

Abstract
The N-glycan-dependent quality control of glycoprotein folding prevents endoplasmic reticulum to Golgi exit of folding intermediates, irreparably misfolded glycoproteins and not completely assembled multimeric complexes. It also enhances folding efficiency by preventing aggregation and facilitating formation of proper disulfide bonds. The control mechanism essentially involves four components, resident lectin-chaperones that recognize monoglucosylated polymannose glycans, a lectin-associated oxidoreductase acting on monoglucosylated glycoproteins, a glucosyltransferase and a glucosidase that creates monoglucosylated epitopes in glycans transferred in protein N-glycosylation or removes the glucose units added by the glucosyltransferase. This last enzyme is the only mechanism component sensing glycoprotein conformations as it creates monoglucosylated glycans exclusively in not properly folded species or in not completely assembled complexes The purpose of the review is to describe the most significant recent findings on the mechanism of glycoprotein folding and assembly quality control and to discuss the main still unanswered questions.
Keywords: Glycoprotein folding, Quality control, Endoplasmic reticulum, Lectins, Glucosyltransferase
1. Introduction
The accepted model for the glycoprotein folding quality control mechanism was proposed by Ari Helenius and co-workers several years ago [24]. It was a robust proposal as, with the addition of a few players as calreticulin (CRT), a soluble lectin highly homologous to calnexin (CNX) and ERp57, a glycoprotein specific protein oxidoreductase, the model is still valid today (Fig. 1). Briefly, immediately after a nascent polypeptide chain entering the endoplasmic reticulum (ER) lumen is N- glycosylated by the “en bloc” addition of Glc3Man9GlcNAc2 (Fig. 2), processing of the glycan starts by the glucosidase I-mediated removal of the outermost glucose residue (residue n, Fig. 2). This is followed by removal of the middle glucose (residue m, Fig. 2) by glucosidase II (GII), thus exposing the innermost glucose unit (residue l, Fig. 2). The epitope Glc1Man9GlcNAc2 may be then recognized by two ER resident lectins, CNX, a membrane bound protein, or by its soluble homologue, CRT. Further removal of residue l by GII liberates glycoproteins from the lectin anchors. The glycan Man9GlcNAc2 may be then reglucosylated by the UDP-Glc:glycoprotein glucosyltransferase (GT for glucosyltransferase), an UDP-Glc-dependent enzyme, thus recreating the monoglucosylated epitope and triggering the reassociation of glycoproteins with the lectins. GT behaves as a sensor of glycoprotein conformation as it only glucosylates glycoproteins not displaying their native 3D structures. In addition, it glucosylates incompletely assembled multimeric complexes containing properly folded subunits. Lectin-glycoprotein binding and unbinding, catalyzed by the opposing activities of GT and GII, continues until glycoproteins either acquire their native structures or, alternatively are recognized by cells as irreparably misfolded species or as complexes unable to acquire the full subunit complement. The first situation results in their permanent liberation as GT is unable to reglucosylate properly folded glycoproteins. Alternatively, irreparably misfolded species or incompletely assembled complexes are pulled out from the so called CNX/CRT cycle by a still ill defined mechanism and routed for degradation (ERAD, for ER associated degradation). CNX/CRT-glycoprotein interaction prevents Golgi exit of folding intermediates, of irreparably misfolded species or of incompletely assembled multimeric complexes and although it slows the folding rate it increases glycoprotein folding efficiency by preventing aggregation and facilitating formation of proper disulfide bonds. This last task is reserved to ERp57, an enzyme of the protein disulfide isomerase (PDI) family loosely associated with CNX/CRT. GT is the only element of the mechanism recognizing protein conformations. CNX and CRT are unconventional chaperones, directly sensing N-glycan and not protein structures as classical chaperones do. Energy required for the CNX/CRT cycle is not directly provided by ATP, as in folding mechanisms assisted by classical chaperones, but by a nucleotide sugar, UDP-Glc. As there are excellent comprehensive reviews on the subject, the purpose of the present one will be to outline only the most recent findings, with special emphasis on still unanswered queries [26, 43].
Fig. 1.
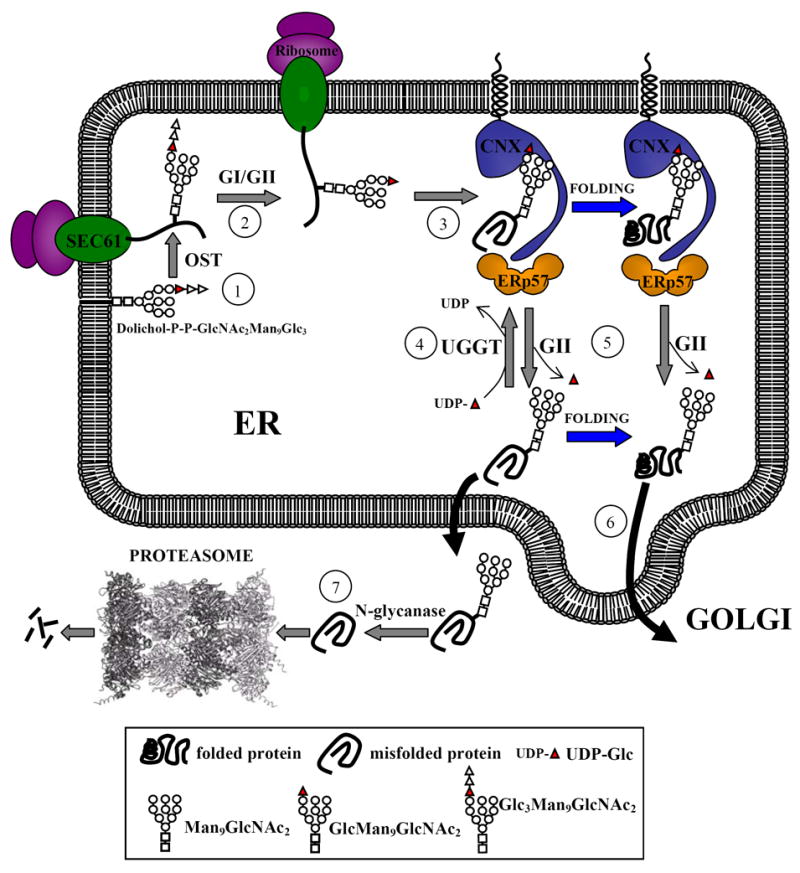
Model proposed for the quality control of glycoprotein folding. Proteins entering the ER are N-glycosylated by the oligosaccharyltransferase (OST) as they emerge from the translocon (Sec61) (1). Two glucoses are removed by the sequential action of glucosidase I and GII to generate monoglucosylated species (2) that are recognized by CNX and/or CRT (only CNX is shown), that are associated with ERp57 (3). The complex between the lectins and folding intermediates/misfolded glycoproteins dissociates upon removal of the last glucose by GII, and is reformed by GT activity (4). Once glycoproteins have acquired their native conformations, either free or complexed with the lectins, GII hydrolyses the remaining glucose residue and releases the glycoproteins from the lectin anchors (5). These species are not recognized by GT and are transported to the Golgi (6). Glycoproteins remaining in misfolded conformations are retrotranslocated to the cytosol where they are deglycosylated and degraded by the proteasome (7). One or more mannose residues may be removed during the whole folding process.
Fig. 2.
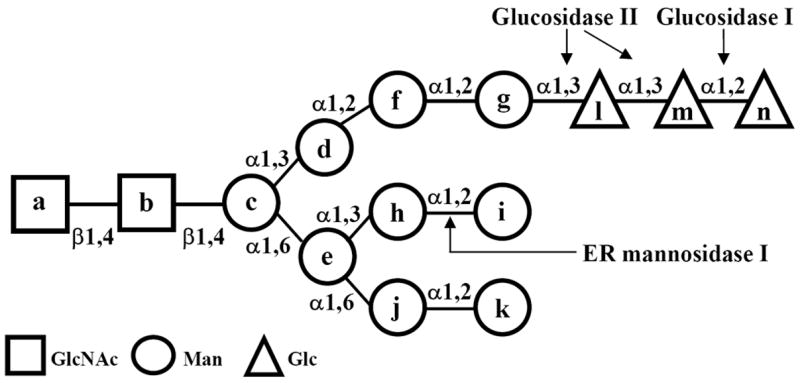
Structure of glycans. Lettering (a-n) follows the order of addition of monosaccharides in the synthesis of Glc3Man9GlcNAc2-P-P-dolichol. Glucosidase I removes residue n and GII residues l and m. GT adds residue l to residue g. The Man8GlcNAc2 isomer A (M8A) lacks residues g and l-n.
2. The UDP-Glc:glycoprotein glucosyltransferase (GT)
The title enzyme is a rather large (about 160 kDa) monomeric protein localized to the ER and in mammalian cells also to the ER-Golgi intermediate compartment (ERGIC) [79]. Unlike most glycosyltransferases of the secretory pathway, it is a soluble protein displaying a KDEL-like ER retention/retrieval signal at its C-terminus, the only exception known so far being the enzyme of the protozoon Trypanosoma cruzi, where it is retained in the ER by an unknown mechanism [8]. Recent advances on four points, namely the protein determinants recognized by GT, the maximal distance between the acceptor glycan and the glycoprotein structural perturbation required for enzymatic activity as well as what is known on the enzyme structure and the requirement of GT for single cell and multicellular organism viability will be developed below.
2.1. Protein determinants recognized by GT
As mentioned above, initial work had shown that GT glucosylated glycoproteins that do not display their native structures [61, 71]. Furthermore, it was reported that practically all glycoproteins were transiently glucosylated by the enzyme in the live cell [21]. This led to the realization that protein determinants recognized by GT could not be specific amino acid sequences. Initial work showed that GT recognized the innermost GlcNAc residue in the glycan (residue a, Fig. 2) [62]. As this residue is occluded in native (but not in denatured) conformations, where it interacts with neighboring amino acids, this result provided a rationale for the requirement of non native conformations for GT activity. Nevertheless, further work showed that the enzyme additionally required exposure of protein determinants as it preferred as substrates molten globule-like structures exposing patches of hydrophobic amino acids, and that these were the structural elements recognized by GT in folding intermediates and irreparably misfolded species. Fragments of a single domain neoglycoprotein, derived from chymotrypsin inhibitor 2, created by linking a Cys residue to the amino group of Asn-GlcNAc2Man9, were assayed as GT substrates [6]. The maximum values of catalytic efficiency (Vmax/Km) were observed for fragments permanently displaying molten globule-like conformations whereas much lower values were observed for those showing random coil or native structures. Moreover, plots of peptide length versus either catalytic efficiency or ANS (8-anilinonaphthalene-1-sulfonic acid) binding were superimposable [6, 7]. As this reagent is a probe for solvent exposure of hydrophobic patches, above mentioned results strongly suggest (if not demonstrate) that GT recognizes those protein determinants. Preference of GT for collapsed structures as those of molten globules over extended ones explains why BiP, a chaperone that recognizes hydrophobic heptapeptide domains in extended polypeptides, interacts with glycoproteins before CNX/CRT in cases in which both N-glycan independent and dependent chaperones participate in the in vivo folding process. Moreover, GT-mediated glucosylation of glycoproteins was also reported to proceed in vivo at the last and not initial folding stages [33]. Recognition of a hydrophobic environment in the vicinity of the glycan by GT was confirmed on assaying the acceptor capacities of glycopeptides displaying a variety of sequences and sizes (6–35 residues) [66].
In agreement with results observed in the live cell, it was shown that GT glucosylates incompletely assembled multimeric complexes on recognition of hydrophobic patches exposed by the absence of the complete subunit complement. Increase in ANS binding caused by the dissociation of soybean agglutinin (a homotetrameric complex having one N-glycan per monomer) upon addition of increasing urea concentrations was shown to parallel an increase in GT mediated glucosylation [31]. Furthermore, reassociation of the monomers upon withdrawing urea also resulted in a concomitant decrease in GT activity.
2.2. Maximal distance between the acceptor glycan and the glycoprotein structural perturbation required for enzymatic activity
There are conflicting reports concerning the distance from which the glycan has to be from the protein structural distortion to be glucosylated by GT. It was initially reported that GT only glucosylates N-glycans in the very near proximity of protein structural perturbations as only the N-glycan attached to the misfolded subunit of an artificial dimer formed by properly folded and misfolded RNase B monomers was glucosylated by GT in in vitro assays. The protein moiety of this enzyme is rather small, having 124 amino acids and a 4.2 nm maximal diameter [54]. Furthermore, based on the fact that elimination of one of the four disulfide bridges in pancreatic RNase A (the enzyme form devoid of glycans) produces a localized structural distortion, the same group introduced several consensus N-glycosylation sequences in the protein [55]. As only the N-glycan closest to the structural modification (N62) was glucosylated in vivo (although all of them were glycosylated), it was concluded that both elements (hydrophobic amino acid patches and the glycan) had to be in very close proximity to allow glucosylation. The design of the experiment, however, had a conceptual mistake: unglucosylated N-glycans corresponded to glycosylation sites present in loops where they were not expected to enhance the structural distortion introduced by the elimination of the disulfide bridge. On the other hand, N62 is in direct contact with the hydrophobic core of the protein. It should be expected a glycan in N62 to produce a significant structural perturbation. That is, the possibility exists that the N62 variant was effectively glucosylated not because of the proximity of the glycan to the structural perturbation but because the addition of the glycan in N62 produced a more drastic perturbation than in other positions. A different group introduced a point mutation (F280S) in a 51 kDa protein, an exo(1,3)-β-glucanase, resulting in a structural distortion that allowed glucosylation of a single glycan present in two distal alternative locations (N165 or N325), one of them close and the other distant to the structural perturbation [65]. As introduced mutations did not affect the enzymatic activity or modified the trypsin sensitivity of the proteins it was concluded that the 3D structures of the mutants were essentially similar to that of the wild type protein and that, therefore, the structural perturbation and the glycan could be in close proximity or far apart for allowing GT-mediated glucosylation. A model was proposed in which the catalytic and folding sensor domains of GT are separated by a flexible linker that can span both long and short distances between exposed hydrophobic patches and glucosylation sites.
2.3. The structure of GT
Although several groups have attempted to crystallize GT, efforts have been so far unsuccessful and, therefore, information about the structure of the enzyme is mainly derived from biochemical results. Sequence analysis of mammalian, insect and yeast GTs suggested that it is composed of at least two domains. The N-terminal domain comprises 80 % of the molecule, has no homology to other known proteins and is presumably involved in non-native conformer recognition and the C-terminal domain that binds [β-32P]5N3UDP-glucose and displays a similar size and significant homology to members of glycosyltransferase family 8 [67]. Members of this family conserve the anomeric configuration of the monosaccharide transferred from a sugar nucleotide, presumably by forming a monosaccharide-enzyme intermediate. All GT C-terminal domains from different species share a significant similarity (65–70 %), but no such similarity occus between the N-terminal domains. For instance, Rattus norvegicus and Drosophila melanogaster GT N-terminal domains share a 32.6 % similarity between them but they only show a respective 15.5 and 16.3 % similarity with the same portion of Schizosaccharomyces pombe GT. Although there are both structural and experimental evidence supporting the idea that the C-terminal domain is the catalytic portion of the enzyme, the often advanced notion that the N-terminal domain is responsible for recognition of non-native conformers has not been firmly established yet. Experimental work also supports the idea of a at least two domain GT structure as it was reported that N- and C-terminal domains from either Rattus norvegicus or S. pombe GTs remained tightly but not covalently bound upon mild proteolytic treatment and could not be separated without loss of enzymatic activity [23]. The notion of a two domain protein was reinforced by the synthesis of an active enzyme on transfection of S. pombe GT null mutants with two expression vectors, each of them encoding one of the domains. Transfection with the C-terminal domain-encoding vector alone yielded an inactive, rapidly degraded protein, thus indicating that the N-terminal domain is required for proper folding of the C-terminal catalytic portion. The picture of the structure of the enzyme that emerges from these results does not convincingly support either the model requiring a close proximity between glycans and structural perturbations for enzymatic activity or that allowing also a relatively long distance between both elements. On one hand, the tight association between N- and C-terminal domains may explain why only N-glycans in close proximity to protein structural perturbations are glucosylated by the enzyme but on the other, the flexible, protease sensitive linker between both domains would conceivably allow glucosylation of distant glycans if separation of both domains occurred exclusively during the glucosylation reaction. Although S. pombe and D. melanogaster GT N-terminal domains display an extremely poor similarity (16.3 %), chimeras containing either yeast N-terminal and fly C-terminal domains or the inverse construction were enzymatically and functionally active in vivo, thus indicating that the N-terminal domains of both GTs shared three-dimensional features [23].
Further insight on the structure of GT was provided by the characterization of the two GT homologues synthesized in human cells, HUGT1 and HUGT2 that share a 55 % identity (83 and 49 % in the C- and the N-terminal domains, respectively) [2]. Whereas both colocalized in the ER, the former but not the latter displayed GT activity and was upregulated under ER stress conditions. Surprisingly, a chimera containing HUGT1 N-terminal and HUGT2 C-terminal domains was enzymatically active and the C-terminal domain was able to bind UDP-Glc. It was speculated that the N-terminal domain somehow activated the C-terminal portion by making the catalytic site accessible to substrates, or alternatively, that the HUGT2 N-terminal domain conferred its own catalytic region specificity for a nucleotide sugar different from UDP-Glc (for instance UDP-GlcNAc). The role of HUGT2 as well as that of Kre5p, the inactive GT homologue present in Saccharomyces cerevisiae is presently unknown. It is evident that further work will be required for determining the structure of the protein and its fine mechanism of conformation recognition as well as the role of apparently enzymatically inactive GT homologues present in several cell types.
2.4. GT is required for viability of multicellular organisms
It was know for some time that GT was apparently not required for single cell viability as shown by mammalian cells in which absence of glucosidase I or GII totally precluded formation of monoglucosylated N-glycans [9, 52, 53]. Furthermore, disruption of the GT encoding gene in the yeast S. pombe revealed that in this microorganism the enzyme was only absolutely required when cells were under conditions of severe ER stress as glycoprotein underglycosylation (caused by transfer of Man9GlcNAc2 instead of Glc3Man9GlcNAc2) plus high temperature [17, 18]. These results strongly suggested that a significant proportion of most cellular glycoproteins may acquire their native structures without the assistance of the CNX/CRT cycle, probably using classical chaperones as BiP for alternative folding assistance and that only a few cellular glycoproteins, involved in very special processes absolutely required interaction with CNX/CRT for proper folding. Contrary to what happens in single cells, it was recently reported that disruption of both GT encoding alleles caused embryonic lethality in mice. The same is true for disruption of genes coding for ERp57 or CRT [22, 37, 39]. Although not embryonically lethal, CNX deletion results in premature death [12]. By comparing the time required for release from the CNX/CRT cycle and folding efficiency of a vesicular stomatitis virus G protein thermosensitive mutant (tsO45) expressed in normal and GT null mouse embryo fibroblasts it was concluded that this particular glycoprotein completed its folding process in a single round of interaction with CNX triggered by partial deglucosylation of the transferred N-glycan [39]. Analysis of additional glycoproteins is required to determine which glycoproteins (probably only a few) absolutely require several rounds of interaction with CNX/CRT, mediated by GT activity, for supporting mouse viability. It is worth mentioning that GT deletion in mouse embryo fibroblasts resulted in ER retention of ts045 G protein by alternative quality control mechanisms, mainly those involving BiP and disulfide-bound protein aggregates [39]. This might be a consequence of profound structural changes triggered in the substrate glycoprotein by the higher temperature because disruption of the GT encoding gene in Arabidopsis thaliana allowed a structurally imperfect yet biochemically competent brassinosteroid receptor mutant protein to reach the plasma membrane [27]. This constitutes an in vivo confirmation of cell free assays showing that GT is able to recognize glycoproteins displaying nearly native conformations [7]. Furthermore, this result suggests that the GT based quality control mechanism might be a disadvantage rather than a benefit in conformational diseases in which mutant glycoproteins are biologically active but displaying minimal structural defects.
3. Glucosidase II (GII)
3.1. The multiple roles of the GIIβ subunit
Unlike many glycosidases of the secretory pathway, GII is a soluble protein localized to the ER. From the yeast S. pombe to mammalian cells it is a heterodimeric complex formed by a catalytic or β subunit (107 kDa in mammalian cells), displaying significant similarity to members of glucosidase families 31 and 9, no KDEL-like sequence at its C-terminus, and a retaining or β subunit carrying a KDEL-like sequence [70]. The latter subunit is not required for the enzymatic activity of the former one when tested in cell free assays [69]. For several years it was assumed that the sole role of the β subunit was that of retaining the catalytic one in the ER but recent work suggests additional, more sophisticated functions. Disruption of either α or β subunit encoding genes in S. pombe totally abolished GII activity when assayed in vitro but although absence of GIIα resulted in the sole formation of diglucosylated glycans in vivo, minute amounts of monoglucosylated compounds were formed in GIIβ minus mutants, thus confirming the catalytic role of the alpha subunit [9]. The partial deglucosylation of the diglucosylated glycans in those mutants was tentatively ascribed to the transient presence of the GIIα subunit in the ER, en route to secretion. Nevertheless, it cannot be excluded that GIIβ is also required for proper GIIα folding and/or stability. A different result was obtained when disrupting a putative GIIβ subunit encoding gene in S. cerevisiae [76]. For several years it was assumed that the budding yeast GII lacked a retainingβ-subunit subunit but it was recently reported that its genome codes for a 702 amino acid protein (called Gtb1p for glucosidase two beta subunit) that shows a 21 % identity with human GIIβ. Moreover, near its C-terminus the protein displays a sequence with 27 % identity to the mannose 6-P receptor homology domain, including three completely conserved residues that have been implicated in mannose binding. Gtb1p localized to the ER and physically interacted with GIIα as revealed by co-immunoprecipitation but neither of them displayed a KDEL-like sequence at their C-termini. Disruption of Gtb1p encoding gene did not affect GIIα localization to the ER or conversion of diglucosylated to monoglucosylated glycans, but total removal of the innermost glucose unit (residue l, Fig. 2) was completely blocked. It was speculated that Gtb1p somehow modified the catalytic and/or substrate binding sites in the catalytic subunit, thus allowing formation of totally deglucosylated glycans.
It was reported several years ago that in many cases only glycoproteins having more than one N-glycan interacted with CNX/CRT [5, 56]. It was then assumed that the lectins were either monomeric with two binding sites per monomer or, alternatively, at least homodimeric complexes with one binding site per monomer. Further characterization of the lectin structure and binding features proved this assumption to be wrong as both CNX and CRT proved to be monomers with a single binding site per monomer [44]. The explanation of the observation, instead, lies in an interesting property of the mammalian cell GII: two glycans in the same glycoprotein molecule are apparently required for in vivo removing the middle glucose unit (residue m, Fig. 2) [13]. Several points should be described for fully understanding the mechanism proposed for explaining this requirement. Mammalian GIIβ has, the same as Gtb1p, a domain with homology to the mannose 6-P receptor. Sequence alignment indicates that all residues involved in mannose binding in the receptor are conserved in the GIIβ domain, except for the residues that interact with the phosphate group. Although GII-mediated cleavages of the middle (first cleavage) and innermost (second cleavage) glucose units display different catalytic rates, pH optima and inhibitor sensitivities, the enzyme has a single catalytic site that recognizes two terminal glucose units [1]. Finally, the NMR structure of the Glc3Man9GlcNAc2 glycan showed that the Glcα3Glc epitope (first cleavage) is exposed to the external face of the 3′ branch (residues d, f and g, Fig. 2), whereas the Glcα 3Man epitope (second cleavage) is on the internal side (that is, facing the 6′ branch, residues e and h-k in Fig. 2) [48]. The need to reorient the substrate probably prevents the consecutive trimming of both residues, thus allowing the glycoprotein to enter the CNX/CRT cycle when in the monoglucosylated form. It was then proposed that the GII has a basal glucosidase activity but that interaction of the 6′ branch of an N-glycan (glycan A) with the mannose 6-P receptor domain would induce a conformational change in the catalytic subunit, thus increasing the enzymatic activity and allowing the first cleavage to proceed at a neighboring N-glycan (glycan B) [13]. It is structurally impossible for the 6′ branch of the same N-glycan (glycan B) bearing the Glcα1,3Glc epitope to be cleaved, to bind the mannose 6-P receptor domain in GIIβ in a productive way because such binding would leave the epitope far from the catalytic site. On the other hand, two glycans would not be required for the second cleavage to proceed because interaction of the 6′ branch of glycan B with the receptor site would allow the second cleavage to occur as such branch and the Glcα1,3Man epitope lie on the same face. Nevertheless, there are exceptions and cases in which glycoproteins bearing a single N-glycan interact with CNX/CRT are known. These may be due to trans-activation of the first cleavage by an N-glycan in different glycoprotein molecules in the crowded ER environment or, for glycoproteins that have a rather long folding process, to GII basal activity. The proposed mechanism allows glycoproteins to enter the CNX/CRT cycle before total deglucosylation, that is, independently from GT activity.
It is remarkable that in S. cerevisiae, an organism that probably does not display a CNX/CRT cycle similar to that in other eukaryotic cells as it lacks both GT and CRT and the role of its CNX remains unclear, the non catalytic subunit is required for the formation of totally deglucosylated glycans. In cells having a proper CNX/CRT cycle, the same subunit promotes and regulates formation of monoglucosylated N-glycans, the key compounds for the entrance of glycoproteins to the cycle. It is also noteworthy that, as a safety device, mammalian (but not yeast) cells display an ERGIC and cis Golgi endomannosidase activity that converts Glc1Man9GlcNAc2 to GlcMan plus Man8GlcNAc2 isomer A (Fig. 2) to ensure that complex type N-glycans can be formed in compounds that have not been totally deglucosylated by GII [80].
4. Calnexin (CNX) and calreticulin (CRT)
Two relevant topics that were developed in recent years will be dealt with below, namely what is known now on the structure and sugar binding features of the lectins and the differential in vivo behavior of CNX and CRT.
4.1. Structure and sugar binding
CNX is a type-I integral membrane protein endowed with a transmembrane segment near its C-terminal end and a RKPRRE ER-localization signal located in a short cytosolic C-terminal tail. The crystal structure of CNX’s ectodomain reveals a globular domain highly resembling that found in the legume lectin family, from which protrudes a 140 Å long arm [58] (Fig. 3A). This last domain is a distinctive feature of this lectin that is shared only with CRT. The globular domain is organized in two antiparallel β sheets, one concave (six strands) and the other convex (seven strands), covering two separate regions of the primary sequence (residues 1–262 and 415–458). The glucose binding site lies on a shallow depression displayed on the surface of the β sheet that faces the arm domain, where the monosaccharide ring rests on Met189 and its hydroxyl groups are hydrogen bonded to Tyr165, Lys186 and Glu217. Several hydrogen bond donors and acceptors located adjacent to this site may contact the mannose residues of the glycan. The arm domain, also known as P-domain, is formed by a proline-rich tandem sequence repeats of four copies of motif 1, IxDP(D/E)(A/D)xKP(E/D)DWD(D/E), and four copies of motif 2, GxWxxPxIxNPxY. These motifs are arranged following a 11112222 pattern that can be divided into four 3D modules, each formed by a copy of motif 1 interacting head-to-tail with a copy of motif 2. The structure of the arm domain of CRT solved by NMR shows a highly flexible hairpin structure similar (but shorter, 110 Å) to that of CNX [16]. Accordingly, this domain displays a similar arrangement, where three tandem repeats of motif 1 and motif 2 are arranged following an 111222 pattern. Electron microscopy shows that the P-domain adopts various curved shapes, also illustrating its high flexibility [64]. Interestingly, the CNX crystal shows that the arm embraces the globular domain of a symmetry-related molecule (Fig. 3B), suggesting a similar disposition for the complex of the lectins with folding glycoproteins. In such arrangement the bound glycoprotein would be protected from interacting with other incompletely folded proteins, thus preventing their aggregation. Presumably the arm domain provides additional points of contact that stabilize the lectin-glycoprotein complex. In this sense, this domain is necessary for optimal lectin activity [34, 73] and also improves its chaperone ability [77]. Noticeably, the tip of the P-domain constitutes an autonomous folding unit, being one of the smallest natural sequences that form a stable non-helical fold in the absence of disulfide bonds or bound metals [15]. In addition, CNX binds a single calcium ion with relatively high affinity (Kd ~ 2 μM) [3]. The ion binding site is located in the globular domain, opposite to the sugar binding site, and presumably fulfills a structural role by stabilizing the lectin, but apparently it does not play a direct role in sugar binding.
Fig. 3.
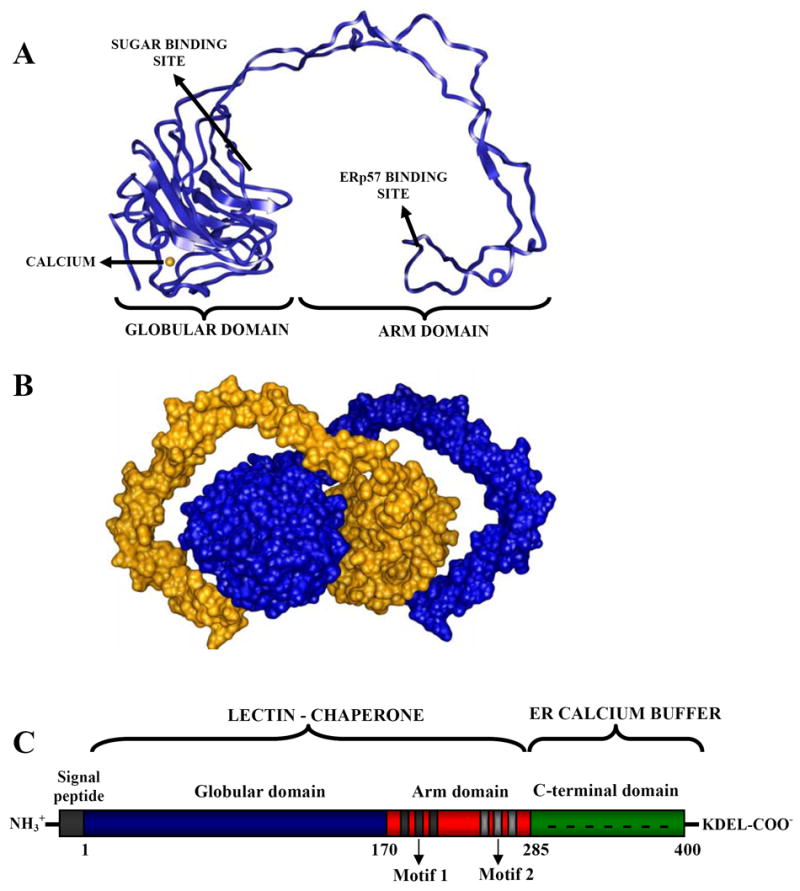
Structure of lectin-chaperones. (A) Crystal structure of the lumenal portion of CNX. (B) The arm domain of CNX embraces the globular domain of a symmetry related molecule. (C) Domain organization of CRT.
Since CRT and CNX share ~40 % sequence identity and present indistinguishable lectin specificity, a similar fold is predicted for CRT. There are two main differences between CRT and the ectodomain of CNX. Besides the above mentioned shorter arm domain, the former lectin has a highly acidic C-terminal domain (residues 285–400) that binds ~20 calcium ions with low affinity (Kd ~ 2 mM). This domain is involved in calcium storage in the lumen of the ER (Fig. 3C) [3] and is not essential for CRT lectin activity [46]. Given its very low hydrophobicity and the high content of charged residues, the C-terminal domain is predicted to adopt a natively unfolded structure. This idea is supported by its high susceptibility to proteases [4] and by direct observation with electron microscopy, as it appears loosely connected to the rest of the protein [64]. Surprisingly, the C-terminal domain has been implicated also in the retention of CRT in the ER, in an unusual mechanism that would operate in conjunction with the KDEL-based retention/retrieval system [60].
CRT and CNX specifically bind monoglucosylated high mannose glycans [75]. Although the terminal glucose is essential for binding, CRT affinity for this monosaccharide is very low. In addition, the following three mannoses present in the α1,3 branch make significant contributions to complex stability (Kb = 2.2 104 M−1, 56.3 104 M−1 and 102 104 M−1 for Glcα1,3Man, Glcα1,3Manα1,2Man and Glcα1,3Manα1,2Manα1,2Man, respectively) (Fig. 2) [30]. As a consequence, mutations in positions that bind the Glc residue (Y109 and D135 in rat CRT) are less tolerated than changes in residues that contact the following mannose residues [29]. CRT is unable to bind 2-deoxygucoseα1,3 mannose, showing the importance of the glucose equatorially oriented 2-hydroxyl group to establish the complex [29]. The α1,6-mannose branch point of the glycan core also contributes to sugar binding, since both lectins display substantial interaction with Glc1Man5GlcNAc2 but they are unable to bind Glc1Man4GlcNAc2 [63, 73]. Probably, the moderate affinity of CRT for Glc1Man9GlcNAc2 (Kb = 4.11 105 M−1) allows the activity of sugar modifying enzymes such as GII during the dissociation phase of the lectin-glycan equilibrium [44]. As expected, CRT and CNX share many amino acids involved in sugar binding, although this conservation is not absolute. For example, residues Y109, K111, Y128 and D317 (numbered following the sequence of rabbit CRT) are vital for both proteins to display lectin activity, but M131 and D160, also present in both lectins, are dispensable for CRT lectin activity [68].
4.2. Differential in vivo behavior of CNX and CRT
Although assays performed with pure proteins and glycans showed no differences in the binding properties of both lectins, glycoprotein interaction with either CNX or CRT may not be equivalent in vivo. Thus, the pattern of glycoproteins precipitated with CNX or CRT antisera from lysed mammalian cells only partially overlapped [47]. This difference might be related to both the soluble or membrane-bound respective status of CRT and CNX and to the relative position of glycans in membrane glycoproteins. It may be speculated that oligosaccharides located in the membrane proximity would more easily interact with CNX whereas those lumenally oriented would preferentially associate with CRT. In fact, similar patterns of glycoproteins were found to interact with CRT and a truncated, soluble CNX fragment, or with the full-length version of this last protein and CRT artificially anchored to the ER membrane by fusion with CNX or with an adenovirus glycoprotein transmembrane domain [11, 74]. Moreover, oligosaccharides located in the top/hinge domain of HA (a protein having 7 N-glycans), that is, in the more lumenally oriented portion of the molecule, associated preferentially with CRT, whereas CNX was less discriminating but mainly bound glycans close to the ER membrane [25]. A detailed study of the time sequence of CNX/CRT interaction with individual glycans showed that HA N-terminus remained membrane associated through nearly all the folding process by the interaction of a glycan in N8 and CNX [10]. Furthermore, apparently CNX and CRT association with the numerous glycans close to the N-terminus sterically prevented formation of a wrong disulfide bond between C14 and several cysteines until its normal partner (C466, near the C-terminus) appeared in the ER lumen. Formation of this disulfide bond was assumed to be facilitated by the proximity of C14 to the ER membrane. It was speculated that the clustering of glycans near the N-terminus has been an evolutionary requirement to ensure proper disulfide bond formation and folding. The notion that CNX preferentially binds membrane-close N-glycans agrees with the fact that interaction of the lectin with the human erythrocyte anion exchanger, a protein having 12–14 transmembrane segments, was not significantly modified when the single N-glycan present in wild type species was moved to other short lumenal loops [51].
Expression of viral and cellular glycoproteins in CRT null mouse embryo fibroblasts and in CNX deficient human T lymphoblastoid cells showed that loss of either CNX- or CRT-glycoprotein interaction or both (by addition of glucosidase inhibitors to the respective wild type cells) affected the process and outcome of glycoprotein production as well as the fidelity of quality control in a variety of ways [38]. Effects were seen in the folding rate (which was accelerated particularly when CRT was absent), in the folding efficiency (which was generally reduced), and in the retention of incompletely folded glycoproteins in the ER (which was affected only when association with both lectins was abolished). CNX seemed to be more important than CRT as folding assistant. Loss of CRT had, in fact, only marginal consequences, while loss of CNX resulted in a dramatic impairment of HA folding and in a more substantial elevation of other alternative ER resident chaperones, a symptom of ongoing ER stress.
Totally unexpected results were obtained on studying the interaction of CNX/CRT and other chaperones with cellular and viral glycoproteins expressed in cells lacking a functional CNX [49]. Three cellular glycoproteins were studied, one of them folding competent, membrane bound and having four N-glycans, and two folding incompetent variants of the same glycoprotein, both displaying two glycans, but one being membrane bound and the other soluble. These glycoproteins, that in wild type cells were CNX substrates, failed to interact with CRT when expressed in CNX null fibroblasts. Instead, they more strongly interacted with BiP. On the contrary, four viral glycoproteins (Semliki forest virus E1 and p62, vesicular stomatitis virus G and influenza virus HA) gave different results. The first two glycoproteins normally interact with both CNX and CRT, but in CNX minus cells they interacted more abundantly with CRT and their maturation proceeded normally. In the case of HA, a glycoprotein deeply dependent on CNX for successful maturation that normally interacts with both CNX and CRT, absence of the former lectin resulted in a persistent interaction with CRT. The most surprising result was obtained with G protein that normally interacts only with CNX. Infection of CNX deficient fibroblasts with VSV (viral infection was also used to express E1, p62 and HA) resulted in the interaction of G protein with CRT. As transfection of G protein failed to trigger its interaction with CRT, it was suggested that viral infection somehow subverted the normal glycoprotein recognition by the ER lectins. This result may explain why total inactivation of the CNX/CRT cycle affects viral replication and infectivity but not viability of mammalian cells. Additional expression of individual glycoproteins, both of cellular and viral origin and in this last case as a result of both viral infection and transfection must be studied to substantiate this very interesting finding.
5. The ERp57 oxidoreductase
CRT and CNX improve protein folding efficiency not only by preventing non-specific interactions but they also favor correct disulfide bridge pairing by presenting substrates to ERp57, a member of the PDI family associated to both lectins [41, 42]. This complex forms independently from ligand binding to the lectins, since it is also detected in cells lacking glucosidase I activity, that is, cells in which no glycoprotein-CNX/CRT interaction can be established [41]. ERp57 forms intermolecular disulfide bridges with glycoproteins bound to CRT or CNX, and usually inhibition of glycoprotein-lectin complex formation abrogates substrate association with ERp57 [14, 28, 40, 72]. Nevertheless, this correlation is not absolute, since in some cases ERp57 has been detected in complexes with ER proteins that are not associated with the lectins [36, 45]. ERp57 isomerase activity is enhanced when its substrates are bound to the lectins. For example, the ability of ERp57 to catalyze disulfide bridge acquisition of denatured monoglucosylated RNAse B in vitro improves in the presence of the lectins. Interestingly, the opposite effect is observed during refolding catalyzed by PDI [78] and while BiP enhances PDI activity, no effect is observed with respect to ERp57. These observations suggest that both enzymes have evolved to operate differently, PDI preferentially with the system centered in BiP, and ERp57 with that based on CNX/CRT [35]. ERp57 is composed of four thioredoxin-like domains (a, b, b’ and a’), in which the a and a’ domains display the active -CXXC- motifs [19, 59] and the b’ domain interacts with the lectins. This last association is mediated through residues 225–251 located on the tip of CRT’s arm [20, 32, 57], and a similar interaction has been shown for CNX [34, 50]. Presumably, the high flexibility of the arm domain allows the enzyme to scan for disulfide bridges located at distant positions. In addition, the moderate stability of the binary ERp57-CRT complex (Kd ~ 9 μnd its fast off-rate (koff >1000 s−1) probably enable ERp57 to rapidly sense for preexisting lectin-glycoprotein complexes [20]. Although the domain architecture of PDI and ERp57 is very similar, they use different strategies to bind their substrates, since the PDI b and b’ domains directly contact its substrates whereas the homologous ERp57 b’ domain has evolved to interact with the arm domains of CNX and CRT, which in turn present most known substrates to ERp57. In this sense, CRT/CNX constitute a system specially fitted to present glycosylated substrates to ERp57
6. Conclusions
Although major advances have been made on several aspects of the mechanism of quality control of glycoprotein folding, there still are significant issues that deserve further study as, for instance, the structure and intimate mechanism of recognition and glucosylation of misfolded glycoproteins by GT, the mechanisms by which apparently GIIβ subunit regulates formation and degradation of monoglucosylated glycans, the differential in vivo recognition of folding intermediates by CNX and CRT and the proposed subversion of the normal differential recognition by viral infection. Finally, another issue is the mechanism by which cells terminate futile cycles of lectin-glycoprotein association and dissociation and divert irreparably misfolded species to degradation. The field has entered into an exciting stage in which further structural and functional studies of the participants involved in quality control will undoubtedly provide answers to the many questions aroused.
Abreviations
- ANS
8-anilinonaphthalene-1-sulfonic acid
- CNX
calnexin
- CRT
calreticulin
- EDEM
Endoplasmic reticulum degradation enhancing α-mannosidase-like proteins
- ER
endoplasmic reticulum
- ERAD
ER associated degradation
- ERGIC
ER-Golgi intermediate compartment
- GII
glucosidase II
- GT, UDP-Glc
glycoprotein glucosyltransferase
- HA
hemagglutinin
- PDI
protein disulfide isomerase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Alonso JM, Santa-Cecilia A, Calvo P. Effect of bromoconduritol on glucosidase II from rat liver. A new kinetic model for the binding and hydrolysis of the substrate. Eur J Biochem. 1993;215:37–42. doi: 10.1111/j.1432-1033.1993.tb18004.x. [DOI] [PubMed] [Google Scholar]
- 2.Arnold SM, Kaufman RJ. The noncatalytic portion of human UDP-glucose:glycoprotein glucosyltransferase I confers UDP-glucose binding and transferase function to the catalytic domain. J Biol Chem. 2003;278:43320–8. doi: 10.1074/jbc.M305800200. [DOI] [PubMed] [Google Scholar]
- 3.Baksh S, Michalak M. Expression of calreticulin in Escherichia coli and identification of its Ca2+ binding domains. J Biol Chem. 1991;266:21458–65. [PubMed] [Google Scholar]
- 4.Bouvier M, Stafford WF. Probing the three-dimensional structure of human calreticulin. Biochemistry. 2000;39:14950–59. doi: 10.1021/bi0019545. [DOI] [PubMed] [Google Scholar]
- 5.Cannon KS, Hebert DN, Helenius A. Glycan-dependent and -independent association of vesicular stomatitis virus G protein with calnexin. J Biol Chem. 1996;271:14280–4. doi: 10.1074/jbc.271.24.14280. [DOI] [PubMed] [Google Scholar]
- 6.Caramelo JJ, Castro OA, Alonso LG, de Prat-Gay G, Parodi AJ. UDP-Glc:glycoprotein glucosyltransferase recognizes structured and solvent accessible hydrophobic patches in molten globule-like folding intermediates. Proc Natl Acad Sci USA. 2003;100:86–91. doi: 10.1073/pnas.262661199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Caramelo JJ, Castro OA, de Prat-Gay G, Parodi AJ. The endoplasmic reticulum glucosyltransferase recognizes nearly native glycoprotein folding intermediate. J Biol Chem. 2004;279:46280–85. doi: 10.1074/jbc.M408404200. [DOI] [PubMed] [Google Scholar]
- 8.Conte I, Labriola C, Cazzulo JJ, Docampo R, Parodi AJ. The interplay between folding facilitating mechanisms in Trypanosoma cruzi endoplasmic reticulum . Mol Biol Cell. 2003;14:3529–40. doi: 10.1091/mbc.E03-04-0228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.D’Alessio C, Fernández F, Trombetta ES, Parodi AJ. Genetic evidence for the heterodimeric structure of glucosidase II. The effect of disrupting the subunit-encoding genes on glycoprotein folding. J Biol Chem. 1999;274:25899–905. doi: 10.1074/jbc.274.36.25899. [DOI] [PubMed] [Google Scholar]
- 10.Daniels R, Kurowski B, Johnson AE, Hebert DN. N-linked glycans direct the cotranslational folding pathway of influenza hemagglutinin. Mol Cell. 2003;11:79–90. doi: 10.1016/s1097-2765(02)00821-3. [DOI] [PubMed] [Google Scholar]
- 11.Danilczyk UG, Cohen-Doyle MF, Williams DB. Functional relationship between calreticulin, calnexin, and the endoplasmic reticulum luminal domain of calnexin. J Biol Chem. 2000;275:13089–97. doi: 10.1074/jbc.275.17.13089. [DOI] [PubMed] [Google Scholar]
- 12.Denzel A, Molinari M, Trigueros C, Martin JE, Velmurgan S, Brown S, Stamp G, Owen MJ. Early postnatal death and motor disorders in mice congenitally deficient in calnexin expression. Mol Cell Biol. 2002;22:7398–404. doi: 10.1128/MCB.22.21.7398-7404.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deprez P, Gautschi M, Helenius A. More than one glycan is needed for ER glucosidase II to allow entry of glycoproteins into the calnexin/calreticulin cycle. Mol Cell. 2005;19:183–95. doi: 10.1016/j.molcel.2005.05.029. [DOI] [PubMed] [Google Scholar]
- 14.Di Jeso B, Park YN, Ulianich L, Treglia AS, Urbanas ML, High S, Arvan P. Mixed-disulfide folding intermediates between thyroglobulin and endoplasmic reticulum resident oxidoreductases ERp57 and protein disulfide isomerase. Mol Cell Biol. 2005;25:9793–805. doi: 10.1128/MCB.25.22.9793-9805.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ellgaard L, Bettendorff P, Braun D, Herrmann T, Fiorito F, Jelesarov I, Guntert P, Helenius A, Wuthrich K. NMR structures of 36 and 73-residue fragments of the calreticulin P-domain. J Mol Biol. 2002;322:773–84. doi: 10.1016/s0022-2836(02)00812-4. [DOI] [PubMed] [Google Scholar]
- 16.Ellgaard L, Riek R, Herrmann T, Guntert P, Braun D, Helenius A, Wuthrich K. NMR structure of the calreticulin P-domain. Proc Natl Acad Sci USA. 2001;98:3133–8. doi: 10.1073/pnas.051630098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fanchiotti S, Fernández F, D’Alessio C, Parodi AJ. The UDP-Glc:Glycoprotein glucosyltransferase is essential for Schizosaccharomyces pombe viability under conditions of extreme endoplasmic reticulum stress. J Cell Biol. 1998;143:625–35. doi: 10.1083/jcb.143.3.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fernández F, Jannatipour M, Hellman U, Rokeach L, Parodi AJ. A new stress protein: synthesis of Schizosaccharomyces pombe UDP--Glc:glycoprotein glucosyltransferase mRNA is induced by stress conditions but the enzyme is not essential for cell viability. EMBO J. 1996;15:705–13. [PMC free article] [PubMed] [Google Scholar]
- 19.Frickel EM, Frei P, Bouvier M, Stafford WF, Helenius A, Glockshuber R, Ellgaard L. ERp57 is a multifunctional thiol-disulfide oxidoreductase. J Biol Chem. 2004;279:18277–87. doi: 10.1074/jbc.M314089200. [DOI] [PubMed] [Google Scholar]
- 20.Frickel EM, Riek R, Jelesarov I, Helenius A, Wuthrich K, Ellgaard L. TROSY-NMR reveals interaction between ERp57 and the tip of the calreticulin P-domain. Proc Natl Acad Sci U S A. 2002;99:1954–9. doi: 10.1073/pnas.042699099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gañán S, Cazzulo JJ, Parodi AJ. A major proportion of N-glycoproteins is transiently glucosylated in the endoplasmic reticulum . Biochemistry. 1991;30:3098–104. doi: 10.1021/bi00226a017. [DOI] [PubMed] [Google Scholar]
- 22.Garbi N, Tanaka S, Momburg F, Hammerling GF. Impaired assembly of the major histocompatibility complex class I peptide-loading complex in mice deficient in the oxidoreductase ERp57. Nat Immunol. 2006;7:93–102. doi: 10.1038/ni1288. [DOI] [PubMed] [Google Scholar]
- 23.Guerin M, Parodi AJ. The UDP-Glc:glycoprotein glucosyltransferase is organized in at least two tightly bound domains from yeasts to mammals. J Biol Chem. 2003;278:20540–6. doi: 10.1074/jbc.M300891200. [DOI] [PubMed] [Google Scholar]
- 24.Hammond C, Braakman I, Helenius A. Role of N-linked oligosaccharide recognition, glucose trimming, and calnexin in glycoprotein folding and quality control. Proc Natl Acad USA. 1994;91:913–7. doi: 10.1073/pnas.91.3.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hebert DN, Zhang JX, Chen W, Foellmer B, Helenius A. The number and location of glycans on influenza hemagglutinin determine folding and association with calnexin and calreticulin. J Cell Biol. 1997;139:613–23. doi: 10.1083/jcb.139.3.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Helenius A, Aebi M. Roles of N-linked glycans in the endoplasmic reticulum. Annu Rev Biochem. 2004;73:1019–49. doi: 10.1146/annurev.biochem.73.011303.073752. [DOI] [PubMed] [Google Scholar]
- 27.Jin H, Yan Z, Nam KH, Li J. Allele-specific suppression of a defective brassinosteroid receptor reveals a physiological role of UGGT in ER quality control. Mol Cell. 2007;26:821–30. doi: 10.1016/j.molcel.2007.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kang SJ, Cresswell P. Calnexin, calreticulin, and ERp57 cooperate in disulfide bond formation in human CD1d heavy chain. J Biol Chem. 2002;277:44838–44. doi: 10.1074/jbc.M207831200. [DOI] [PubMed] [Google Scholar]
- 29.Kapoor M, Ellgaard L, Gopalakrishnapai J, Schirra C, Gemma E, Oscarson S, Helenius A, Surolia A. Mutational analysis provides molecular insight into the carbohydrate-binding region of calreticulin: pivotal roles of tyrosine-109 and aspartate-135 in carbohydrate recognition. Biochemistry. 2004;43:97–106. doi: 10.1021/bi0355286. [DOI] [PubMed] [Google Scholar]
- 30.Kapoor M, Srinivas H, Kandiah E, Gemma E, Ellgaard L, Oscarson S, Helenius A, Surolia A. Interactions of substrate with calreticulin, an endoplasmic reticulum chaperone. J Biol Chem. 2003;278:6194–200. doi: 10.1074/jbc.M209132200. [DOI] [PubMed] [Google Scholar]
- 31.Keith N, Parodi AJ, Caramelo JJ. Glycoprotein tertiary and quarternary structures are monitored by the same quality control mechanism. J Biol Chem. 2005;280:18138–41. doi: 10.1074/jbc.M501710200. [DOI] [PubMed] [Google Scholar]
- 32.Kozlov G, Maattanen P, Schrag JD, Pollock S, Cygler M, Nagar B, Thomas DY, Gehring K. Crystal structure of the bb' domains of the protein disulfide isomerase ERp57. Structure. 2006;14:1331–9. doi: 10.1016/j.str.2006.06.019. [DOI] [PubMed] [Google Scholar]
- 33.Labriola C, Cazzulo JJ, Parodi AJ. Trypanosoma cruzi calreticulin is a lectin that binds monoglucosylated oligosaccharides but not protein moieties of glycoproteins. Mol Biol Cell. 1999;10:1381–96. doi: 10.1091/mbc.10.5.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leach MR, Cohen-Doyle MF, Thomas DY, Williams DB. Localization of the lectin, ERp57 binding, and polypeptide binding sites of calnexin and calreticulin. J Biol Chem. 2002;277:29686–97. doi: 10.1074/jbc.M202405200. [DOI] [PubMed] [Google Scholar]
- 35.Mayer M, Frey S, Koivunen P, Myllyharju J, Buchner J. Influence of the oxidoreductase ERp57 on the folding of an antibody fab fragment. J Mol Biol. 2004;34:1077–84. doi: 10.1016/j.jmb.2004.06.068. [DOI] [PubMed] [Google Scholar]
- 36.McCormick LM, Urade R, Arakaki Y, Schwartz AL, Bu G. Independent and cooperative roles of N-glycans and molecular chaperones in the folding and disulfide bond formation of the low-density lipoprotein (LDL) receptor-related protein. Biochemistry. 2005;44:5794–803. doi: 10.1021/bi047652a. [DOI] [PubMed] [Google Scholar]
- 37.Mesaeli N, Nakamura K, Zvaritch E, Dickie P, Dziak E, Krause KH, Opas M, MacLennan DH, Michalak M. Calreticulin is essential for cardiac development. J Cell Biol. 1999;144:857–68. doi: 10.1083/jcb.144.5.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Molinari M, Eriksson KK, Calanca V, Galli C, Cresswell P, Michalak M, Helenius A. Contrasting functions of calreticulin and calnexin in glycoprotein folding and ER quality control. Mol Cell. 2004;13:125–35. doi: 10.1016/s1097-2765(03)00494-5. [DOI] [PubMed] [Google Scholar]
- 39.Molinari M, Galli C, Vanoni O, Arnold SM, Kaufman RJ. Persistent glycoprotein misfolding activates the glucosidase II/UGT1-driven calnexin cycle to delay aggregation and loss of folding competence. Mol Cell. 2005;20:503–12. doi: 10.1016/j.molcel.2005.09.027. [DOI] [PubMed] [Google Scholar]
- 40.Morrice NA, Powis SJ. A role for the thiol-dependent reductase ERp57 in the assembly of MHC class I molecules. Curr Biol. 1998;8:713–6. doi: 10.1016/s0960-9822(98)70279-9. [DOI] [PubMed] [Google Scholar]
- 41.Oliver JD, Roderick HL, Llewellyn DH, High S. ERp57 functions as a subunit of specific complexes formed with the ER lectins calreticulin and calnexin. Mol Biol Cell. 1999;10:2573–82. doi: 10.1091/mbc.10.8.2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oliver JD, van der Wal FJ, Bulleid NJ, High S. Interaction of the thiol-dependent reductase ERp57 with nascent glycoproteins. Science. 1997;275:86–8. doi: 10.1126/science.275.5296.86. [DOI] [PubMed] [Google Scholar]
- 43.Parodi AJ. Protein glucosylation and its role in protein folding. Annu Rev Biochem. 2000;69:69–93. doi: 10.1146/annurev.biochem.69.1.69. [DOI] [PubMed] [Google Scholar]
- 44.Patil AR, Thomas CJ, Surolia A. Kinetics and the mechanism of interaction of the endoplasmic reticulum chaperone, calreticulin, with monoglucosylated (Glc1Man9GlcNAc2) substrate. J Biol Chem. 2000;275:24348–56. doi: 10.1074/jbc.M003102200. [DOI] [PubMed] [Google Scholar]
- 45.Peaper DR, Wearsch PA, Cresswell P. Tapasin and ERp57 form a stable disulfide-linked dimer within the MHC class I peptide-loading complex. EMBO J. 2005;24:3613–23. doi: 10.1038/sj.emboj.7600814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Peterson JR, Helenius A. In vitro reconstitution of calreticulin-substrate interactions. J Cell Sci. 1999;112:2775–84. doi: 10.1242/jcs.112.16.2775. [DOI] [PubMed] [Google Scholar]
- 47.Peterson JR, Ora A, Van PN, Helenius A. Transient, lectin-like association of calreticulin with folding intermediates of cellular and viral glycoproteins. Mol Biol Cell. 1995;6:1173–84. doi: 10.1091/mbc.6.9.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Petrescu AJ, Butters TD, Reinkensmeier G, Petrescu S, Platt FM, Dwek RA, Wormald MR. The solution NMR structure of glucosylated N-glycans involved in the early stages of glycoprotein biosynthesis and folding. EMBO J. 1997;16:4302–10. doi: 10.1093/emboj/16.14.4302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pieren M, Galli C, Denzel A, Molinari M. The use of calnexin and calreticulin by cellular and viral glycoproteins. J Biol Chem. 2005;280:28265–71. doi: 10.1074/jbc.M501020200. [DOI] [PubMed] [Google Scholar]
- 50.Pollock S, Kozlov G, Pelletier MF, Trempe JF, Jansen G, Sitnikov D, Bergeron JJ, Gehring K, Ekiel I, Thomas DY. Specific interaction of ERp57 and calnexin determined by NMR spectroscopy and an ER two-hybrid system. EMBO J. 2004;23:1020–9. doi: 10.1038/sj.emboj.7600119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Popov M, Reithmeier RA. Calnexin interaction with N-glycosylation mutants of a polytopic membrane glycoprotein, the human erythrocyte anion exchanger 1 (band 3) J Biol Chem. 1999;274:17635–42. doi: 10.1074/jbc.274.25.17635. [DOI] [PubMed] [Google Scholar]
- 52.Ray MK, Yang J, Sundaram S, Stanley P. A novel glycosylation phenotype expressed by Lec23, a Chinese hamster ovary mutant deficient in α-glucosidase I. J Biol Chem. 1991;266:22818–25. [PubMed] [Google Scholar]
- 53.Reitman ML, Trowbridge LS, Kornfeld S. A lectin-resistant mouse lymphoma cell line is deficient in glucosidase II, a glycoprotein-processing enzyme. J Biol Chem. 1982;257:10357–63. [PubMed] [Google Scholar]
- 54.Ritter C, Helenius A. Recognition of local glycoprotein misfolding by the ER folding sensor UDP-glucose:glycoprotein glucosyltransferase. Nat Struct Biol. 2000;7:278–80. doi: 10.1038/74035. [DOI] [PubMed] [Google Scholar]
- 55.Ritter C, Quirin K, Kowarik M, Helenius A. Minor folding defects trigger local modification of glycoprotein by the ER folding sensor GT. EMBO J. 2005;24:1730–8. doi: 10.1038/sj.emboj.7600645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rodan AR, Simons JF, Trombetta ES, Helenius A. N-linked oligosaccharides are necessary and sufficient for association of glycosylated forms of bovine RNase with calnexin and calreticulin. EMBO J. 1996;15:6921–30. [PMC free article] [PubMed] [Google Scholar]
- 57.Russell SJ, Ruddock LW, Salo KE, Oliver JD, Roebuck QP, Llewellyn DH, Roderick HL, Koivunen P, Myllyharju J, High S. The primary substrate binding site in the b' domain of ERp57 is adapted for endoplasmic reticulum lectin association. J Biol Chem. 2004;279:18861–9. doi: 10.1074/jbc.M400575200. [DOI] [PubMed] [Google Scholar]
- 58.Schrag JD, Bergeron JJ, Li Y, Borisova S, Hahn M, Thomas DY, Cygler M. The structure of calnexin, an ER chaperone involved in quality control of protein folding. Mol Cell. 2001;8:633–44. doi: 10.1016/s1097-2765(01)00318-5. [DOI] [PubMed] [Google Scholar]
- 59.Silvennoinen L, Myllyharju J, Ruoppolo M, Orru S, Caterino M, Kivirikko KI, Koivunen P. Identification and characterization of structural domains of human ERp57: association with calreticulin requires several domains. J Biol Chem. 2004;279:13607–15. doi: 10.1074/jbc.M313054200. [DOI] [PubMed] [Google Scholar]
- 60.Sonnichsen B, Fullekrug J, Nguyen Van P, Diekmann W, Robinson DG, Mieskes G. Retention and retrieval: both mechanisms cooperate to maintain calreticulin in the endoplasmic reticulum. J Cell Sci. 1994;107:2705–17. doi: 10.1242/jcs.107.10.2705. [DOI] [PubMed] [Google Scholar]
- 61.Sousa M, Ferrero-García MA, Parodi AJ. Recognition of the oligosaccharide and protein moieties of glycoproteins by the UDP-Glc:glycoprotein glucosyltransferase. Biochemistry. 1992;31:97–105. doi: 10.1021/bi00116a015. [DOI] [PubMed] [Google Scholar]
- 62.Sousa M, Parodi AJ. The molecular basis for the recognition of misfolded glycoproteins by the UDP-Glc:glycoprotein glucosyltransferase. EMBO J. 1995;14:4196–203. doi: 10.1002/j.1460-2075.1995.tb00093.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Spiro RG, Zhu Q, Bhoyroo V, Soling HD. Definition of the lectin-like properties of the molecular chaperone, calreticulin, and demonstration of its copurification with endomannosidase from rat liver Golgi. J Biol Chem. 1996;271:11588–94. doi: 10.1074/jbc.271.19.11588. [DOI] [PubMed] [Google Scholar]
- 64.Tan Y, Chen M, Li Z, Mabuchi K, Bouvier M. The calcium- and zinc-responsive regions of calreticulin reside strictly in the N-/C-domain. Biochim Biophys Acta. 2006;1760:745–53. doi: 10.1016/j.bbagen.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 65.Taylor SC, Ferguson AD, Bergeron JJM, Thomas DY. The ER protein folding sensor UDP-glucose glycoprotein-glucosyltransferase modifies substrates distant to local changes in glycoprotein conformation. Nat Struct Mol Biol. 2004;11:128–34. doi: 10.1038/nsmb715. [DOI] [PubMed] [Google Scholar]
- 66.Taylor SC, Thibaut P, Tessier DC, Bergeron JJM, Thomas DY. Glycopeptide specificity of the secretory protein folding sensor UDP-glucose glycoprotein:glucosyltransferase. EMBO Rep. 2003;4:405–11. doi: 10.1038/sj.embor.embor797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tessier DC, Dignard D, Zapun A, Radominska-Pandya A, Parodi AJ, Bergeron JJM, Thomas DY. Cloning and characterization of mammalian UDP-glucose glycoprotein:glucosyltransferase and the development of a specific substrate for the enzyme. Glycobiology. 2000;10:403–12. doi: 10.1093/glycob/10.4.403. [DOI] [PubMed] [Google Scholar]
- 68.Thomson SP, Williams DB. Delineation of the lectin site of the molecular chaperone calreticulin. Cell Stress Chaperon. 2005;10:242–51. doi: 10.1379/CSC-126.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Trombetta ES, Fleming KG, Helenius A. Quaternary and domain structure of glycoprotein processing glucosidase II. Biochemistry. 2001;40:10717–22. doi: 10.1021/bi010629u. [DOI] [PubMed] [Google Scholar]
- 70.Trombetta ES, Simons JF, Helenius A. Endoplasmic reticulum glucosidase II is composed of a catalytic subunit, conserved from yeast to mammals, and a tightly bound noncatalytic HDEL-containing subunit. J Biol Chem. 1996;271:27509–16. doi: 10.1074/jbc.271.44.27509. [DOI] [PubMed] [Google Scholar]
- 71.Trombetta S, Bosch M, Parodi AJ. Glucosylation of glycoproteins by mammalian, plant, fungal and trypanosomatid protozoa microsomal membranes. Biochemistry. 1989;28:8108–16. doi: 10.1021/bi00446a022. [DOI] [PubMed] [Google Scholar]
- 72.Van der Wal FJ, Oliver JD, High S. The transient association of ERp57 with N-glycosylated proteins is regulated by glucose trimming. Eur J Biochem. 1998;256:51–59. doi: 10.1046/j.1432-1327.1998.2560051.x. [DOI] [PubMed] [Google Scholar]
- 73.Vassilakos A, Michalak M, Lehrman MA, Williams DB. Oligosaccharide binding characteristics of the molecular chaperones calnexin and calreticulin. Biochemistry. 1998;37:3480–90. doi: 10.1021/bi972465g. [DOI] [PubMed] [Google Scholar]
- 74.Wada I, Imai S, Kai M, Sakane F, Kanoh H. Chaperone function of calreticulin when expressed in the endoplasmic reticulum as the membrane-anchored and soluble forms. J Biol Chem. 1995;270:20298–304. doi: 10.1074/jbc.270.35.20298. [DOI] [PubMed] [Google Scholar]
- 75.Ware FE, Vassilakos A, Peterson PA, Jackson MR, Lehrman MA, Williams DB. The molecular chaperone calnexin binds Glc1Man9GlcNAc2 oligosaccharide as an initial step in recognizing unfolded glycoproteins. J Biol Chem. 1995;270:4697–704. doi: 10.1074/jbc.270.9.4697. [DOI] [PubMed] [Google Scholar]
- 76.Wilkinson BM, Purswani J, Stirling CJ. Yeast GTB1 encodes a subunit of glucosidase II required for glycoprotein processing in the endoplasmic reticulum. J Biol Chem. 2005;281:6325–33. doi: 10.1074/jbc.M510455200. [DOI] [PubMed] [Google Scholar]
- 77.Xu X, Azakami H, Kato A. P-domain and lectin site are involved in the chaperone function of Saccharomyces cerevisiae calnexin homologue. FEBS Lett. 2004;570:155–60. doi: 10.1016/j.febslet.2004.06.039. [DOI] [PubMed] [Google Scholar]
- 78.Zapun A, Darby NJ, Tessier DC, Michalak M, Bergeron JJ, Thomas DY. Enhanced catalysis of ribonuclease B folding by the interaction of calnexin or calreticulin with ERp57. J Biol Chem. 1998;273:6009–12. doi: 10.1074/jbc.273.11.6009. [DOI] [PubMed] [Google Scholar]
- 79.Zuber C, Fan JY, Guhl B, Parodi A, Fessler JH, Parker C, Roth J. Immunolocalization of UDP-glucose:glycoprotein glucosyltransferase indicates involvement of pre-Golgi intermediates in protein quality control. Proc Natl Acad Sci USA. 2001;98:10710–15. doi: 10.1073/pnas.191359198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zuber C, Spiro MJ, Guhl B, Spiro RG, Roth J. Golgi apparatus immunolocalization of endomannosidase suggests post-endoplasmic reticulum glucose trimming: implications for quality control. Mol Biol Cell. 2000;11:4227–40. doi: 10.1091/mbc.11.12.4227. [DOI] [PMC free article] [PubMed] [Google Scholar]