IRF-1 expression was not detected in IRF-1−/− mice with clinical EAE (Fig. 5, lane 6, score 2.5; lane 7, score 3.5). However, iNOS mRNA was observed in an IRF-1−/− mouse with moderate EAE (Fig. 5, lane 6), which suggests that iNOS can be induced by an IRF-1-independent pathway. Caution should be exercised in interpreting this result, since >30 cycles of PCR were required to detect expression. In the case of CIA, similar levels of IRF-1 and iNOS mRNA expression were observed in four normal and two arthritic (severity grade 2) IRF-1+/− mice. However, no iNOS signal could be detected in joints from the three arthritic IRF-1−/− mice (two grade 1 and one grade 2).
Figure 5.

RT-PCR study of IRF-1 (A) and iNOS (C) mRNA expression in spinal cords from IRF-1+/− (lanes 2–5) and IRF-1−/− (lanes 6 and 7) PL/J mice with (lanes 4–7) or without (lanes 2 and 3) clinical EAE. Lane 1 is a DNA size marker (φx174 DNA digested with HaeIII). IRF-1 expression was detected in normal spinal cord (lanes 2 and 3) and was increased in the spinal cords of IRF-1+/− mice with EAE (lanes 4 and 5). No PCR product was observed in IRF-1−/− samples (lanes 6 and 7). iNOS mRNA was detected in spinal cords from IRF-1+/− (lanes 4 and 5) and IRF-1−/− (lane 6) mice with EAE. PCR reactions were run 26 cycles for IRF-1 and β-actin, and 36 cycles for iNOS. Clinical scores at the time of tissue preparation were the following: lane 4, grade 3.5; lane 5, grade 4; lane 6, grade 2.5; lane 7, grade 3.5.
Discussion
IRF-1 was originally identified by its ability to induce the transcription of type I IFN and IFN-induced genes. Although the expression of type I IFN and IFN-induced genes was generally unimpaired in IRF-1-deficient mice (6, 11), recent studies have revealed that IRF-1 is involved in the regulation of other important genes and cellular functions, such as iNOS and NO production (9, 10), and ICE- and DNA damage–induced apoptosis (21). In the present study, we investigated the role of IRF-1 in inflammation and autoimmunity in two antigen-induced autoimmune disease models, CIA and EAE. Our data show that the incidence of CIA was significantly decreased in IRF-1−/− DBA/1 mice; furthermore, arthritic IRF-1−/− mice showed a milder disease than did IRF-1+/− mice. The suppression of CIA in IRF-1−/− mice was more dramatic than that reported in CD4- or CD8-deficient mice (24) or in TNF-receptor p55-deficient mice (our unpublished observation). In the EAE model, IRF-1−/− PL/J mice were less susceptible to EAE than IRF-1+/− PL/J mice, yet the EAE that developed in IRF-1−/− mice was as severe as that in the control mice. These results suggest that IRF-1 functions as a proinflammatory factor in these disease models.
In this study, IRF-1−/− DBA/1 mice exhibited decreased IFN-γ production in LN cells after immunization with CII. This result could explain the lower incidence of CIA in IRF-1−/− mice, although it is not clear whether this low IFN-γ response was attributable to T cells or to antigen-presenting cells. To bypass the primary immune responses against CII, we adoptively transferred disease into naive IRF-1+/− and IRF-1−/− mice by injecting a CIIspecific T cell line together with sera from arthritic mice, thereby mimicking the cellular and humoral responses against against CII immunization. The results showed that disease development in IRF-1−/− mice after adoptive transfer was still impaired. These data suggest that effector cells other than CII-specific helper T cells, or cellular components of the autoimmune target organ, may limit disease progression.
It has been shown that neither CD4+ T cells (30, 31) nor TCRαβ+ T cells (31) are required to maintain CIA after its onset. These results taken together with the finding that the majority of infiltrating cells in inflamed joints are macrophages (32, 33), suggest that the principal force driving CIA after onset may be derived from macrophages. We speculate that the decreased disease severity in IRF-1−/− mice after adoptive transfer may be partly due to decreased proinflammatory functions of effector cells, presumably macrophages, in IRF-1-deficient mice. Cytokine secretion of peritoneal macrophages from IRF-1−/− mice in response to LPS, or LPS with IFN-γ, was normal. Although this result does not necessarily indicate that cytokine production in macrophages is intact in IRF-1−/− mice with CIA, normal levels of proinflammatory cytokines were produced in response to certain stimuli. It is also possible that activation of synovial cells or endothelial cells resulting in cytokine production or adhesion molecule expression might be deregulated in the absence of IRF-1. In contrast with the mild disease exhibited in the CIA experiment, IRF-1−/− mice that developed EAE showed severities similar to those in IRF-1+/− mice, although only a small number of mice developed clinical disease. This suggests that the role of IRF-1 may be less important in the disease progression of EAE than in CIA.
NO exerts a plethora of effects on a variety of cells and tissues, including the modification of enzymes and transcription factors that result in modulated cellular responses (12, 34). A role for NO in rheumatoid arthritis was suggested by the finding that the NO catabolite nitrite is increased in the synovial fluid and sera of patients (35). In addition, NO and iNOS were identified in the spinal cords of mice with EAE (18–20). These results are confirmed in this study because iNOS was expressed in the spinal cord of mice with EAE and in the joint tissue of mice with CIA. Inhibitors of iNOS can suppress some autoimmune disease models in rodents, including streptococcal cell wall fragment–induced rat arthritis (15), murine lupus (16), and murine EAE (17). It has been shown previously that IRF-1 regulates iNOS, and that macrophages from IRF-1−/− mice produce little NO (6, 11). Therefore, it is conceivable that the decreased iNOS mRNA induction and subsequent NO production observed in macrophages of IRF-1−/− mice contribute to the suppression of CIA and EAE disease in these animals.
IRF-1 mRNA expression was found to be increased in the spinal cords of IRF-1+/− mice with EAE. Upregulation of IRF-1 mRNA in the spinal cord suggests the activation of infiltrating inflammatory cells and/or resident glial cells. Because NO in the spinal cord is thought to be derived from microglia and infiltrating macrophages (36–38), upregulation of IRF-1, presumably in response to cytokines from T cells and macrophages, might lead to the expression of iNOS mRNA and subsequent NO production in these cells. However, we also observed iNOS expression in an IRF-1−/− mouse with EAE, suggesting that iNOS can be induced by an IRF-1-independent pathway.
ICE plays a role in IL-1β secretion by processing the precursor form of IL-1β intracellularly to mature bioactive IL-1β (39, 40). ICE-deficient mice showed no IL-1β secretion and markedly reduced IL-1α secretion in monocytes and macrophages (22, 23). In this study, no differences in IL-1α, IL-1β, and TNF-α secretion between IRF-1−/− and IRF-1+/− macrophages were observed. These data seem to contradict a previous report in which IRF-1 was found to regulate the induction of ICE (21). Although the precise mechanism remains to be elucidated, basal expression of ICE might be sufficient to induce secretion of mature IL-1β in macrophages. Alternatively, the use of different cells and stimulants in these experiments (splenocytes versus peritoneal macrophages, Con A versus LPS and IFN-γ) might produce different activation profiles.
It has been shown that IRF-1-deficient mice have only 70% of the number of CD8+ T cells present in IRF-1+/− mice (6). However, it is unlikely that the low number of CD8+ T cells prevented the development of CIA and EAE disease in IRF-1-deficient mice. In previous experiments with CD8-deficient mice, the incidence of EAE was found to be normal (25). The incidence of CIA in CD8-deficient mice was decreased (50%) relative to controls (84.2%), but the severity of the disease was similar (24).
Since the critical role of IRF-1 in iNOS induction was discovered, IRF-1 has been targeted as a therapeutic agent with an eye to regulating NO production and NO-induced toxicity. Our data provide further evidence that IRF-1 is important in inflammation and autoimmunity in in vivo murine disease models. The loss of IRF-1 affects both the antigen-priming phase and the effector phase in CIA disease. The proinflammatory function of IRF-1 may not be limited to iNOS induction and may include the regulation of other molecules. Although the exact mechanisms have yet to be determined, these results indicate that the modulation of IRF-1 could be an effective strategy in mitigating inflammation and autoimmune disease.
Acknowledgments
We thank Drs. J. Penninger, H.-W. Mittruecker, and M. Saunders for critical reading of the manuscript.
Footnotes
This study is supported by Medical Research Council of Canada.
1 Abbreviations used in this paper: CIA, collagen-induced arthritis; CII, type II collagen; EAE, experimental allergic encephalomyelitis; ICE, interleukin-1β-converting enzyme; iNOS, inducible nitric oxide synthase; IRF-1, interferon regulatory factor-1; ISRE, IFN-stimulated response element; NO, nitric oxide; NOD, nonobese diabetes.
References
- 1.Miyamoto M, Fujita T, Kimura Y, Maruyama M, Harada H, Sudo Y, Miyata T, Taniguchi T. Regulated expression of a gene encoding a nuclear factor, IRF-1, that specifically binds to IFN-β gene regulatory elements. Cell. 1988;54:903–913. doi: 10.1016/s0092-8674(88)91307-4. [DOI] [PubMed] [Google Scholar]
- 2.Harada H, Fujita T, Miyamoto M, Kimura Y, Maruyama M, Furia A, Miyata T, Taniguchi T. Structurally similar but functionally distinct factors, IRF-1 and IRF-2, bind to the same regulatory elements of IFN and IFN-inducible genes. Cell. 1989;58:729–739. doi: 10.1016/0092-8674(89)90107-4. [DOI] [PubMed] [Google Scholar]
- 3.Friedman RL, Stark GR. α-interferon-induced transcription of HLA and metallothionein genes containing homologous upstream sequences. Nature (Lond) 1985;314:637–639. doi: 10.1038/314637a0. [DOI] [PubMed] [Google Scholar]
- 4.Levy DE, Kessler DS, Pine R, Reich N, Darnell JE., Jr Interferon-induced nuclear factors that bind a shared promoter element correlate with positive and negative transctiptional control. Genes & Dev. 1988;2:383–393. doi: 10.1101/gad.2.4.383. [DOI] [PubMed] [Google Scholar]
- 5.Tanaka N, Kawakami T, Taniguchi T. Recognition DNA sequences of interferon regulatory factor 1 (IRF-1) and IRF-2, regulators of cell growth and the interferon system. Mol Cell Biol. 1993;13:4531–4538. doi: 10.1128/mcb.13.8.4531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matsuyama T, Kimura T, Kitagawa M, Pfeffer K, Kawakami T, Watanabe N, Kundig TM, Amakawa R, Kishihara K, Wakeham A, et al. Targeted disruption of IRF-1 or IRF-2 results in abnormal type I IFN gene induction and aberrant lymphocyte development. Cell. 1993;75:83–97. [PubMed] [Google Scholar]
- 7.Tanaka N, Ishihara M, Kitagawa M, Harada H, Kimura T, Matsuyama T, Lamphier MS, Aizawa S, Mak TW, Taniguchi T. Cellular commitment to oncogene-induced transformation or apoptosis is dependent on the transcription factor IRF-1. Cell. 1994;77:829–839. doi: 10.1016/0092-8674(94)90132-5. [DOI] [PubMed] [Google Scholar]
- 8.Harada H, Kitagawa M, Tanaka N, Yamamoto H, Harada K, Ishihara M, Taniguchi T. Anti-oncogenic and oncogenic potentials of transcriptional regulators IRF-1 and -2. Science (Wash DC) 1993;259:971–974. doi: 10.1126/science.8438157. [DOI] [PubMed] [Google Scholar]
- 9.Kamijo R, Harada H, Matsuyama T, Bosland M, Gerecitano J, Shapiro D, Le J, Koh SI, Kimura T, Green SJ, Mak TW, Taniguchi T, Vicek J. Requirement for transcription factor IRF-1 in NO synthase induction in macrophages. Science (Wash DC) 1994;263:1612–1615. doi: 10.1126/science.7510419. [DOI] [PubMed] [Google Scholar]
- 10.Martin E, Nathan C, Xie Q. Role of interferon regulatory factor 1 in induction of nitric oxide synthase. J Exp Med. 1994;180:977–984. doi: 10.1084/jem.180.3.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reis LFL, Ruffner H, Stark G, Aguet M, Weissman C. Mice devoid of interferon regulatory factor 1 (IRF-1) show normal expression of type I interferon genes. EMBO (Eur Mol Biol Organ) J. 1994;13:4798–4806. doi: 10.1002/j.1460-2075.1994.tb06805.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harald HH, Schmidt W, Walter U. NO at work. Cell. 1994;78:919–925. doi: 10.1016/0092-8674(94)90267-4. [DOI] [PubMed] [Google Scholar]
- 13.MacMicking JD, Nathan C, Hom G, Chartrain N, Fletcher DS, Trumbauer M, Stevens K, Xie Q-w, Sokol K, Hutchinson N, Chen H, Mudgett JS. Altered responses to bacterial infection and endotoxic shock in mice lacking inducible nitric oxide synthase. Cell. 1995;81:641–650. doi: 10.1016/0092-8674(95)90085-3. [DOI] [PubMed] [Google Scholar]
- 14.Wei X-q, Charies IG, Smith A, Ure J, Feng G-J, Huang F-P, Xu D, Muller W, Moncada S, Liew FY. Altered immune responses in mice lacking inducible nitric oxide synthase. Nature (Lond) 1995;375:408–411. doi: 10.1038/375408a0. [DOI] [PubMed] [Google Scholar]
- 15.McCartney-Francis N, Allen JB, Mizel DE, Albina JE, Xie Q, Nathan CF, Wahl SM. Suppression of arthritis by an inhibitor of nitric oxide synthase. J Exp Med. 1993;178:749–754. doi: 10.1084/jem.178.2.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weinberg JB, Granger DL, Pisetsky DS, Seldin MF, Misukonis MA, Mason SN, Pippen AM, Ruiz P, Wood ER, Gilkeson GS. The role of nitric oxide in the pathogenesis of spontaneous murine autoimmune disease: Increased nitric oxide production and nitric oxide synthase expression in MRL-lpr/lpr mice, and reduction of spontaneous glomerulonephritis and arthritis by orally administered NG-Monomethyl-L-Arginine. J Exp Med. 1994;179:651–660. doi: 10.1084/jem.179.2.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cross AH, Misko TP, Lin RF, Hickey WF, Trotter JL, Tilton RG. Aminoguanidine, an inhibitor of inducible nitric oxide synthase, ameliorates experimental autoimmune encephalomyelitis in SJL mice. J Clin Invest. 1994;93:2684–2690. doi: 10.1172/JCI117282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koprowski H, Zheng YM, Heber-Katz E, Fraser N, Rorke L, Fu ZF, Hanlon C, Dietzschold B. In vivo expression of inducible nitric oxide synthase in experimentally induced neurologic diseases. Proc Natl Acad Sci USA. 1993;90:3024–3027. doi: 10.1073/pnas.90.7.3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin RF, Lin T-S, Tilton RG, Cross AH. Nitric oxide localized to spinal cords of mice with experimental allergic encephalomyelitis: An electron paramagnetic resonance study. J Exp Med. 1993;178:643–648. doi: 10.1084/jem.178.2.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hopper DC, Ohnishi ST, Kean R, Numagami Y, Dietzschold B, Koprowski H. Local nitric oxide production in viral and autoimmune diseases of the central nervous system. Proc Natl Acad Sci USA. 1995;92:5312–5316. doi: 10.1073/pnas.92.12.5312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tamura T, Ishihara M, Lamphier MS, Tanaka N, Oishi I, Aizawa S, Matsuyama T, Mak TW, Taki S, Taniguchi T. An IRF-1-dependent pathway of DNA damage–induced apoptosis in mitogen-activated T lymphocytes. Nature (Lond) 1995;376:596–599. doi: 10.1038/376596a0. [DOI] [PubMed] [Google Scholar]
- 22.Li P, Allen H, Banerjee S, Franklin S, Herzog L, Johnston C, McDowell J, Paskind M, Rodman L, Salfeld J, et al. Mice deficient in IL-1β-converting enzyme are defective in production of mature IL-1β and resistant to endotoxic shock. Cell. 1995;80:401–411. doi: 10.1016/0092-8674(95)90490-5. [DOI] [PubMed] [Google Scholar]
- 23.Kuida K, Lippke JA, Ku G, Harding MW, Livingston DJ, Su MSS, Flavell RA. Altered cytokine export and apoptosis in mice deficient in interleukin-1β converting enzyme. Science (Wash DC) 1995;267:2000–2003. doi: 10.1126/science.7535475. [DOI] [PubMed] [Google Scholar]
- 24.Tada Y, Ho A, Koh DR, Mak TW. Collagen-induced arthritis in CD4- or CD8-deficient mice: CD8+T cells play a role in the initiation and regulate the recovery phase of arthritis. J Immunol. 1996;156:4520–4526. [PubMed] [Google Scholar]
- 25.Koh DR, Fung WP -leung, A. Ho, D. Gray, H. Acha-orbea, and T.W. Mak. Less mortality but more relapses in experimental allergic encephalomyelitis in CD8−/−mice. Science (Wash DC) 1992;256:1210–1213. doi: 10.1126/science.256.5060.1210. [DOI] [PubMed] [Google Scholar]
- 26.Rottenberg ME, Sporrong L, Persson I, Wigzell H, Örn A. Cytokine gene expression during infection of mice lacking CD4 and/or CD8 with Trypanosoma cruzi. Scand J Immunol. 1995;41:164–170. doi: 10.1111/j.1365-3083.1995.tb03549.x. [DOI] [PubMed] [Google Scholar]
- 27.Hom JT, Bendele AM, Carlson DG. In vivo administration with IL-1 accelerates the development of collagen-induced arthritis in mice. J Immunol. 1988;141:834–841. [PubMed] [Google Scholar]
- 28.Thorbecke GJ, Shah R, Leu CH, Kuruvilla AP, Hardison AM, Palladino MA. Involvement of endogenous tumor necrosis factor α and transforming growth factor β during induction of collagen type II arthritis mice. Proc Natl Acad Sci USA. 1992;89:7375–7379. doi: 10.1073/pnas.89.16.7375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Williams RO, Feldmann M, Maini RN. Antitumor necrosis factor ameliorates joint disease in murine collagen-induced arthritis. Proc Natl Acad Sci USA. 1992;89:9784–9788. doi: 10.1073/pnas.89.20.9784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goldschmidt TJ, Holmdahl R. Anti-T cell receptor antibody treatment of rats with established autologous collagen-induced arthritis: suppression of arthritis without reduction of anti-type II collagen autoantibody levels. Eur J Immunol. 1991;21:1327–1330. doi: 10.1002/eji.1830210536. [DOI] [PubMed] [Google Scholar]
- 31.Yoshino S, Cleland LG. Depletion of α/β T cells by a monoclonal antibody against the α/β T cell receptor suppresses established adjuvant arthritis, but not established collagen-induced arthritis in rats. J Exp Med. 1992;175:907–915. doi: 10.1084/jem.175.4.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holmdahl R, Jonsson R, Larsson P, Klareskog L. Early appearance of activated CD4+T lymphocytes and class II antigen-expressing cells in joints of DBA/1 mice immunized with type II collagen. Lab Invest. 1988;58:53–60. [PubMed] [Google Scholar]
- 33.Dejoy SQ, Ferguson KM, Oronsky AL, Kerwar SS. Studies on the synergy between collagen and adjuvant arthritis in rats. Cell Immunol. 1988;113:117–129. doi: 10.1016/0008-8749(88)90011-1. [DOI] [PubMed] [Google Scholar]
- 34.Stamier JS. Redox signaling: Nitrosylation and related target interactions of nitric oxide. Cell. 1994;78:931–936. doi: 10.1016/0092-8674(94)90269-0. [DOI] [PubMed] [Google Scholar]
- 35.Farrell AJ, Blake DR, Palmer RMJ, Moncada S. Increased concentrations of nitrite in synovial fluid and serum samples suggest increased nitric oxide synthesis in rheumatic diseases. Ann Rheum Dis. 1992;51:1219–1222. doi: 10.1136/ard.51.11.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Merrill JE, Ignarro LJ, Sherman MP, Melinek J, Lane TE. Microglial cell cytotoxicity of oligodendrocytes is mediated through nitric oxide. J Immunol. 1993;151:2132–2141. [PubMed] [Google Scholar]
- 37.Boje KM, Arora PK. Microglia-produced nitric oxide and reactive nitrogen oxides mediate neuronal cell death. Brain Res. 1992;587:250–256. doi: 10.1016/0006-8993(92)91004-x. [DOI] [PubMed] [Google Scholar]
- 38.Chao CC, Hu S, Molitor TW, Shaskan EG, Peterson PK. Activated microglia mediated neuronal cell death injury via a nitric oxide mechanism. J Immunol. 1992;149:2736–2741. [PubMed] [Google Scholar]
- 39.Black RA, Kronheim SR, Cantrell M, Deeley MC, March CJ, Prickett KS, Wignall J, Conlon PJ, Cosman D, Hopp TP, Mochizuki DY. Generation of biologically active interleukin-1β by proteolytic cleavage of the inactive precursor. J Biol Chem. 1988;263:9437–9442. [PubMed] [Google Scholar]
- 40.Kostura MJ, Tocci MJ, Limjuco G, Chin J, Cameron P, Hillman AG, Chartrain NA, Schmidt JA. Identification of a monocyte specific pre-interleukin 1β convertase activity. Proc Natl Acad Sci USA. 1989;86:5227–5231. doi: 10.1073/pnas.86.14.5227. [DOI] [PMC free article] [PubMed] [Google Scholar]