

Abstract
Multiple sclerosis is an autoimmune disease thought to be mediated by CD4+ T helper cells (Th). Experimental autoimmune encephalomyelitis is a rodent model of multiple sclerosis and has been used extensively to explore a variety of immunotherapies using soluble protein or peptide antigens. The underlying mechanisms of such therapy have been attributed to induction of T cell anergy, a switch in Th1 to Th2 responses, or peripheral deletion of autoreactive T cells. In this study, we have developed transgenic mice expressing a T cell receptor (TCR) specific for the NH2-terminal peptide Ac1-11 of the autoantigen myelin basic protein to explore the mechanism of soluble peptide therapy. T cells from these mice are highly skewed toward the CD4 population and have an abnormal thymic architecture, a phenomenon found in other TCR transgenic mice that exhibit a highly skewed CD4/CD8 ratio. Soluble Ac1-11 or the analogues Ac1-11[4A] or Ac1-11[4Y] (which bind to the major histocompatibility complex [MHC] class II molecule I-Au with increasing affinities) given intravenously activates T cells, rendering cells hyperresponsive in vitro for at least two days after injection. Concomitantly, T cells apoptose in the periphery, the degree of which correlates with the affinity of the peptide for the MHC. In addition, a shift in the T helper phenotype of the surviving T cells occurs such that the low affinity peptide, Ac1-11, induces primarily a Th1 response, whereas the highest affinity peptide, Ac1-11[4Y], induces primarily a Th2 type response. These data show that both the nature and the presumed number of the peptide–MHC complexes formed during specific peptide therapy affect both the degree of peripheral programmed cell death as well as the outcome of the T helper subset response in vivo, leading to amelioration of disease.
In an immune response, encounter with a foreign antigen may trigger an inflammatory cell–mediated or a primarily humoral response, each of which is characterized by a subset of CD4+ T helper cells, Th1 and Th2, respectively. These T cell subsets secrete a distinct set of cytokines which influence cytolytic function and antibody isotype production (1–3). Many organ-specific autoimmune diseases are thought to be initiated by Th1 responses, whereas protection, or recovery, is thought to be mediated by Th2 responses (4). Experimental autoimmune encephalomyelitis (EAE)1 is a Th1-mediated rodent model of multiple sclerosis that can be induced with the NH2-terminal peptide of myelin basic protein (MBP) Ac1-11, in PL/J or (PL/J × SJL/J)F1 mice (5). EAE has been successfully treated with the immunodominant epitope of MBP, Ac1-11, as well as analogues in which position four is changed from the native lysine to an alanine (Ac1-11[4A]) or tyrosine (Ac1-11[4Y]) (6–9). Ac1-11[4A] and Ac1-11[4Y] bind to the MHC with ∼50 and 1,500 times higher affinity than does Ac1-11, and both peptides stimulate most Ac1-11–specific T cells more efficiently than does Ac1-11 (10, 11). The affinities of these peptides for I-Au correlate with the half-lives of each of the peptides complexed to I-Au; Ac1-11/I-Au has an immeasurably short half-life, Ac1-11[4A] has a half-life of ∼10 min, and Ac1-11[4Y]/I-Au can be detected for as long as 10 h (6, 12). The efficacy of treatment of EAE with these three peptides correlates with the affinity of the peptides for I-Au. The mechanism of this treatment may be due to anergy, deletion, a switch in Th subset, or a combination of these phenomena (6–9, 13, 14). In other systems, soluble superantigen or peptide has been shown to activate T cells whose initial expansion is followed by massive deletion (15–17), whereas administration of soluble antigen has been shown to induce Th2 type responses (18–21).
To explore more fully the mechanism of peptide therapy in EAE, we developed transgenic mice expressing a TCR specific for Ac1-11 and restricted to I-Au, which was derived from an encephalitogenic T cell clone (22). In two lines of transgenic mice that were established, the CD4/ CD8 ratio is increased at least fivefold, and at least 60% of CD4+ T cells express the Ac1-11–specific αβ-TCR. Injection of Ac1-11, Ac1-11[4A], or Ac1-11[4Y] into TCR transgenic mice induces thymic deletion and peripheral activation and apoptosis, the degree of which correlates directly with the affinity of the peptide for I-Au.
Materials and Methods
Mice.
DNA encoding a genomic construct was derived by amplifying cDNA from the 1934.4 hybridoma, which expresses Vα4 and Vβ8.2 (10), and slotting the cDNAs into genomic shuttle vectors for the α and β chains as described (23). Transgenic mice were established in (B10.S × SJL/J)F1 embryos by using standard techniques (24). Two founders were backcrossed to PL/J mice (purchased from Jackson Laboratories, Bar Harbor, ME) several times. Mice used in these experiments were backcrossed at least two times to PL/J (Jackson Laboratories), and no differences were observed between generations N2 and N6. A few experiments were performed using TCR transgenic mice backcrossed one or two times to B10.PL (Jackson Laboratories). TCR transgenic lines 2B4 and 5C.C7, both specific for cytochrome c and I-Ek, were provided by Dr. Yueh-hsiu Chien (Stanford University, Stanford, CA), and TCR transgenic line D011.10, specific for ovalbumin and I-Ad, was provided by Dr. Anne O'Garra (DNAX Research Institute, Palo Alto, CA). TCR transgenic mice were also backcrossed onto mice homozygous for targeted disruption of the IL-4 gene (25).
Peptides.
Peptides were synthesized using Fmoc chemistry on a peptide synthesizer (model 431A; Applied Biosystems, Foster City, CA) and purified by HPLC to >95% pure. Sequence composition was confirmed by amino acid analysis, and molecular weight was confirmed by mass spectrometry by the University of California at San Francisco Mass Spectrometry Facility. The sequences of the peptides are as follows: Ac1-11, Ac-ASQKRPSQRHG; Ac1-11[4A], Ac-ASQARPSQRHG; Ac1-11[4Y], Ac-ASQYRPSQRHG. Peptides were dissolved in PBS and injected intravenously into TCR transgenic mice 4–10-wk-old in volumes <200 μl. Mice were killed at various times after injection, and the phenotype and functional state of cells from the thymus, lymph nodes (inguinal, popliteal, brachial, axillary, cervical, and mesenteric), and spleen were ascertained.
Antibodies.
TCR expression was ascertained by staining with F23.2, an antibody specific for Vβ8.2 (26), and counterstaining with a biotinylated goat anti–mouse IgG (CALTAG Labs., South San Francisco, CA). T cells were assayed for expression of CD69, CD25, CD62L, CD44, and CD45RB using biotinylated antibodies (PharMingen, San Diego, CA) and counterstaining with streptavidin-conjugated Texas red (Boehringer Mannheim, Indianapolis, IN) or streptavidin-conjugated phycoerythrin (CALTAG Labs.). The number of CD4+ and CD8+ T cell subpopulations were ascertained using phycoerythrin- and FITC-conjugated antibodies (CALTAG Labs.), respectively, while the number of macrophages was determined with a biotinylated antibody specific for Mac-1 (CALTAG Labs.). The number of B cells was determined by staining with a phycoerythrin-conjugated antibody to B220 (PharMingen) or FITC-conjugated 40.N, an antibody specific for I-Ak,u (27). Cells stained with these antibodies were analyzed for median fluorescence and percentage of expression by flow cytometry as described (28). Detection of Fas ligand (FasL), membrane-bound TNF-α (mTNF), and lymphotoxin (LT)-αβ was determined by incubating cells with recombinant Fas/Fc, TNF receptor (TNFR)/Fc, and LT-βR/Fc fusion proteins (Fas/Fc and TNFR/Fc proteins were a gift of D. Lynch, Immunex Corp., Seattle, WA; and LT-βR/Fc protein was a gift of R. Ettinger (Stanford University, Stanford, CA), C.M. Ambrose and C. Hession (Biogen Corp., Boston, MA), and W. Force and C. Ware (La Jolla Institute for Allergy and Immunology, La Jolla, CA), and counterstaining with a biotinylated mouse anti–human IgG Fc-specific antibody (Jackson ImmunoResearch Labs., Inc., West Grove, PA) and visualized with streptavidin–Texas red. Anti–IL-4 antibody BVD-4 was used to block endogenous IL-4 production by naive T cells stimulated in vitro (29). Anti–B7-2 antibody GL-1 was administered in vivo to block co-stimulatory activity by B7-2 and was purchased from PharMingen (30). IL-4 was provided by Dr. Anne O'Garra (DNAX Research Institute). Anti–class II antibody 10-3.6 was used to block peptide-MHC-TCR interactions in in vitro cultures (31).
T Cell Stimulation In Vitro.
To determine the in vitro proliferative response, 2.5 × 104 lymph node cells from PBS- or peptide-injected TCR transgenic mice were incubated with 3 × 105 irradiated (30 Gy) nontransgenic syngeneic splenocytes, or 3 × 105 splenocytes from naive or peptide-injected TCR transgenic mice were incubated in flat-bottom 96-well plates in 0.2 ml/well of RPMI 1640 medium (GIBCO BRL, Gaithersburg, MD) supplemented with 5 × 10−5 M 2-mercaptoethanol, pencillin, streptomycin, 200 mM glutamine, and 5% heat-inactivated fetal bovine serum. 24–48 h later, cultures were pulsed with 0.5 μCi [3H]thymidine per well for 18–24 h, harvested, and counted. Cytokine profiles were assayed by culturing 105 lymph node cells with 3 × 105 irradiated (30 Gy) nontransgenic syngeneic splenocytes or 3 × 105 splenocytes from PBS-injected or peptide-injected TCR transgenic mice in 96-well round-bottom plates for 48 h. Cultures were frozen at −80°C, and the levels of IL-2, IL-4, IL-10, and IFN-γ in the supernatants were determined by ELISA using paired antibodies purchased from PharMingen. Fas, FasL, mTNF, and LT-αβ levels were determined on splenocytes derived from either PBS- or peptide-injected Ac1-11–specific or influenza hemagglutinin–specific TCR transgenic mice, which were cultured in T162 tissue culture flasks at 10–100 μM Ac1-11 or 0.2 μg/ml hemagglutinin peptide 126-138, respectively, in complete medium for 48 h.
Detection of Apoptosis.
Inguinal and brachial lymph nodes from PBS-injected or peptide-injected MBP-specific TCR transgenic mice were fixed at least 24 h in 3.7% formaldehyde in PBS and embedded in paraffin. The TUNEL reaction, which detects nicked DNA in nuclei of apoptotic cells, was performed as described (32) except that diaminobenzidine was used as the substrate for the streptavidin-conjugated peroxidase.
Thymic Architecture.
Thymi from naive or peptide-injected mice were fixed in 3.7% formaldehyde for 24 h and embedded in paraffin. Sections were first stained with hematoxylin and eosin to assess thymic architecture. Specific staining for medullary epithelial cells was determined as described using frozen sections (33). In brief, tissue was frozen in Tissue Tek, and 5 μm sections were cut and placed on gelatin-coated slides and air dried. Next, sections were dipped in acetone, air dried, and immersed in PBS containing 0.5% horse serum and 0.01% Tween-20. Sections were then incubated with antibody ERTR5, washed, and incubated with an anti–Ig antibody conjugated to horseradish peroxidase. After washing, ERTR5+ cells were visualized by incubating sections with diaminobenzidine. Sections were then rinsed, mounted, and photographed.
Results
A High CD4/CD8 Ratio Results in Abnormal Thymic Architecture.
Two lines of transgenic mice were established. In one line, both the α and β transgenes were integrated onto the X chromosome. In this line, >90% of T cells from males expressed Vβ8.2, whereas only 50% of T cells from females express Vβ8.2, presumably due to random inactivation of the X chromosome on a cell by cell basis. In males, the CD4/CD8 ratio is increased by ∼15-fold over that of normal, whereas in females, the CD4/CD8 ratio is ∼1.5-fold over that of normal. In the second line, the transgenes were integrated onto an autosomal chromosome, and >90% of T cells from both males and females express Vβ8.2. The CD4/CD8 ratio in the second line is increased by fivefold over that of normal. In males of the first line and all mice of the second line, at least 60% of CD4+ T cells express the Ac1-11–specific αβ-TCR as judged by the number of CD4+ T cells that flux calcium in response to splenocytes presenting Ac1-11 (data not shown). Lymph node or spleen cells from either line when cultured in vitro with Ac1-11, Ac1-11[4A], or Ac1-11[4Y] spontaneously proliferate (data not shown). Routine hematoxylin and eosin staining of sections of the thymus from both lines of our TCR transgenic mice revealed that the thymi do not have a clearly defined cortex and medulla, an observation first noted in two other independent Ac1-11– specific TCR transgenic mice (33a). Interestingly, the thymus from the line with a higher CD4/CD8 ratio has a more severe defect than the line with a lower CD4/CD8 ratio. Specific staining of sections from thymi from the second MBP-specific TCR transgenic line that has a lower CD4/CD8 ratio shows that medullary epithelial cells are scattered throughout the thymus (Fig. 1). The cortex has a decreased size, while macrophage and dendritic cell populations appear normal (data not shown). To determine whether this defect was specific to these particular MBPspecific TCR transgenic mouse lines, we examined whether other class II–restricted TCR transgenic lines that have a high CD4/CD8 ratio have abnormal thymic architecture. All three different TCR transgenic lines examined so far, 2B4 (34) and 5C.C7 (35), both cytochrome c-specific and I-Ek– restricted, and DO11.10 (36), specific for ovalbumin peptide 323-339 and restricted to I-Ad, show a similar defect when examined by hematoxylin and eosin stains (data not shown and Table 1). These data suggest that a normal medullary and cortical epithelial architecture is disrupted by the increased numbers of single positive (SP) and/or decreased numbers of double positive (DP) cells found in these TCR transgenic mice.
Figure 1.


Thymic architecture of MBP-specific TCR transgenic mice and a normal nontransgenic littermate, as illustrated by ERTR5, an antibody that is specific for medullary epithelium. (A) Thymus from a MBPspecific TCR transgenic. (B) Thymus from a nontransgenic littermate. Thymi were from the MBP-specific TCR transgenic line that has a CD4/ CD8 ratio of ∼5:1 and a less pronounced thymic defect than the line with the higher CD4/CD8 ratio. Mice were (PL/J × B10.PL)F1. c, cortex; m, medulla.
Table 1.
Characteristics of Class II–restricted TCR Transgenic Mice
| HNT(15) | D011.10(16) | MBP-1 | MBP-2 | 2B4(33) | 5C.C7(31) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Peptide* | HA126-138 | OVA323-339 | Ac1-11 | Ac1-11 | cyt. c | cyt. c | ||||||
| 84-103 | 84-103 | |||||||||||
| Restriction‡ | I-Ad | I-Ad | I-Au | I-Au | I-Ek | I-Ek | ||||||
| CD4/Cd8§ | 3:1 | 7:1 | 6–10:1 | 15–25:1 | 9–13:1 | 15–20:1 | ||||||
| Thymic | ||||||||||||
| abnormality‖ | None | Mild | Moderate | Severe | Severe | Severe | ||||||
| Peripheral | ||||||||||||
| AICD¶ | Yes | Yes | Yes | Yes | Yes | Yes | ||||||
| Decline in | ||||||||||||
| No. T cells** | Yes | Yes | No | No | Yes | Yes | ||||||
| Th subset | ||||||||||||
| Low dose†† | nd | Th1 and Th 2¶¶, ** | Th1 | Th1 | nd | nd | ||||||
| High dose§§ | nd | Th2 | Th2 | Th2 | nd | nd | ||||||
| Background‖ ‖ | B10.D2 | BALB/c | PL/J | PL/J | B10.BR | B10.A |
MBP-1 refers to the Ac1-11–specific TCR transgenic line in which the α and β transgenes are integrated autosomally. MBP-2 refers to the Ac1-11– specific TCR transgenic line in which the α and β transgenes are integrated on the X chromosome.
The peptide for which the TCR is specific.
The class II molecule for which the TCR is restricted.
The ratio of CD4+ cell to CD8+ cells. Normal ratio is 2:1.
The extent of the thymic abnormality observed as determined by hematoxylin and eosin staining.
Whether T cells in the periphery undergo AICD after soluble peptide administration.
Whether a decline in the number of CD4+ T cells in the periphery occurs after soluble peptide administration.
Th subset induced in peripheral CD4+ T cells after soluble low dose (or low affinity) peptide administration.
Th subset induced in peripheral CD4+ T cells after soluble high dose (or affinity) peptide administration.
∥
∥ Background strain on which experiments were performed.
Experiments were performed in vitro (38).
At very low doses, Th2 responses were observed (38). nd, not determined.
Soluble Ac1-11, Ac1-11[4A], and Ac1-11[4Y] Induce Thymic Apoptosis.
A single large dose of Ac1-11, Ac1-11[4A], or Ac1-11[4Y] induced varying degrees of deletion of CD4+CD8+ DP thymocytes (data not shown). 4 h after injection of 2.4 mg of peptide, there was no difference in the proportion of CD4+ SP, DP, double negative, or CD8+ SP cells; by 24 h, however, the number of DP cells after injection of peptide was reduced 80% by Ac1-11[4Y], 50–75% by Ac1-11[4A], and 25–50% by Ac1-11 (data not shown). By 6 d after injection of peptide, the total number of thymocytes was reduced up to 10-fold after injection of Ac1-11[4Y], two- to fivefold with Ac1-11[4A], and up to twofold with Ac1-11. A single dose of 240 μg of peptide induced similar profiles (data not shown). These results show that despite the abnormal thymic architecture, thymic T cell deletion can occur normally; similar results have been shown for the DO11.10 and 2B4 TCR transgenic models (36, 37).
Soluble Peptide Induces Peripheral T Cell Activation.
As in the thymus, the affinity of the peptide for the MHC plays an important role in the periphery in regulating the development of mature, naive T cells. After intravenous injection of PBS or peptide, cells from lymph nodes were pooled and analyzed by flow cytometry for expression of TCR and T cell activation and memory markers. Table 2 summarizes the median fluorescence intensity of these markers on CD4+Vβ8.2+ T cells. Within 4 h after injection of 2.4 mg of Ac1-11, Ac1-11[4A], or Ac1-11[4Y], TCR expression, as measured by expression of Vβ8.2, was downregulated by about threefold in ∼80% of CD4+ T cells from Ac1-11[4Y]–treated mice, about twofold in Ac111[4A]–treated mice, and not at all in Ac1-11–treated mice. TCR expression of cells from Ac1-11[4A]–injected mice returned to normal within 24 h, while TCR expression of cells from Ac1-11[4Y]–injected mice remained downregulated at 24 h but returned to almost normal by day 2.
Table 2.
Median fluroescent Intensity of Vβ8.2, CD69, CD25, CD62L, and CD44 on CD4+ T Cells from Lymph Nodes after Injection of 2.4 mg of Soluble Peptide
| Treatment | 4 h | 24 h | 72 h | 144 h | ||||
|---|---|---|---|---|---|---|---|---|
| Vβ8.2 | ||||||||
| PBS | 24.8 | 19.9 | 30.1 | 17.4 | ||||
| Ac1-11 | 24.1 | 19.3 | 26.4 | 17.3 | ||||
| Ac1-11[4A] | 10.6 | 18.0 | 27.3 | 16.7 | ||||
| Ac1-11[4Y] | 8.2 | 7.7 | 30.9 | 14.1 | ||||
| CD69 | ||||||||
| PBS | 0.5 | 0.5 | 0.7 | 0.4 | ||||
| Ac1-11 | 31.6 | 15.2 | 1.0 | 0.5 | ||||
| Ac1-11[4A] | 33.0 | 27.0 | 1.0 | 0.4 | ||||
| Ac1-11[4Y] | 32.8 | 39.6 | 1.2 | 0.4 | ||||
| CD25 | ||||||||
| PBS | 0.5 | 0.4 | 0.7 | 0.4 | ||||
| Ac1-11 | 10.1 | 4.6 | 1.9 | 0.7 | ||||
| Ac1-11[4A] | 9.4 | 13.1 | 1.4 | 1.0 | ||||
| Ac1-11[4Y] | 8.5 | 25.4 | 1.2 | 0.5 | ||||
| CD62L | ||||||||
| PBS | 24.0 | 16.5 | 35.0 | 23.6 | ||||
| Ac1-11 | 1.7 | 4.2 | 25.9 | 16.7 | ||||
| Ac1-11[4A] | 1.2 | 3.5 | 29.4 | 19.2 | ||||
| Ac1-11[4Y] | 1.4 | 2.7 | 58.4 | 17.9 | ||||
| CD44 | ||||||||
| PBS | 2.2 | 2.5 | 6.9 | 3.4 | ||||
| Ac1-11 | 2.0 | 4.0 | 23.0 | 8.4 | ||||
| Ac1-11[4A] | 2.2 | 5.9 | 29.3 | 9.3 | ||||
| Ac1-11[4Y] | 2.1 | 4.8 | 16.3 | 8.4 |
CD69, an early activation marker, and CD25, the alpha chain of the IL-2 receptor, were upregulated on ∼80% of CD4+ T cells in the periphery within 4 h and remained high for at least 24 h (Table 2). CD62L, or L-selectin, a homing receptor that is expressed at high levels on naive T cells, was downregulated within 4 h on ∼80% of CD4+ T cells and remained low for at least 24 h. By 72 h, however, most T cells from peptide-injected mice had upregulated CD62L to levels similar to those found in naive mice.
CD44, a marker of memory T cells, was unchanged at 4 h after injection, but by 24 h it was upregulated approximately threefold over that expressed by T cells from PBSinjected mice (Table 2). Cells from Ac1-11[4Y]–injected mice consistently expressed slightly lower levels of CD44 at 6 d after injection. No significant changes were detected in the level of expression of CD45RB, another putative naive cell marker, at any time point (data not shown). Four daily injections of any of the three peptides induced high expression of CD44, whereas CD62L and CD45RB remained high at day 6 after the first injection (data not shown). B cells upregulated the co-stimulation molecule B7-2, but not B7-1, within 24 h, and the expression level of the co-stimulation molecule B7-2 on B cells correlated with the affinity of the peptide (data not shown). In addition, macrophages, but not B cells, from peptide-injected mice upregulated B7-1 (data not shown). Thus, soluble peptide induces T cell activation, and the degree and length of the activated state as measured by CD25, CD69, and CD62L depended on the affinity of the peptide for the MHC.
Proliferative Responses Are Enhanced after Peptide Injection.
The proliferative responses of T cells from lymph nodes from TCR trangenic mice treated with soluble PBS, Ac1-11, Ac1-11[4A], or Ac1-11[4Y] cultured with irradiated nontransgenic syngeneic splenocytes were enhanced after peptide treatment through day 2 (Fig. 2). In some experiments, proliferative responses increased with the affinity of the peptide for I-Au (data not shown). Similar results were found when splenocytes were cultured, except that cells from Ac1-11[4Y]–injected mice proliferated less well than or about equal to those from PBS-injected mice (data not shown). Approximately 60% of CD4+ T cells from lymph nodes from PBS-injected TCR transgenic mice fluxed calcium in response to Ac1-11 presented by nontransgenic syngeneic splenocytes; 24 h after injection, up to 90% of CD4+ T cells from lymph nodes from mice injected with either Ac1-11 or Ac1-11[4A] fluxed calcium, whereas only 20–30% of CD4+ T cells from mice injected with Ac111[4Y] fluxed calcium (data not shown). By 48 h, however, the number of cells from Ac1-11[4Y]–injected mice that fluxed calcium was equal to that of cells from PBSinjected mice, and both lymph node cells and splenocytes from Ac1-11–, Ac1-11[4A]–, and Ac1-11[4Y]–injected mice proliferated in an enhanced fashion (data not shown and Fig. 2). These results suggest that only Ac1-11[4Y] induces a transient hyporesponsiveness in the first 24 h and that this can be overcome by the presence of irradiated antigen presenting cells. By day 6, T cells from peptide-injected mice responded equally to those from PBS-injected mice, indicating that peptide-injection does not induce any longterm anergy (Fig. 2).
Figure 2.

Peripheral T cells from mice injected with Ac1-11, Ac1-11[4A], or Ac1-11[4Y] hyperproliferate for the first 48 h after injection, but proliferate approximately as well as those from PBS-injected mice by day 6. The proliferative responses to Ac1-11, Ac1-11[4A], and Ac1-11[4Y] of cells from PBS-injected mice (closed squares), Ac1-11–injected mice (open squares), Ac1-11[4A]–injected mice (closed circles), and Ac1-11[4Y]–injected mice (open circles) were determined in vitro as described in Materials and Methods.
The CD4+ T Cell Population Does Not Decrease after Peptide Injection.
Despite the fact that T cells are hyperresponsive after peptide injection, the total number of CD4+ T cells increased only 1.5–2-fold over those of PBSinjected controls by day 2 and 3, and no significant net changes in the numbers of CD4+ T cells in the spleen or the lymph nodes were detected by day 6 (Fig. 3). When TCR transgenic mice were given four daily doses of 24 μg per dose, on average there was approximately a two- to threefold increase in the number of CD4+ T cells by day 6 (data not shown). Thus, significant numbers of responsive Ac1-11–specific T cells remain in the periphery, unlike CD4+ T cells in the 2B4, HNT-TCR, and DO11.10 TCR transgenic systems after peptide injection (15, 16, 35). Preliminary results using TCR transgenic mice on the B10.PL background indicate that each of the three peptides are able to induce CD4+ deletion, the degree of which correlates with the peptide affinity for the MHC (data not shown), suggesting that genetic background is important in determining the extent of peripheral deletion. In both the PL/J and B10.PL backgrounds, the numbers of CD8+ T cells, B cells, and macrophages do increase, however, by approximately twofold (data not shown). Presumably, CD8+ T cells are activated through cytokines produced by the CD4+ T cells, whereas B cells and macrophages are activated through interaction of the TCR with peptide/I-Au complex.
Figure 3.

No net change in the numbers of CD4+ T cells in the periphery is observed after peptide injection. The numbers of CD4+ and CD8+ cells from lymph nodes and spleen from TCR transgenic mice injected with PBS, Ac1-11, Ac1-11[4A], or Ac1-11[4Y] are shown 1, 2, and 6 d after a single administration of 2.4 mg of peptide. Black bars represent the numbers of CD4+ T cells in the spleen, dark cross-hatched bars represent the numbers of CD4+ T cells in lymph nodes, shaded bars represent the numbers of CD8+ T cells in the spleen, and light cross-hatched bars represent the numbers of CD8+ T cells in the lymph nodes. Results are pooled from three experiments for days 1 and 6, and two experiments for day 2. In one experiment from day 3, CD4+ T cells had increased in number by 1.5–2-fold in peptide-injected mice (data not shown).
T Cell Death in the Periphery after Peptide Injection.
Despite the lack of significant reduction in the numbers of CD4+ T cells in TCR transgenic mice on the PL/J background, apoptotic cells are detected in lymph nodes 24 h after injection of peptide (Fig. 4). The amount of apoptosis correlates with the affinity of the peptide such that Ac1-11 induces the least amount of apoptosis, Ac1-11[4A] induces an intermediate level, and Ac1-11[4Y] induces the greatest degree of apoptosis. Thus, T cells are apparently dying off rapidly enough to counteract any increase due to proliferation. Activation-induced cell death (AICD) accompanied by cytokine production was detected at 500 μM Ac1-11, 100 μM Ac1-11[4A], and 20 μM Ac1-11[4Y] in purified CD4+ T cells cultured in vitro (data not shown). These data differ from those of Critchfield et al., who detected AICD of Ac1-11–specific T cells at 100 μM MBP (9). T cells from our MBP TCR transgenic mice may have a higher threshold for activation-induced apoptosis due to a lower level of TCR expression on these T cells compared with the level on T cells from those TCR transgenic mice studied by Critchfield et al. (9).
Figure 4.

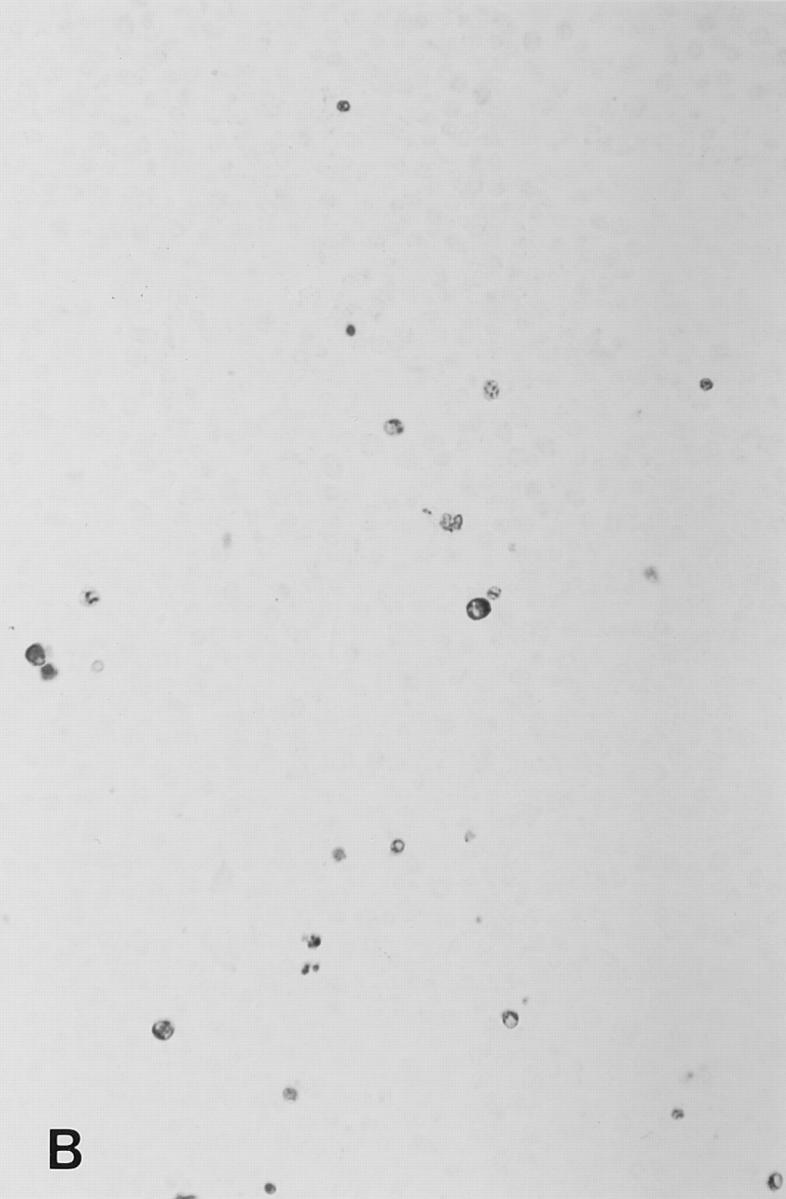
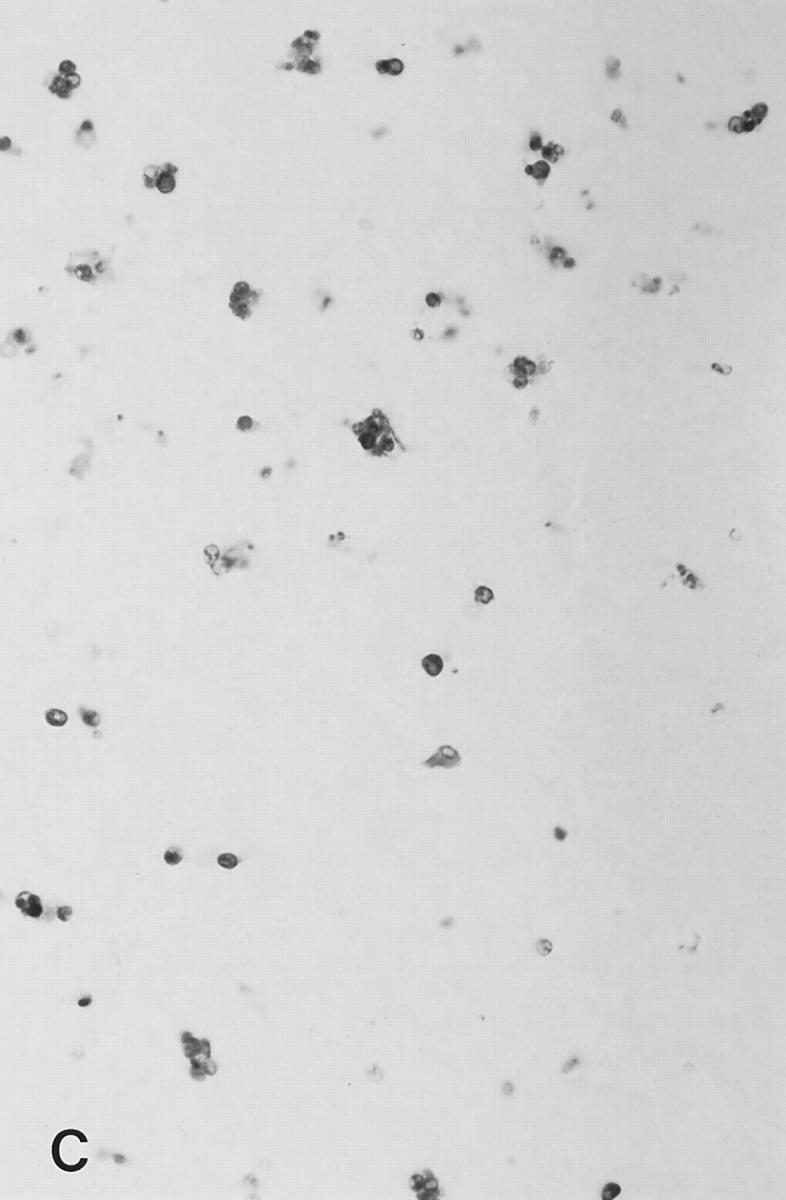
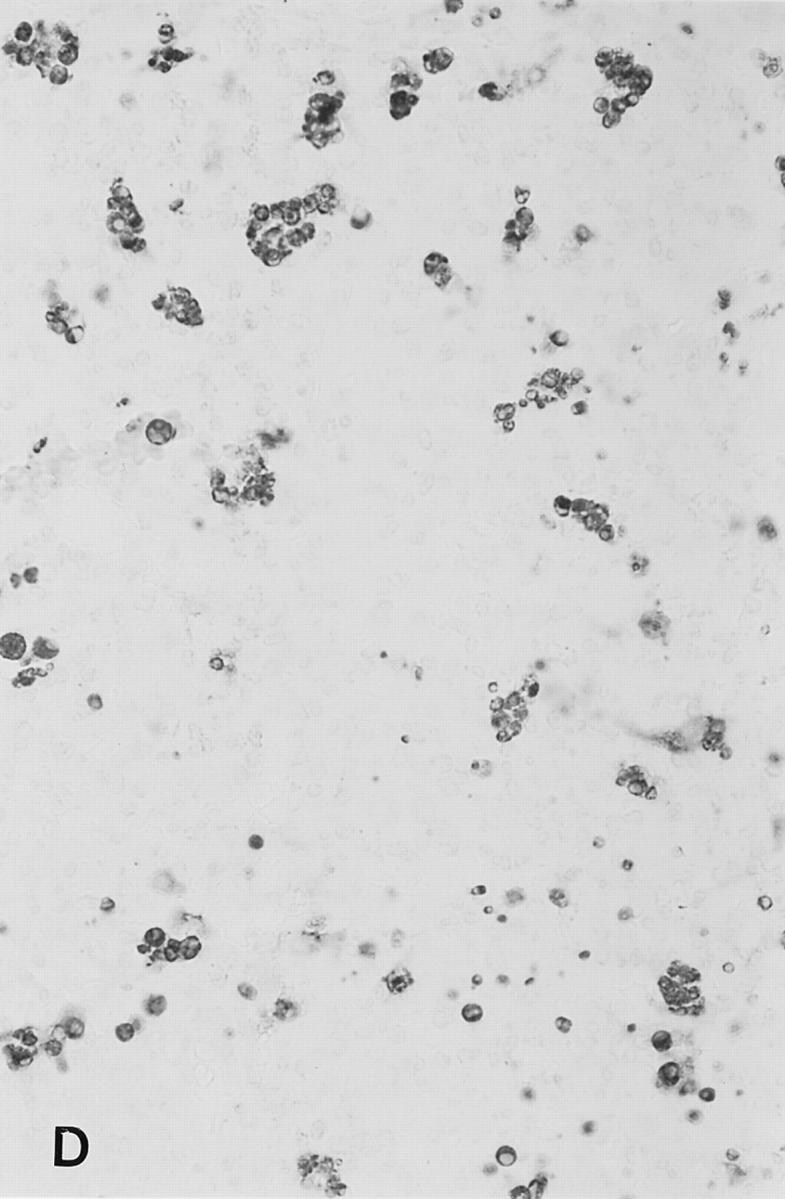
Soluble peptide induces apoptosis in lymph nodes. Sections from brachial or inguinal lymph nodes from Ac1-11–specific TCR transgenic mice removed 24 h after intravenous injection with (A) PBS, (B) 2.4 mg of Ac1-11, (C) 2.4 mg of Ac1-11[4A], and (D) 2.4 mg of Ac1-11[4Y]. Apoptotic cells were visualized using the TUNEL reaction as described in Materials and Methods section and appear as dark spots.
Expression of Fas, Fas Ligand, and TNF-α by CD4+ T Cells.
Peripheral T cells have been shown to be deleted after soluble antigen (37, 38) or anti-CD3 (39) exposure through the Fas/FasL and TNF/TNFR pathways. To ascertain whether molecules involved in these pathways can be induced in the MBP TCR system, we compared the levels of Fas, FasL, membrane TNF, and lymphotoxin-αβ on CD4+ T cells from the MBP-specific TCR transgenic line to those from an influenza hemagglutinin (HA)–specific TCR transgenic line (HNT TCR; reference 16) in which up to 80% of CD4+ T cells are deleted after soluble peptide treatment. Splenocytes from each type of transgenic line were cultured in vitro with their respective cognate peptides. Both Fas and FasL levels are comparable to those seen in the HA-specific TCR transgenic system (Fig. 5). Lower levels of membrane TNF and LTαβ were detected on CD4+ MBP-specific T cells activated with Ac1-11 in vitro. The levels, however, are at least half of those detected on HNT CD4+ T cells activated in vitro with HA peptide (Fig. 5).
Figure 5.

CD4+ T cells from MBP-specific TCR transgenic mice express less membrane TNF-α and LT-αβ than do those from HA-specific TCR transgenic mice. Expression of CD69, Fas, FasL, mTNF, and LT-αβ on splenocytes from either the MBP-specific TCR transgenic line cultured with 50 μM Ac1-11 (Ac1-11) or the hemagglutinin-specific TCR transgenic line (HA) cultured in vitro with 0.2 μg/ml HA peptide 126-138 for 48 h. Live cells from the 48-h cultures were collected on a Ficoll gradient and washed two times. Cells were then stained with a biotinylated antibody to CD69, a phycoerythrin-conjugated antibody to Fas, or Fas/Fc, TNFR/Fc, or LT-βR/Fc fusion proteins as described in Materials and Methods. Histograms display only large, blasting cells as determined by forward scatter. For CD69 and Fas, dashed lines in plots indicating levels of CD69 or Fas, respectively, of naive cells from PBS-injected MBP-specific TCR transgenic mice. For FasL, mTNF, and LT-αβ expression, dashed lines indicate fluorescence intensity of cells incubated with only the biotinylated secondary antibody and streptavidin-conjugated Texas red as a negative control.
Cytokine Profiles of Surviving T Cells Differ with Affinity of Peptide.
Since a large population of antigen-responsive T cells survive the single injection of Ac1-11, Ac1-11[4A], or Ac1-11[4Y], we examined whether these peptides differed in induction of T helper subsets. TCR transgenic spleen cells when cultured in vitro with 100 μM Ac1-11[4A] or Ac1-11[4Y] (the concentration at which T cells proliferate equally) produce >200 U/ml IL-2, <50 ng/ml IFN-γ, and little or no detectable IL-4 or IL-10, which is consistent with a Th0 profile (Fig. 6). Lymph node cells (105) cultured with irradiated syngeneic splenocytes (5 × 105) displayed similar cytokine profiles (data not shown). Within 24 h after injection of 2.4 mg of peptide, spleen or lymph node cells from Ac1-11– or Ac1-11[4A]–injected mice produce more IL-2 and up to 10 times more IFN-γ. Little or no IL-4 is detectable, and <1 ng/ml IL-10 is produced (Fig. 6 A). In contrast, cells from Ac1-11[4Y]–injected mice make the least amount of IL-2, IFN-γ at levels comparable to those produced by cells from naive mice, detectable levels of IL-4 (up to 1 ng/ml), and 2–6 times more IL-10 than that produced by cells from Ac1-11– and Ac1-11[4A]– injected mice (Fig. 6 A). By day 6, IFN-γ production was comparable between the different treatment groups. Cells from Ac1-11[4Y] consistently produce the least amount of IFN-γ. The amount of IL-4 or IL-10 produced increased with the affinity of the peptide for I-Au (Fig. 6 B). Thus, a single dose of low or medium affinity peptides, Ac1-11 and Ac1-11[4A], induces differentiation into the Th1 subset, while a single dose of a high affinity peptide, Ac1-11[4Y], induces differentiation into the Th2 subset. Similar results were obtained after a single dose of 240 μg of peptide (data not shown).
Figure 6.

Cytokine profiles of splenocytes from mice injected with PBS, Ac1-11, Ac1-11[4A], or Ac1-11[4Y] 1 d after administration. Increasing amounts of Th2 type cytokines IL-4 and IL-10 were detected, while decreasing amounts of the Th1 type cytokine IFN-γ were detected depending on the peptide injected. Cells were cultured at 3–5 × 105 cells per well in 96-well round-bottom plates with either medium only, Ac1-11, Ac1-11[4A], or Ac1-11[4Y]. Supernatants were collected 45–52 h later, and the amount of IL-2, IFN-γ, IL-4, and IL-10 was determined by ELISA. Black bars, cytokine production in response to Ac1-11 stimulation; shaded bars, cytokine production in response to Ac1-11[4A]; and cross-hatched bars, cytokine production in response to Ac1-11[4Y]. (A) Cytokine profiles of splenocytes from mice injected with PBS or 2.4 mg of Ac1-11, Ac1-11[4A], or Ac1-11[4Y] 1 d after administration. (B) Cytokine profiles of splenocytes from mice injected with PBS or 2.4 mg of Ac1-11, Ac1-11[4A], or Ac1-11[4Y] 6 d after administration. Similar to day 1, increasing amounts of Th2 type cytokines IL-4 and IL-10 were detected, while decreasing amounts of the Th1 type cytokines IL-2 and IFN-γ were detected depending on the peptide injected. No cytokines were detected in supernatants from cells incubated with medium only.
To test whether any of these cytokine responses were the result of differences in APC activation induced in vivo by peptide administration, CD4+ T cells were purified (>95%) from lymph nodes from mice injected four times with 600 μg of Ac1-11, 600 μg of Ac1-11[4A], or 24 μg of Ac111[4Y], and stimulated in vitro with irradiated nontransgenic splenocytes. The cytokine profiles were very similar to those derived from stimulation of whole lymph node cells with irradiated splenocytes as APCs, indicating that any differences lie in the T cells rather than in APCs (data not shown).
T Helper Differentiation In Vitro by Ac1-11, Ac1-11[4A], or Ac1-11[4Y].
To ascertain whether Ac1-11 and Ac1-11 [4A] uniformly induce Th1 differentiation, and whether Ac1-11[4Y] uniformly induces Th2 differentiation, the cytokine profiles of purified naive CD4+ T cells in response to a secondary stimulation after a primary stimulation in vitro with various doses of either Ac1-11, Ac1-11[4A], or Ac1-11[4Y] were determined. Ac1-11 induced primarily Th1 type responses, whereas Ac1-11[4A] and Ac1-11[4Y] induced both Th1 and Th2 type responses, depending on the dose of peptide in the primary stimulation (Fig. 7). IL-2 production was inversely correlated with the concentration of peptide in the primary stimulation. Both high and low concentrations of the three peptides induced the most IFN-γ, with the exception that Ac1-11[4Y], regardless of dose, did not induce a large IFN-γ response. IL-4 production was highest at intermediate concentrations, corresponding to those that induced the least amount of IFN-γ. IL-10 production of cells stimulated by Ac1-11 in the primary culture was detected only in cells activated by the highest concentration of Ac1-11. IL-10 production by cells stimulated by Ac1-11[4A] and Ac1-11[4Y] followed a dose curve–response similar to that of IL-4 production.
Figure 7.

The induction of Th1 and Th2 type cells in vitro. Lymph node cells from an MBP-specific TCR transgenic mouse were stained with CD8, B220, Mac-1, CD69, and CD44, and the negative cells were collected by flow cytometry as naive, CD4+ T cells (reanalysis of cells by flow cytometry indicated a population >98% CD4+). These cells were cultured in vitro at a concentration of 105 CD4+ cells and 106 irradiated nontransgenic syngeneic splenocytes per well in 24-well plates with varying concentrations of Ac1-11, Ac1-11[4A], or Ac1-11[4Y]. 10 d later, cells were washed several times and restimulated at a concentration of 5 × 104 T cells plus 3 × 105 irradiated splenocytes per well in 96-well round-bottom plates with 10 μM Ac1-11, Ac111[4A], or Ac1-11[4Y]. Supernatants were collected at 48 h, and cytokines were detected by ELISA. Black bars, cytokine response to Ac1-11 in secondary challenge; shaded bars, cytokine response to Ac1-11[4A] in secondary challenge; cross-hatched bars, cytokine response to Ac1-11[4Y] in secondary challenge. In general, Ac1-11 induced Th1 type responses regardless of primary concentration, Ac1-11[4A] induced Th2 type responses at high concentrations and Th1 type responses at low concentrations, and Ac1-11[4Y] induced primarily Th2 type responses, except at the lowest two doses. nd, not determined. Previous experiments indicated that too few live cells were recovered at the doses for which cytokine levels were not determined to test a secondary response.
In general, strong Th1 responses were induced at low concentrations, whereas Th2 responses were induced at high concentrations. Furthermore, the dose–response curves for each peptide were shifted in correlation with their affinity for I-Au such that about 5–25-fold more Ac1-11 than Ac1-11 [4A] was required to induce the least amount of IFN-γ and the most IL-4, whereas approximately fivefold more Ac1-11 [4A] than Ac1-11[4Y] was required to induce a strong Th2 response. These experiments emphasize that the ability to induce a specific Th response correlates with the dose and the affinity of the peptide for the MHC, and they show that Ac1-11[4Y] is much more potent at inducing Th2 type responses in vitro, paralleling our findings in vivo.
Inhibition of MHC-Peptide-TCR Interaction In Vitro by Anti–class II Antibody.
The different abilities of each of the three peptides to induce Th1 and Th2 responses may stem from the individual affinity of the peptide–MHC for the TCR or from differing numbers of peptide–MHC ligands interacting with TCR at the site of APC–T cell contact. To differentiate between these possibilities, two different concentrations of anti–class II antibody (10-3.6) were added to three different doses of Ac1-11[4Y] in the primary stimulation. At 12.5 μg/ml, 10-3.6 reduced the primary IL-2 production by 30–50%, whereas 3.125 μg/ml 10-3.6 reduced the primary IL-2 production by 5–30% (Fig. 8 A). Interestingly, suppression of the primary response by anti–class II antibody at 12.5 μg/ml induced stronger Th2 responses in the secondary stimulation at all three primary doses of Ac1-11[4Y] than did addition of no antibody (Fig. 8, B and C). Addition of 3.125 μg/ml 103.6 in the primary stimulation correlated with about twofold increased production of both IFN-γ and IL-4 in the secondary stimulation but did not change the ratio of IFN-γ to IL-4. In contrast, addition of 3.125 μg/ml 10-3.6 to a primary stimulation of 50 μM Ac1-11 inhibited primary IL-2 production by 30% and correlated with increases in IFN-γ responses of 20-fold in the secondary stimulation, while addition of 12.5 μg/ml 10-3.6 inhibited primary IL-2 production by ∼75% and correlated with increases in IFN-γ production of >200-fold in the secondary stimulation (data not shown). The production of IL-4 was similar between 12.5 μg/ml, 3.125 μg/ml, and no antibody in the Ac1-11 primary stimulation. These data indicate that the difference in affinity between Ac1-11 and Ac1-11[4Y] accounts for their differential ability to induce Th1 or Th2 responses.
Figure 8.

The effect of the addition of anti–class II antibody (10-3.6) on in vitro differentiation of naive TCR transgenic mice T cells by 50, 5, and 0.5 μM Ac1-11[4Y]. Black bars represent responses of T cells cultured with 12.5 μg/ml 10-3.6 and Ac1-11[4Y], shaded bars those of T cells cultured with 3.125 μg/ml 10-3.6 and Ac111[4Y], and cross-hatched bars those of T cells cultured with Ac1-11[4Y] alone. Sorted naive T cells were cultured in 24-well plates with irradiated splenocytes as APCs with various amounts of 10-3.6 and Ac1-11[4Y]. IL-2 production in the primary stimulation was determined on culture supernatant 48 h after initial stimulation with Ac1-11[4Y] and 10-3.6. After a total of 7 d, viable cells were then washed and cultured with 10 μM Ac1-11[4Y] or medium only in 96-well round-bottom plates at 5 × 104 cells/well and 5 × 105 APCs/well for 48 h. IFN-γ and IL-4 production in the secondary stimulation was determined as described. (A) IL-2 production by naive T cells in the primary stimulation is decreased by addition of 10-3.6. (B) IFN-γ production by T cells in the secondary stimulation does not change significantly by addition of 10-3.6. (C) IL-4 production by T cells in the secondary stimulation increases 2–20-fold, depending on the initial concentration of Ac1-11[4Y].
Addition of IL-4 or Anti–IL-4 Antibody to Primary Stimulations.
Addition of IL-4 to Ac1-11 or Ac1-11[4Y] primary stimulations in vitro induced strong Th2 responses regardless of the dose of Ac1-11 or Ac1-11[4Y] (Table 3). Conversely, addition of anti–IL-4 antibody (BVD-4) to the primary stimulations induced strong Th1 responses regardless of the dose of peptide in the primary stimulation (Table 3). These results parallel other findings that show that exogenous IL-4 will induce Th2 responses, whereas anti–IL-4 will induce Th1 responses (1). Thus, despite the results reported above, neither Ac1-11 nor Ac1-11[4Y] carries an intrinsic quality that prevents Th2 or Th1 responses, respectively. When Ac1-11[4Y] is administered to TCR transgenic mice given 200 μg of anti–IL-4 on days −1 and 0 or to mice homozygous for a disrupted IL-4 gene, however, production of IL-2, IFN-γ, and IL-10 resembles that seen in normal TCR transgenic mice (data not shown), indicating that endogenous IL-4 production in vivo is not solely responsible for the Th2 responses that are induced after soluble peptide administration.
Table 3.
Production of IFN-γ and IL-4 in Secondary Cultures of T Cells Stimulated with Varying Doses of Ac1-11 or Ac1-11[4Y] with or without Exogenous IL-4 or Anti–IL-4 Antibody
| Ac1-11 Condition | IFN-γ | IL-4 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Primary dose | ||||||||||||
| 500 μM | 5 μM | 500 μM | 5 μM | |||||||||
| ng/ml | ||||||||||||
| None | 4.23 | 22.82 | 0.70 | 0.18 | ||||||||
| IL-4 | 0.08 | <0.05 | 5.11 | 5.21 | ||||||||
| Anti–IL-4 | 20.97 | 7.38 | <0.03 | <0.03 | ||||||||
| Ac1-11[4Y] Condition | Primary dose | |||||||||||
| 500 μM | 5 μM | 0.05 μM | 500 μM | 5 μM | 0.5 μM | |||||||
| ng/ml | ||||||||||||
| None | 7.38 | 0.53 | 3.39 | 0.31 | 1.30 | 2.33 | ||||||
| IL-4 | 0.04 | 0.39 | 0.10 | 5.80 | 9.70 | 4.10 | ||||||
| Anti–IL-4 | 6.08 | 16.71 | 3.91 | <0.03 | <0.03 | 0.06 | ||||||
Anti–B7-2 Downregulates In Vivo Differentiation of Th2 Responses by Ac1-11[4Y].
Previous studies have indicated that co-stimulatory molecules influence T helper subset differentiation (see references 46 and 47). To test whether blocking upregulation of B7-2 on B cells would downregulate Th2 differentiation observed after Ac1-11[4Y] administration in vivo, 100 μg of anti–B7-2 antibody was given on day −1, and both antibody and 2.4 mg of Ac1-11[4Y] were given on day 0. 1 d after peptide administration, B7-1 was upregulated on macrophages, but not on B cells in both PBS- and anti–B7-2–treated mice given Ac1-11[4Y]. B7-2, in contrast, was upregulated on B cells in mice given Ac1-11[4Y] only, but not in mice given both Ac1-11[4Y] and anti–B7-2 antibody (Fig. 9 A). IL-2 production by splenocytes from Ac1-11[4Y]–treated mice having either no treatment or anti–B7-2 did not show any significant differences. In contrast, IFN-γ production by cells from anti– B7-2– and Ac1-11[4Y]–treated mice increased by approximately twofold, while IL-4 and IL-10 production decreased two- to fourfold relative to that by cells from Ac1-11[4Y]– treated mice (Fig. 9 B).
Figure 9.


The effect of anti–B7-2 on the in vivo expression of B7-1 and B7-2 and the development of Th2 responses in vivo after injection of Ac111[4Y]. TCR transgenic mice were injected intraperitoneally with either PBS or 100 μg of anti–B7-2 antibody GL-1 on days −1 and 0. On day 0, mice were injected intravenously with either PBS or 2.4 mg of Ac1-11[4Y], and mice were killed on day 1. (A) The expression of B7-1 and B7-2 on B cells and macrophages was determined as described in Materials and Methods. Solid lines, expression of B7-1 or B7-2 on cells from mice injected intravenously with PBS; dashed lines, expression on cells from mice injected with Ac1-11[4Y]. B7-1 was upregulated on macrophages from Ac1-11[4Y]– injected mice treated intraperitoneally with either PBS or anti–B7-2, while B7-2 was upregulated on B cells from Ac1-11[4Y]–injected mice given PBS intraperitoneally, but not on cells from Ac1-11[4Y]–injected mice given anti–B7-2 intraperitoneally. (B). The cytokine profile of splenocytes in response to 10 μM Ac1-11, Ac1-11[4A], or Ac1-11[4Y] was determined by ELISA as described. Black bars, cytokine response to Ac1-11 in secondary challenge; shaded bars, cytokine response to Ac1-11[4A] in secondary challenge; cross-hatched bars, cytokine response to Ac1-11[4Y] in secondary challenge. IL-2 production was similar between mice given PBS or Ac1-11[4Y] intravenously, regardless of antibody treatment. IFN-γ production was increased approximately twofold in mice given both Ac1-11[4Y] and anti–B7-2 over that of cells from mice given only Ac1-11[4Y], while IL-4 and IL-10 production by cells from mice given both Ac1-11[4Y] and anti–B7-2 was reduced by about two- to fourfold relative to production by cells from mice given Ac111[4Y] only.
Discussion
Many studies have demonstrated the benefits of peptide therapy of autoimmune disease, yet little is understood about the precise mechanisms that underlie these benefits. Several studies have suggested that autoimmune T cells are deleted, anergized, or switched from a deleterious Th subset to a protective one (6–9, 13, 14, 40). We have developed a transgenic model in which most T cells express a T cell receptor specific for the NH2-terminal peptide of myelin basic protein, Ac1-11, and restricted to the class II MHC molecule I-Au, as a model for easily following potentially encephalitogenic T cells in the periphery after soluble peptide therapy. Injection of a single dose of Ac1-11, Ac111[4A], or Ac1-11[4Y] deletes CD4+CD8+ DP thymocytes and activates peripheral T cells. Although AICD was detected in lymph nodes 24 h after peptide injection, the numbers of CD4+ T cells in the periphery did not decline. Surviving cells from peptide-injected mice proliferated in vitro and had differing cytokine profiles. Specifically, peptides with low and intermediate affinities for the MHC, Ac1-11, and Ac1-11[4A], induced Th1 responses, while Ac1-11[4Y], which had the highest affinity for the MHC, induced a Th2 response. In addition, the thymus of both lines have no discrete cortex and medulla, an observation first noted in two other TCR transgenic mice that have the same MBP specificity (33a).
We observed similar thymic abnormalities in three other class II–restricted TCR transgenic lines that have a high CD4/CD8 ratio, indicating that the skewing toward a CD4 population, rather than the specificity of the TCR, results in the thymic abnormality. The reduced cortical size and disorganized medullary epithelium may be due to the over- or underproduction of a growth factor needed for normal organization of the cortex and medulla. A paucity of DP thymocytes, which normally reside in the cortex, and/or an overabundance of CD4+ SP cells, which reside in the medulla, could account for the under- or overproduction of such a factor.
Despite this abnormality, however, deletion of DP thymocytes was readily induced by injection of soluble peptide, as has been shown for other TCR transgenic systems (34, 35). The efficacy of deletion correlated well with the affinity of the peptide for the MHC. In a previous study using similar TCR transgenic mice, Ac1-11 was unable to induce any significant deletion (41). Our results may differ because (a) a higher dose of Ac1-11 was used in our studies and/or (b) there are fewer DP cells in our TCR transgenic mice. Our results and those of Liu et al. (41) reflect the importance of affinity of the selecting peptide for the MHC. Furthermore, the fact that Ac1-11 is a relatively poor mediator of negative selection provides support for the hypothesis that low affinity ligands may allow self-reactive T cells to mature and emigrate from the thymus into the periphery, as has been suggested (41).
The nature of the TCR-peptide-MHC interaction plays a critical role in the induction of deletion in the thymus, as it does in activation of the T cell in the periphery. Multiple stimulations through the TCR are thought to first activate T cells and then result in apoptosis or AICD (22). Although apoptosis is detected within 24 h after injection of Ac1-11, Ac1-11[4A], or Ac1-11[4Y], many cells survive. In the case of Ac1-11, the relative resistance to efficient deletion in the periphery of T cells from MBP TCR mice may stem from the immeasurably short half-life of the Ac1-11/ I-Au complex (12). Thus, these T cells may receive enough repetitive stimulations through the TCR to become activated, but only a small percentage receive a large enough signal to apoptose. This phenomenon is highlighted by another study in which 1.4 mg of Ac1-11 given eight times intravenously was required to delete Ac1-11–specific trangenic T cells from a different TCR transgenic mouse in a transfer system (9). The Ac1-11[4A]/I-Au complex has a longer half-life of ∼10 min (12), and accordingly, more apoptotic cells are observed in the lymph node 24 h after Ac1-11[4A] injection. Ac1-11[4Y], which when complexed to I-Au has a half-life of at least 4 h (12), stimulates the largest amount of apoptosis, although a significant fraction still survives. Because there are no net decreases in the numbers of CD4+ T cells in the periphery after injection of any of the three peptides, we conclude that the number of cells that are deleted through AICD is replaced by an approximately equal number of cells, presumably through proliferation in vivo. Thus, the proliferative and apoptotic responses induced in vivo by soluble peptide correlate directly with the peptide affinity for the MHC, resulting in no net loss of CD4+ T cells in the periphery.
Our findings that cell numbers do not decline after a single dose of peptide differ from other studies using similar regimens (15, 16, 35, 37). This discrepancy cannot simply be a result of these T cells developing a thymus that has an abnormal architecture, as peripheral CD4+ T cells are deleted in other TCR transgenic systems that have a similar thymic abnormality (see Table 1). One possible explanation is that genetic background may be important in determining the extent of deletion, as preliminary data indicate that after peptide injection, the number of CD4+ T cells declines in direct correlation with the affinity of the peptide for the MHC in our MBP-specific TCR transgenic mice on the B10.PL background rather than the PL/J background.
The surviving cells from peptide-injected mice do not differ in their in vitro proliferative response but do differ in their Th subset. Differentiation into Th1 or Th2 cells is influenced by the nature and level of expression of peptide/ MHC complexes (42–45), the costimulatory molecules with which the T cells interact (46, 47), and the route and dose of the antigen (1). In addition to these previous studies, we have shown that the induction of Th2 type cytokines IL-4 and IL-10 correlates with the peptide affinity for the MHC both in vivo and in vitro. These observations agree with other studies in which high antigen dose leads to Th2 type responses, whereas low antigen dose leads to Th1 type responses (42, 43). Our in vivo results differ from recent data that indicate that high affinity analogues of Ac1-11 induce Th1 responses (48). In this study, however, responses were analyzed after immunization in complete Freund's adjuvant, which is used to induce EAE.
The in vitro data in this study show that the three distinct peptides, Ac1-11, Ac1-11[4A], and Ac1-11[4Y], induce different relative levels of IFN-γ, IL-4, and IL-10, regardless of the primary stimulating dose. Inhibition of MHCpeptide-TCR interactions by the addition of anti–class II antibody to primary cultures stimulated with Ac1-11[4Y] enhanced Th2 responses, whereas addition of antibody to those stimulated with Ac1-11 enhanced Th1 responses. These data suggest that the differential ability of these peptides to induce different Th subsets lies within the affinity of the individual peptide for the MHC. These data also suggest that Ac1-11[4A] and Ac1-11[4Y] may act as altered peptide ligands (APL), which can stimulate a different type of T cell response than does the wild-type peptide (49). In addition to increasing the affinity of the peptide for the MHC and the half-life of the peptide–MHC complex, the changes of lysine to alanine and tyrosine at position four in Ac1-11 may subtly alter the conformation of the peptide bound to the MHC, altering the affinity of the TCR for the peptide–MHC complex. Such an altered interaction could lead to a qualitatively different T cell response, which in this case would be enhanced stimulatory capacity and enhanced ability to induce Th2 responses. Other studies have used APL in treatment (13, 14) or prevention (40) of EAE and shown that they preferentially induce Th2 type responses.
How antigen dose affects T helper subset differentiation is unknown. We have used Ac1-11, Ac1-11[4A], and Ac111[4Y] to vary the effective antigen dose. Our data support the hypothesis that the individual peptide–MHC complex is the critical factor in determining Th subset. In fact, however, affinity as well as concentration, half-life, and TCR affinity for the peptide–MHC complex determine the actual numbers of peptide–MHC complexes and ultimately the effective antigen dose at any one time. Thus, the addition of a peptide that has a very low affinity and very short half-life, such as Ac1-11, results in effective doses that favor Th1 type differentiation, whereas addition of a peptide that has a very high affinity and very long half-life, such as Ac111[4Y], results in effective doses that favor Th2 type differentiation.
The results presented here provide evidence that increasing the dose or signals through the TCR favors certain pathways of differentiation. Consequently, weak or strong signals will favor up- or downregulation of molecules that influence T helper status. Examples of such molecules are the co-stimulatory molecules B7-1 and B7-2, which are upregulated on B cells and other antigen-presenting cells after TCR ligation of peptide–MHC complex or encountering various cytokines (50, 51). One day after injection of peptide in vivo, the level of expression of B7-2 on B cells increases with increasing affinity of the peptide. The combination of upregulation of B7-2 on B cells may be particularly relevant for Th2 responses, as B cells and B7-2 have been shown separately to induce Th2 responses (46, 47, 52). In accordance, blocking B7-2 upregulation, but not B7-1, in Ac1-11[4Y]–injected mice correlates with increased IFN-γ and decreased IL-4 and IL-10 production. Thus, Ac111[4Y] favors upregulation of B7-2 on B cells through a strong TCR signal which in turn favors Th2 type responses. B7-1, however, is also upregulated on macrophages from Ac1-11[4Y]–injected mice, suggesting that B7-2 is not solely responsible for the development of Th2 responses in vivo.
The presence of IL-4 dominates IFN-γ during in vitro differentiation of T helper subsets (1). The data presented here suggest that production of endogenous IL-4 is sensitive to the strength of signal through the TCR such that enough IL-4 dominates during differentiation of T cells receiving strong signals (Ac1-11[4Y]) but does not during differentiation of T cells receiving weak signals (Ac1-11). Although the in vitro experiments show that endogenous IL-4 is required for secondary IL-4 responses, in vivo experiments using mice with a disrupted IL-4 gene or mice treated with anti–IL-4 antibody indicate that the presence of endogenous IL-4 is not required for other features characteristic of Th2 responses, such as the downregulation of IL-2 and IFN-γ and the upregulation of IL-10 by Ac111[4Y]. Thus, differential upregulation of B7-2 and IL-4 by Ac1-11, Ac1-11[4A], or Ac1-11[4Y] represent only two of many molecules that influence T helper subset differentiation in vivo.
In addition to these factors, genetic background plays a role in whether a Th1 or Th2 response predominates. A number of studies indicate that T cells from mice on the B10 background are more likely to develop into Th1 cells, while those from BALB/c mice are more likely to develop into Th2 cells (53, 54). This difference has a profound effect on the outcome of immune responses to autoantigens and parasites (55, 56). Recent experiments suggest that the Th2 pathway is the default pathway, and a Th1 type response depends on the strength of signaling of IL-12, a potent inducer of Th1 cells, such that BALB/c mice have a less efficient IL-12 response (57, 58). In addition, IL-1 and TNF-α, both Th1 type cytokines, are required for maximal IFN-γ production (59). Indeed, B10 mice express higher endogenous levels of TNF than do BALB/c mice, and LPSinduced TNF production follows a similar pattern (Cope, A., and H. McDevitt, unpublished observations). Moreover, production of membrane TNF and LTαβ on cells from MBP-specific TCR transgenic mice on the PL/J background was lower than that seen on cells from HA-specific TCR transgenic mice on the B10.D2 background. Recent findings indicate that TNF-α initiates T cell apoptosis in both the CD8+ and CD4+ compartments (38, 39). Furthermore, other experiments suggest that Th1 cells are more susceptible to Fas-induced apoptosis than are Th2 cells (60). Thus, in our system, the PL/J background may resemble the BALB/c background such that IL-12 and TNF induction may be lower than that found in the B10 background, leading to stronger Th2 responses that are more resistant to Fas-induced apoptosis, and less apoptosis mediated by a weaker TNF response.
The induction of tolerance by soluble antigen has long been observed, but only recently have studies shown that soluble antigen can induce Th2 responses while suppressing Th1 type responses (18–21). Although some studies support the concept that T cells are deleted or anergized after peptide therapy (6, 8, 41), others have shown that EAE therapy with MBP or proteolipid protein peptide analogues that are TCR antagonists induce Th2 type cytokines (13, 14, 40). This is the first study that demonstrates that a peptide with high affinity for the MHC and which efficiently treats EAE induces both apoptosis and a protective Th2 subset differentiation of autoimmune T cells in vivo. However, multiple high doses of Ac1-11 and Ac1-11[4A], which induce Th1 responses in our TCR transgenic mice, can also treat EAE in nontransgenic mice (6, 7). In these studies, many different T cells are involved, which may produce Th1 or Th2 responses at different antigen doses. The effects of therapeutic peptide analogs in treating autoimmunity therefore appear to be determined by both peptide affinity and TCR repertoire variation. Depending on as yet undefined genetic background influences and the strength of signal received by the TCR from the peptide–MHC complex, a particular balance is struck between induction of Th1 and Th2 type responses, proliferation in vivo, and activation-induced cell death.
Acknowledgments
We thank P. Sullivan, S. Phillips, and M. Vadeboncoeur for production of transgenic mice and technical assistance, J. Sun and T. Knaak at the Stanford Shared FACS® Facility, and Drs. R.S. Liblau, A. Cope, R. Ettinger, P. Patten, and H.-K. Sytwu for reagents, helpful discussions, and critical readings of the manuscript.
This work was supported by National Institutes of Health grant AI 07757.
Footnotes
1 Abbreviations used in this paper: AICD, activation-induced cell death; APL, altered peptide ligands; DP, double positive; EAE, experimental autoimmune encephalomyelitis; FasL, Fas ligand; HA, hemagglutinin; LT, lymphotoxin; MBP, myelin basic protein; mTNF, membrane-bound TNF-α; SP, single positive; TNFR, TNF receptor.
References
- 1.O'Garra A, Murphy K. Role of cytokines in determining T-lymphocyte function. Curr Opin Immunol. 1994;6:458–466. doi: 10.1016/0952-7915(94)90128-7. [DOI] [PubMed] [Google Scholar]
- 2.Fitch FW, McKisic MD, Lancki DW, Gajewski TF. Differential regulation of murine T lymphocyte subsets. Annu Rev Immunol. 1993;11:29–48. doi: 10.1146/annurev.iy.11.040193.000333. [DOI] [PubMed] [Google Scholar]
- 3.Sher A, Coffman RL. Regulation of immunity to parasites by T cells and T cell–derived cytokines. Annu Rev Immunol. 1992;10:385–409. doi: 10.1146/annurev.iy.10.040192.002125. [DOI] [PubMed] [Google Scholar]
- 4.Liblau RS, Singer SM, McDevitt HO. Th1 and Th2 CD4+ T cells in the pathogenesis of organ-specific autoimmune diseases. Immunol Today. 1995;16:34–38. doi: 10.1016/0167-5699(95)80068-9. [DOI] [PubMed] [Google Scholar]
- 5.Zamvil SS, Steinman L. The T lymphocyte in experimental allergic encephalomyelitis. Annu Rev Immunol. 1990;8:579–621. doi: 10.1146/annurev.iy.08.040190.003051. [DOI] [PubMed] [Google Scholar]
- 6.Samson MF, Smilek DE. Reversal of acute experimental autoimmune encephalomyelitis and prevention of relapses by treatment with a myelin basic protein peptide analogue modified to form long-lived peptide-MHC complexes. J Immunol. 1995;155:2737–2746. [PubMed] [Google Scholar]
- 7.Metzler B, Wraith DC. Inhibition of experimental autoimmune encephalomyelitis by inhalation but not oral administration of the encephalitogenic peptide: influence of MHC binding affinity. Int Immunol. 1993;5:1159–1165. doi: 10.1093/intimm/5.9.1159. [DOI] [PubMed] [Google Scholar]
- 8.Gaur A, Wiers B, Liu A, Rothbard J, Fathman CG. Amelioration of autoimmune encephalomyelitis by myelin basic protein synthetic peptide-induced anergy. Science (Wash DC) 1992;258:1491–1494. doi: 10.1126/science.1279812. [DOI] [PubMed] [Google Scholar]
- 9.Critchfield JM, Racke MK, Zuniga-Pflucker JC, Cannella B, Rainse CS, Goverman J, Lenardo M. T cell deletion in high antigen dose therapy of autoimmune encephalomyelitis. Science (Wash DC) 1994;263:1139–1143. doi: 10.1126/science.7509084. [DOI] [PubMed] [Google Scholar]
- 10.Wraith DC, Smilek DE, Mitchell DJ, Steinman L, McDevitt HO. Antigen recognition in autoimmune encephalomyelitis and the potential for peptide-mediated immunotherapy. Cell. 1989;59:247–255. doi: 10.1016/0092-8674(89)90287-0. [DOI] [PubMed] [Google Scholar]
- 11.Fugger L, Liang J, Gautam A, Rothbard JB, McDevitt HO. Quantitative analysis of peptides from myelin basic protein binding to the MHC class II protein, I-Au, which confers susceptibility to experimental allergic encephalomyelitis. Mol Med. 1996;2:181–188. [PMC free article] [PubMed] [Google Scholar]
- 12.Fairchild PJ, Wildgoose R, Atherton E, Webb S, Wraith DC. An autoantigenic T cell epitope forms unstable complexes with class II MHC: a novel route for escape from tolerance induction. Int Immunol. 1993;5:1151–1158. doi: 10.1093/intimm/5.9.1151. [DOI] [PubMed] [Google Scholar]
- 13.Karin N, Mitchell DJ, Brocke S, Ling N, Steinman L. Reversal of experimental autoimmune encephalomyelitis by a soluble peptide variant of a myelin basic protein epitope: T cell receptor antagonism and reduction of interferon γ and tumor necrosis factor α production. J Exp Med. 1994;180:2227–2237. doi: 10.1084/jem.180.6.2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brocke S, Gijbels K, Allegretta M, Ferber I, Piercy C, Blankenstein T, Martin R, Utz U, Karin N, Mitchell D, et al. Treatment of experimental encephalomyelitis with a peptide analogue of myelin basic protein. Nature (Lond) 1996;379:343–346. doi: 10.1038/379343a0. [DOI] [PubMed] [Google Scholar]
- 15.Liblau RS, Tisch R, Shokat K, Yang X-D, Dumont N, Goodnow CC, McDevitt HO. Intravenous injection of soluble antigen induces thymic and peripheral T-cell apoptosis. Proc Natl Acad Sci USA. 1996;93:3031–3036. doi: 10.1073/pnas.93.7.3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kearney ER, Pape KA, Loh DY, Jenkins MK. Visualization of peptide-specific T cell immunity and peripheral tolerance induction in vivo. Immunity. 1994;1:327–339. doi: 10.1016/1074-7613(94)90084-1. [DOI] [PubMed] [Google Scholar]
- 17.Webb SR, Morris C, Sprent J. Extrathymic tolerance of mature T cells: clonal elimination as a consequence of immunity. Cell. 1990;63:1249–1256. doi: 10.1016/0092-8674(90)90420-j. [DOI] [PubMed] [Google Scholar]
- 18.De Wit D, Van Mechelen M, Ryelandt M, Figueiredo AC, Abramowicz D, Goldman M, Bazin H, Urbain J, Leo O. The injection of deaggregated gamma globulins in adult mice induces antigen-specific unresponsiveness of T helper type 1 but not type 2 lymphocytes. J Exp Med. 1992;175:9–14. doi: 10.1084/jem.175.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van Mechelen M, De Wit D, Ryelandt M, Hjulstrom S, Heynderickx M, Bazin H, Urbain J, Leo O. Induction of Th2 responses to soluble proteins is independent of B cell tolerance status. Int Immunol. 1995;7:199–205. doi: 10.1093/intimm/7.2.199. [DOI] [PubMed] [Google Scholar]
- 20.Burstein HJ, Abbas AK. In vivo role of interleukin 4 in T cell tolerance induced by aqueous protein antigen. J Exp Med. 1993;177:457–463. doi: 10.1084/jem.177.2.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peterson, J.D., W.J. Karpus, R.J. Clatch, and S.D. Miller. Split tolerance of Th1 and Th2 cells in tolerance to Theiler's murine encephalomyelitis virus. Eur. J. Immunol. 23:46–55. [DOI] [PubMed]
- 22.Liblau RS, Pearson CI, Shokat K, Tisch R, Yang X-D, McDevitt HO. High-dose soluble antigen: peripheral T-cell proliferation or apoptosis. Immunol Rev. 1994;142:193–208. doi: 10.1111/j.1600-065x.1994.tb00890.x. [DOI] [PubMed] [Google Scholar]
- 23.Patten PA, Rock EP, Sonoda T, Fazekas de St B, Groth, Jorgensen JL, Davis MM. Transfer of putative complementarity-determining region loops of T cell receptor V domains confers toxin reactivity but not peptide/MHC specificity. J Immunol. 1993;150:2281–2294. [PubMed] [Google Scholar]
- 24.Hogan, B., F. Constantini, and E. Lacy. 1986. Manipulating the Mouse Embryo: A Laboratory Manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY.
- 25.Kopf M, Le Gros G, Bachmann M, Lamers MC, Bluethmann H, Kohler G. Disruption of the murine IL-4 gene blocks Th2 cytokine responses. Nature (Lond) 1993;362:245–248. doi: 10.1038/362245a0. [DOI] [PubMed] [Google Scholar]
- 26.Kappler JW, Staerz U, White J, Marrack PC. Self-tolerance eliminates T cells specific for Mls-modified products of the major histocompatibility complex. Nature (Lond) 1989;332:35–40. doi: 10.1038/332035a0. [DOI] [PubMed] [Google Scholar]
- 27.Devaux C, Pierres M. Clonal analysis of B and T cell responses to Ia antigens. III. Characterization of 12 xenogeneic anti-idiotypic antisera to A.TH-derived anti–I-Ak and anti–I-Ekmonoclonal antibodies. J Immunol. 1982;128:751–757. [PubMed] [Google Scholar]
- 28.Hayakawa K, Hardy HH, Parks DR, Herzenberg LA. The “Ly-1 B” cell subpopulation in normal, immunodefective, and autoimmune mice. J Exp Med. 1983;157:202–218. doi: 10.1084/jem.157.1.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sander B, Hoiden I, Andersson U, Moller E, Abrams J. Similar frequencies and kinetics of cytokine producing cells in murine peripheral blood and sera. J Immunol Methods. 1993;166:201–214. doi: 10.1016/0022-1759(93)90361-a. [DOI] [PubMed] [Google Scholar]
- 30.Larsen CP, Ritchie SC, Hendrix R, Linsley PS, Hathcock KS, Hodes RJ, Lowry RP, Pearson TC. Regulation of immunostimulatory function and costimulatory molecule (B7-1 and B7-2) expression on murine dendritic cells. J Immunol. 1994;152:5208–5219. [PubMed] [Google Scholar]
- 31.Oi VT, Jones PP, Goding JW, Herzenberg LA, Herzenberg LA. Properties of monoclonal antibodies to mouse Ig allotypes, H-2 and Ia antigens. Curr Top Microbiol Immunol. 1978;81:115–120. doi: 10.1007/978-3-642-67448-8_18. [DOI] [PubMed] [Google Scholar]
- 32.Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van Ewijk W, Kisielow P, Von Boehmer H. Immunohistology of T cell differentiation in the thymus of H-Y–specific T cell receptor α/β transgenic mice. Eur J Immunol. 1990;20:129–137. doi: 10.1002/eji.1830200119. [DOI] [PubMed] [Google Scholar]
- 33a.Brabb, T., E.S. Huseby, T.M. Morgan, D.B. Sant' Angelo, J. Kirchner, A.G. Farr, and J. Goverman. 1997. Thymic stromal organization is regulated by the specificity of T cell receptor/ MHC interaction. Eur. J. Immunol. In press. [DOI] [PubMed]
- 34.Berg LJ, Fazekas de St B, Groth, Pullen AM, Davis MM. Phenotypic differences between αβ versus β T-cell receptor transgenic mice undergoing negative selection. Nature (Lond) 1989;340:559–562. doi: 10.1038/340559a0. [DOI] [PubMed] [Google Scholar]
- 35.Wikstrom, M., K. Scott, M. Cook, and B. Fazekas de St. Groth. 1995. Peripheral tolerance in T cell receptor transgenic mice. J. Cell. Biochem. 21A(Suppl.):109.
- 36.Murphy KM, Heimberger AB, Loh DY. Induction by antigen of intrathymic apoptosis of CD4+CD8+ TCRlothymocytes in vivo. Science (Wash DC) 1990;250:1720–1723. doi: 10.1126/science.2125367. [DOI] [PubMed] [Google Scholar]
- 37.Singer GG, Abbas AK. The Fas antigen is involved in peripheral but not thymic deletion of T lymphocytes in T cell receptor transgenic mice. Immunity. 1994;1:365–371. doi: 10.1016/1074-7613(94)90067-1. [DOI] [PubMed] [Google Scholar]
- 38.Sytwu H-K, Liblau RS, McDevitt HO. Effect of the lpr/lpr mutant Fas gene and tumor necrosis factor (TNF) on antigen induced programmed cell death in HNTTCR transgenic mice. Immunity. 1996;5:17–30. doi: 10.1016/s1074-7613(00)80306-4. [DOI] [PubMed] [Google Scholar]
- 39.Zheng L, Fisher G, Miller RE, Peschon J, Lynch DH, Lenardo MJ. Induction of apoptosis in mature T cells by tumour necrosis factor. Nature (Lond) 1995;377:348–351. doi: 10.1038/377348a0. [DOI] [PubMed] [Google Scholar]
- 40.Nicholson LB, Greer JM, Sobel RA, Lees MB, Kuchroo VK. An altered peptide ligand mediates immune deviation and prevents autoimmune encephalomyelitis. Immunity. 1995;3:397–405. doi: 10.1016/1074-7613(95)90169-8. [DOI] [PubMed] [Google Scholar]
- 41.Liu GY, Fairchild PJ, Smith RM, Prowle JR, Kioussis D, Wraith DC. Low avidity recognition of self-antigen by T cells permits escape from central tolerance. Immunity. 1995;3:407–415. doi: 10.1016/1074-7613(95)90170-1. [DOI] [PubMed] [Google Scholar]
- 42.Hosken N, Shibuya K, Heath AW, Murphy KM, O'Garra A. The effect of antigen dose on CD4+T helper cell phenotype development in a T cell receptor–αβ–transgenic model. J Exp Med. 1995;182:1579–1584. doi: 10.1084/jem.182.5.1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bretscher PA, Wei G, Menon JN, BielefeldtOhmann H. Establishment of stable, cell-mediated immunity that makes “susceptible” mice resistant to Leishmania major. . Science (Wash DC) 1992;257:539–542. doi: 10.1126/science.1636090. [DOI] [PubMed] [Google Scholar]
- 44.Murray JS, Pfeiffer C, Madri J, Bottomly K. Major histocompatibility complex (MHC) control of CD4 T cell subset activation. II. A single peptide induces either humoral or cell-mediated responses in mice of distinct MHC genotype. Eur J Immunol. 1992;22:559–565. doi: 10.1002/eji.1830220239. [DOI] [PubMed] [Google Scholar]
- 45.Constant S, Pfeiffer C, Woodard A, Pasqualini T, Bottomly K. Extent of T cell receptor ligation can determine the functional differentiation of naive CD4+T cells. J Exp Med. 1995;182:1591–1596. doi: 10.1084/jem.182.5.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kuchroo VK, Das MP, Brown JA, Ranger AM, Zamvil SS, Sobel RA, Weiner HL, Nabavi N, Glimcher LH. B7-1 and B7-2 costimulatory molecules activate differentially the Th1/Th2 developmental pathways: application to autoimmune disease therapy. Cell. 1995;80:707–718. doi: 10.1016/0092-8674(95)90349-6. [DOI] [PubMed] [Google Scholar]
- 47.Freeman GJ, Boussiotis VA, Anumanthan A, Bernstein GM, Ke XY, Rennert PD, Gray GS, Gribben JG, Nadler LM. B7-1 and B7-2 do not deliver identical costimulatory signals, since B7-2 but not B7-1 preferentially costimulates the initial production of IL-4. Immunity. 1995;2:523–532. doi: 10.1016/1074-7613(95)90032-2. [DOI] [PubMed] [Google Scholar]
- 48.Kumar V, Bhardwaj V, Soares L, Alexander J, Sette A, Sercarz E. Major histocompatibility complex binding affinity of an antigenic determinant is crucial for the differential secretion of interleukin 4/5 or interferon γ by T cells. Proc Natl Acad Sci USA. 1995;92:9510–9514. doi: 10.1073/pnas.92.21.9510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sloan-Lancaster J, Allen PM. Significance of T-cell stimulation by altered peptide ligands in T cell biology. Curr Opin Immunol. 1995;7:103–109. doi: 10.1016/0952-7915(95)80035-2. [DOI] [PubMed] [Google Scholar]
- 50.Nabavi N, Freeman GJ, Gault A, Godfrey D, Nadler LM, Glimcher LH. Signalling through the MHC class II cytoplasmic domain is required for antigen presentation and induces B7 expression. Nature (Lond) 1992;360:266–268. doi: 10.1038/360266a0. [DOI] [PubMed] [Google Scholar]
- 51.Lenschow DJ, Su GH, Zuckerman LA, Nabavi N, Jellis CL, Gray GS, Miller J, Bluestone JA. Expression and functional significance of an additional ligand for CTLA-4. Proc Natl Acad Sci USA. 1993;90:11054–11058. doi: 10.1073/pnas.90.23.11054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Day MJ, Tse AG, Puklavec M, Simmonds SJ, Mason DW. Targeting autoantigen to B cells prevents the induction of a cell-mediated autoimmune disease in rats. J Exp Med. 1992;175:655–659. doi: 10.1084/jem.175.3.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hsieh C-S, Macatonia SE, O'Garra A, Murphy KM. T cell genetic background determines default T helper phenotype development in vitro. J Exp Med. 1995;181:713–721. doi: 10.1084/jem.181.2.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guler ML, Gorham JD, Hsieh C-S, Mackey AJ, Steen RG, Dietrich WF, Murphy KM. Genetic susceptibility to Leishmania: IL-12 responsiveness in Th1 cell development. Science (Wash DC) 1996;271:984–987. doi: 10.1126/science.271.5251.984. [DOI] [PubMed] [Google Scholar]
- 55.Scott B, Liblau R, Degerman S, Marconi AL, Caton AJ, McDevitt HO, Lo D. A role for non-MHC genetic polymorphism in susceptibility to spontaneous autoimmunity. Immunity. 1994;1:72–83. doi: 10.1016/1074-7613(94)90011-6. [DOI] [PubMed] [Google Scholar]
- 56.Reiner SL, Locksley RM. Cytokines in the differentiation of Th1/Th2 CD4+subsets in leishmaniasis. J Cell Biochem. 1993;53:323–328. doi: 10.1002/jcb.240530409. [DOI] [PubMed] [Google Scholar]
- 57.Szabo SJ, Jacobson NG, Dighe AS, Gubler U, Murphy KM. Developmental commitment to the Th2 lineage by extinction of IL-12 signaling. Immunity. 1995;2:665–675. doi: 10.1016/1074-7613(95)90011-x. [DOI] [PubMed] [Google Scholar]
- 58.Paul WE, Seder RA. Lymphocyte responses and cytokines. Cell. 1994;76:241–251. doi: 10.1016/0092-8674(94)90332-8. [DOI] [PubMed] [Google Scholar]
- 59.O'Garra A, Murphy K. Role of cytokines in development of Th1 and Th2 cells. Chem Immunol. 1996;63:1–13. doi: 10.1159/000319475. [DOI] [PubMed] [Google Scholar]
- 60.Ramsdell F, Seaman MS, Miller RE, Picha KS, Kennedy MK, Lynch DH. Differential ability of Th1 and Th2 T cells to express Fas ligand and to undergo activation-induced cell death. Int Immunol. 1994;6:1545–1553. doi: 10.1093/intimm/6.10.1545. [DOI] [PubMed] [Google Scholar]