

Abstract
We tested for antigen recognition and T cell receptor (TCR)–ligand binding 12 peptide derivative variants on seven H-2Kd–restricted cytotoxic T lymphocytes (CTL) clones specific for a bifunctional photoreactive derivative of the Plasmodium berghei circumsporozoite peptide 252– 260 (SYIPSAEKI). The derivative contained iodo-4-azidosalicylic acid in place of PbCS S-252 and 4-azidobenzoic acid on PbCS K-259. Selective photoactivation of the N-terminal photoreactive group allowed crosslinking to Kd molecules and photoactivation of the orthogonal group to TCR. TCR photoaffinity labeling with covalent Kd–peptide derivative complexes allowed direct assessment of TCR–ligand binding on living CTL. In most cases (over 80%) cytotoxicity (chromium release) and TCR–ligand binding differed by less than fivefold. The exceptions included (a) partial TCR agonists (8 cases), for which antigen recognition was fivetenfold less efficient than TCR–ligand binding, (b) TCR antagonists (2 cases), which were not recognized and capable of inhibiting recognition of the wild-type conjugate, (c) heteroclitic agonists (2 cases), for which antigen recognition was more efficient than TCR–ligand binding, and (d) one partial TCR agonist, which activated only Fas (CD95), but not perforin/granzymemediated cytotoxicity. There was no correlation between these divergences and the avidity of TCR–ligand binding, indicating that other factors than binding avidity determine the nature of the CTL response. An unexpected and novel finding was that CD8-dependent clones clearly incline more to TCR antagonism than CD8-independent ones. As there was no correlation between CD8 dependence and the avidity of TCR–ligand binding, the possibility is suggested that CD8 plays a critical role in aberrant CTL function.
CD4+ and CD8+ T lymphocytes recognize antigenic peptides bound to MHC molecules on APC or target cells by their α/β TCR (1). TCR per se have no signaling capabilities but are associated with CD3 components and ζ chains, which mediate TCR signaling (1, 2). In addition, the coreceptors CD4 or CD8 are associated with the src tyrosine kinase p56lck, which plays a critical role in T cell activation (1, 2). In normal T cell activation, intercellular TCR– ligand interactions promote phosphorylation of ζ chains, which then bind the tyrosine kinase ZAP-70, leading to the activation of the ZAP-70–NFAT pathway (1, 2). As originally observed by Allen and coworkers, amino acid substitutions in antigenic peptides can result in aberrant TCR signaling, i.e., altered lymphokine production, lack of cell proliferation, or total anergy of the T cells (3–5). Subsequent studies by several groups indicated that aberrant T cell activation is a general phenomenon and can be observed on T cell hybridomas, CD4+ and CD8+ T cells, and is not limited to peptide modification, but can also occur upon mutations of MHC molecules (6–8).
As to the mechanism, it has been shown that TCR antagonists give rise to monophosphorylation of ζ chains, which fail to bind and activate ZAP-70, thus perturbing the ZAP-70–NFAT pathway of T cell activation (4, 9, 10). Two different concepts have been put forward to explain these phenomena. According to the first, ligand modification results in accelerated dissociation of TCR–ligand complexes, which implies shorter TCR ligation times, resulting in changes of the size and/or composition of clusters of TCR and associated signaling molecules (11–13). Alternatively, according to the second concept, ligand modifications confer via the TCR conformational changes, which qualitatively alter the signaling by TCR-associated molecules (8, 14). Evidence supporting both concepts has been reported, but it is not clear whether either of these concepts explains these phenomena. Actually, there are indications suggesting that there may be no unifying principle for aberrant TCR signaling. For example, it has been shown in several systems that TCR antagonists require large molar excesses to efficiently inhibit T cell responses of the wildtype ligand (3, 6, 9, 13). In contrast, other studies indicated that antagonists can effectively inhibit CTL responses at very low concentrations, which according to these reports allows fast mutating viruses to escape immune recognition (15, 16).
There exist other forms of aberrant TCR signaling as well, such as heteroclitic T cell responses (e.g., a ligand modification increases T cell response) (17) or T cell anergy induced by TCR agonists in the absence of a costimulatory signal (4). Heteroclitic T cell responses have been attributed to increased MHC–peptide binding or TCR–ligand binding (17). However, as the latter has not been directly measured, it cannot be excluded that, actually, the efficiency of TCR signaling was increased. Moreover, for CTL responses peptide variants have been described that activate Fas, but not perforin/granzyme-mediated cytotoxicity (18, 19). These studies suggest that selective activation of Fas-mediated apotosis may play a role in eliminating selfreactive CTL. While the signaling pathway leading to Fas ligand expression is emerging (20, 21), it is not clear how it can be selectively activated via the TCR.
A major difficulty in studying aberrant TCR signaling consists in accurately controlling and measuring key parameters that initiate signaling, such as MHC–peptide interactions, TCR–ligand binding, kinetics and the contribution of the coreceptor to TCR–ligand binding. Although different reliable in vitro assays exist to measure MHC–peptide interactions, vigorous control of MHC–peptide binding on living cells under physiological conditions is complicated by dissociation of MHC–peptide complexes (22, 23). As to the assessment of TCR–ligand interactions, a direct TCR– ligand binding assay has been introduced, in which radiolabeled soluble ligand was incubated with CTL and cell-associated ligand measured after separation of unbound ligand (24). More recently, soluble TCR and ligand molecules have been used in binding assays based on plasmon resonance (13, 25, 26). Although this assay is very precise for high affinity TCR–ligand interactions, detection problems were encountered when studying low affinity ligand variants. In addition, this assay does not take into account factors that influence TCR–ligand interactions on living cells, such as the coreceptor or TCR downmodulation (27, 28).
We have previously introduced a system that allows assessment of TCR–ligand interactions on living cells by TCR photoaffinity labeling (23, 27, 29). To this end, the Plasmodium berghei circumsporozoite peptide PbCS 252– 260 (SYIPSAEKI) was modified by replacing S-252 with photoreactive iodo-4-azidosalicylic acid (IASA)1 and conjugation of the TCR contact residue K-259 with 4-azidobenzoic acid (ABA). Seven Kd-restricted CTL clones were derived from mice immunized with this conjugate. They recognized this peptide derivative and the one lacking the IASA group, but not the derivative lacking the ABA group (29). These clones exhibited all the hallmarks of antigen recognition by conventional CTL, but had the unique features that the peptide derivative can be covalently attached to Kd molecules by selective photoactivation of the IASA group and that TCR–ligand interactions can be assessed by TCR–photoaffinity labeling (23, 29). The TCR photoaffinity labeling with soluble ligand directly reflected TCR– ligand binding and its dependence on CD8 (23, 27).
In the present study, we tested 12 variants of this peptide derivative on seven CTL clones for antigen recognition (chromium release assay) and TCR–ligand binding by TCR photoaffinity labeling with soluble ligand. In 80% of the cases TCR–ligand binding and antigen recognition correlated well. Among the exceptions (⩾fivefold divergences between these two parameters), the most frequent cases were partial agonists for which TCR–ligand binding was more efficient than antigen recognition. However, cases in which the recognition was more efficient than TCR– ligand binding were observed as well. Only two antagonists were found, e.g., derivatives that were not recognized and could inhibit the recognition of the wild-type epitope. Remarkably, the relative efficiency of recognition of epitope variant did not correlate with TCR–ligand binding avidity. Data are also presented indicating that CD8-dependent clones are more susceptible to TCR antagonism than CD8independent ones, suggesting that CD8 can interefere with CTL activation.
Materials and Methods
Synthesis and Characterization of Photoreactive PbCS Peptide Derivatives.
Chemicals for peptide and conjugate synthesis were obtained from Sigma Chemie (Buchs, Switzerland), Neosystems (Strasbourg, France), and Bachem Finechemical AG (Bubendorf, Switzerland). The synthesis, purification, and analysis of IASAYIPSAEK(ABA)I and its derivatives were performed as described (23, 29, 30). In brief, all conjugates were synthesized on an ABI 431 peptide synthesizer (Applied Biosystems Instruments, Foster City, CA) using Fmoc for transient NH2-terminal protection. K(ABA) was incorporated as Fmoc-K(ABA)-OH and Y as Fmoc-Y(PO3H2). After blocking the NH2 terminus with 4-azidosalicylic acid (ASA), the conjugates were deprotected and cleft from the resin. Purification was by reverse-phase HPLC on a C-18 column (1 × 25 cm, 5-μm particle size, Marcherey & Nagel, Oensingen, Switzerland) using a Waters 600 E HPLC system and an in-line 1000 S diode-array photospectrometer (Applied Biosystems Instruments, Foster City, CA). The column was eluted by a linear gradient of acetonitrile in 0.01% trifluoroacetic acid rising within 1 h from 0–75%. The purified conjugates containing ASA and Y(PO3H2) were iodinated with 125I iodine (New England Nuclear, Boston, MA; specific radioactivity ∼2,000 Ci/mMol) or nonradioactive iodine followed by enzymatic dephosphorylation and purified by HPLC. The elution times were as follows: IASA–YIPSAEK (ABA) I ∼44 min; I254A, ∼41 min; P255A, ∼44 min; S256A, ∼45 min; E258A, ∼46 min; K259(ASA), ∼43 min; K259(BA), ∼42 min; P255L, ∼48 min; P255S, ∼43 min; P255N, ∼44 min; P255D, ∼43 min; P255K, ∼41 min; and P255H, ∼42 min. All conjugates displayed the expected spectra and molecular mass (as assessed on a LDI 7000 mass spectrometer; Linear Scientific, Reno, CA).
Cells and Antibodies.
The P815 mastocytomas, A20 and A20 Fas− B lymphomas (31), were maintained in DMEM supplemented with FCS (5%) and Hepes (10 mM). The CTL clones and their culture conditions have been described previously (29). They were stimulated weekly by irradiated BALB/c splenocytes and irradiated P815 cells pulsed with IASA–YIPSAEK(ABA)I in conditioned medium. Hybridomas producing the mAb used were obtained from the American Type Culture Collection (ATCC, Rockville, MD).
Cytolytic Assays.
51Cr-labeled P815 or A20 cells were used as targets in a cytolytic assay as described (23, 29). In brief, target cells (2 × 103 per well) were incubated in microtiter plates containing threefold dilutions of peptide derivative in DMEM supplemented with FCS (5%) and Hepes (10 mM). CTL (6 × 103 per well) were added after 15 min and after 4 h or 8 h for A20 and A20 Fas+ cells of incubation at 37°C, the 51Cr content of supernatants was determined. The specific lysis was calculated as 100 × ([experimental − spontaneous release]/[total − spontaneous release]). The relative antigenic activities were calculated by dividing the concentration of IASA-YIPSAEK(ABA)I required for half-maximal lysis, by that required for the variant peptide derivatives. The Kd competitor activity, reflecting Kd binding of the test compounds, was assessed in a recognition-based competition assay and normalized relative to the one of IASA-YIPSAEK(ABA)I, which was defined as 1, as described (23, 29). For sake of comparison, the relative antigenic activities of the compounds were normalized by dividing the relative antigenic activity by the corresponding relative Kd competitor activities (29). To test the ability of the conjugates to inhibit recognition of IASA– YIPSAEK(ABA)I, P815 target cells were labeled with 51Cr in the presence of 20–120 pM of IASA-YIPSAEK(ABA)I for 1 h at 37°C. The cells were irradiated for 20 s at ⩾350 nm with a 3,000 W Supuva Sun 3,000 UV irradiator (Mutzhas AG, Luzern, Switzerland). The cells were incubated with 10 μM PbCS 252–260 peptide for 30 min and washed again (three times), and resuspended at 2 × 105 cells/ml. Aliquots (104 cells in 50 μl) were plated in round-bottomed microtiter plates containing threefold dilutions of the test compound. After preincubation for 30 min at 37°C, CTL (3 × 104 in 50 μl) were added and the plates incubated for 4 h at 37°C. The suboptimal concentration of IASA– YIPSAEK(ABA)I antigenic peptide was dosed such that a specific lysis of 20–50% was obtained in the absence of a test compound. Spontaneous 51Cr release was ⩽5% in all experiments. Each experiment was performed in triplicates and repeated at least twice.
Kd and TCR Photoaffinity Labeling.
All photoaffinity labeling procedures were performed essentially as described (23, 29, 30). In brief, purified soluble Kd was incubated with 125I-labeled peptide derivatives and β2-microglobulin (Sigma) for 2 h at ambient temperature. After UV irradiation at ⩾350 nm, PbCS peptide 252–260 was added (10 μM) and after overnight incubation at 4°C the covalent Kd-peptide derivative complexes were purified by gel filtration FPLC. Cloned CTL (1-ml aliquots containing 1 × 106 cells) were incubated with Kd–peptide derivative complexes (3–10 × 106 cpm/incubation) for 3 h at 0–4°C. After UV irradiation at 312 ± 40 nm with a 90 W mercury fluorescence lamp (Bioblock Scientific, Illkirch, France) for 30 s the cells were washed, detergent lysed, and the immunoprecipitated TCRligand complexes analyzed by SDS-PAGE.
Immunoprecipitation, Gel Electrophoresis, and Quantitative Analysis.
Immunoprecipitation with the anti-TCR mAb H57-597 and SDS PAGE analysis (10%, reducing conditions) were performed following published procedures (23, 29, 30). The dried gels were exposed to Kodak 5 XAR x ray films in Kodak Super Rapid (Kodak x-Omatic; Rochester, NY) cassettes at −80°C or evaluated by phospho-imaging using a Phospho Imager SF and Image Quant software (Molecular Dynamics). Standard deviations were calculated according to the Student's t test from data of at least three different experiments, each performed in triplicates. The detection limit of TCR photoaffinity labeling was ∼1% for clones S4, S14, S17, and T1, 5% for S18 and S1, and 10% for S15. Relative TCR photoaffinity labeling was calculated by dividing the labeling intensity of the ligand variant by the one of the wildtype ligand. The TCR binding of Kd–“125IASA”-YIPSAEK(BA)I was titrated in threefold dilution up to a 50-fold molar excess. The concentration required for 50% inhibition was determined by extrapolation and used to calculate the relative TCR binding.
TCR-Ligand Binding Assay.
CTL were incubated with covalent soluble radiolabeled Kd–peptide derivative complexes as described above. After 3 h of incubation, the cells were centrifuged in 400-μl tubes (10–15 s, Beckman Microfuge; Beckman Instrs., Palo Alto, CA) through a layer of silicon (84%) and paraffin oil (16%) to separate cell-bound and free Kd–peptide complex, counting each in a Packard Gamma Counter. Nonspecific binding of the soluble ligand complex was 20–30% observed in the presence of anti-Kd mAb 20-8-4S (10 μg/ml). Percentage of binding was calculated by dividing the value of the corresponding ligand complex by the one of Kd-IASA-YIPSAEK(ABA)I, which was defined as 100%.
Results
Photoreactive Peptide Derivative Variants Under Study.
To assess the correlation between TCR–ligand binding and antigen recognition by cloned CTL, we examined 12 variants of the PbCS peptide derivative IASA-YIPSAEK (ABA)I listed in Table 1. Four of these variants contained single Ala substitutions in positions known to be potentially relevant for antigen recognition but not Kd binding (32). Two further variants contained modifications of the ABA group, namely K259(BA), which lacks the azido substituent and K259(ASA), which had an additional hydroxy substituent. The remaining six variants contained L, S, N, D, K, or H in place of PbCS P-255. The ability of these conjugates to bind to Kd was assessed in a recognition based competition assay and the Kd competitor activities were normalized to the one of IASA-YIPSAEK(ABA)I, which was defined as 1. The relative Kd competitor activities varied between 0.07 (P255D) and 0.7 (E258A), e.g., bound to Kd 14–1.4-fold less efficiently than IASA-YIPSAEK (ABA)I.
Table 1.
Kd Binding of IASA-YIPSAEK (ABA) I Variants Under Study

| No. | Name | Sequence | Relative Kd Competitor Activity* | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | wt | IASA | Y | I | P | S | A | E | K (ABA) | I | 1 | |||||||||||
| 2 | 1254A | − | − | A | − | − | − | − | − | − | 0.2 | |||||||||||
| 3 | P255A | − | − | − | A | − | − | − | − | − | 0.3 | |||||||||||
| 4 | S256A | − | − | − | − | A | − | − | − | − | 0.2 | |||||||||||
| 5 | E258A | − | − | − | − | − | − | A | − | − | 0.7 | |||||||||||
| 6 | K259 (ASA) | − | − | − | − | − | − | − | K (ASA) | − | 0.5 | |||||||||||
| 7 | K259 (BA) | − | − | − | − | − | − | − | K(BA) | − | 0.6 | |||||||||||
| 8 | P255L | − | − | − | L | − | − | − | K (ABA) | − | 0.1 | |||||||||||
| 9 | P255S | − | − | − | S | − | − | − | − | − | 0.2 | |||||||||||
| 10 | P255N | − | − | − | N | − | − | − | − | − | 0.2 | |||||||||||
| 11 | P255D | − | − | − | D | − | − | − | − | − | 0.07 | |||||||||||
| 12 | P255K | − | − | − | K | − | − | − | − | − | 0.1 | |||||||||||
| 13 | P255H | − | − | − | H | − | − | − | − | − | 0.2 |
The relative Kd competitor activity indicates the ability of a test compound to bind to Kd. It was determined in a recognition based compeitition assay and was normalized relative to one of IASA-YIPSAEK (ABA) (see Materials and Methods).
Antigen Recognition by S14 CTL.
The recognition of the different conjugates by cloned CTL was assessed in a 51Cr release cytolytic assay. As shown for a representative experiment in Fig. 1 A, half-maximal lysis of P815 cells by S14 CTL was observed at about 8 × 10−12 M IASA-YIPSAEK(ABA)I. The variant P255A showed the same efficiency of recognition, but serine substitution in this position (P255S) resulted in an ∼7-fold decrease and aspartic acid substitution (P255D) in a 2,000-fold decrease. Histidine or leucine substitution (P255H and P255L) obliterated recognition by S14 CTL. For sake of comparison and as shown in Fig. 1 B, the normalized antigenic activities were calculated for the different compounds from the antigenic activities (concentrations required for half-maximal lysis) and the corresponding relative Kd competitor activities (Table 1) (see Materials and Methods).
Figure 1.


Recognition of IASA-YIPSAEK(ABA)I and variants by cloned S14 CTL. (A) 51Cr-labeled P815 cells were incubated with S14 cells in the presence of the indicated concentrations of IASA-YIPSAEK(ABA)I (•), P255A (○), P255S (▴), P255D (♦), P255L (▾) and P255H (▪) at an E/T ratio of 3:1. After 4 h of incubation the specific lysis was determined from the relased chomium. (B) The relative antigenic activities calculated from A were normalized with the relative Kd competitor activities (Table 1) (see Materials and Methods). By definition, the normalized antigenic activity of IASA-YIPSAEK(ABA)I is 1.
TCR–Ligand Binding on S14 CTL.
The same peptide derivatives were radiolabeled and photocross-linked to soluble monomeric Kd. These complexes were incubated with cloned S14 cells and after photoactivation of the ABA group, the immunoprecipitated TCR were analyzed by SDS-PAGE under reducing conditions and autoradiography. As shown for a representative experiment in Fig. 2 A, the trimolecular complexes (MHC–peptide–TCR α or β chain) migrated with an apparent M r of ∼90 kD. Intense TCR photoaffinity labeling was observed in the case of the wild-type ligand (Fig, 2 A, lane 1), which was abolished in the presence of the anti-Kd mAb 20-8-4S, which blocks Kd–TCR interactions (lane 2) (29). Strong labeling was also observed with the ligand variants Kd-P255A and Kd-P255S (Fig. 2 A, lanes 3 and 4), whereas the variants Kd-P255D and KdP255L produced only trace labeling (lanes 5 and 6). No labeling was detectable with the ligand Kd-P255H (Fig. 2 A, lane 7).
Figure 2.


TCR–ligand binding assessed by TCR photoaffinity labeling and a direct binding assay. (A) Cloned S14 CTL were incubated with equal amounts of radiolabeled soluble covalent complexes of Kd and peptide derivatives IASA-YIPSAEK(ABA)I (lane 1 without and lane 2 with antiKd mAb 20-8-4S), P255A (lane 3), P255S (lane 4), P255D (lane 5), P255L (lane 6), and P255H (lane 7) at 0–4°C, and after UV irradiation, the immunoprecipated TCR–ligand complexes were analyzed by SDS-PAGE and autoradiography. (B) The gels were evaluated by phospho- imaging and represented as bar graphs. (C) Alternatively, the UV irradiation was omitted and cell-associated and free ligand were separated by centrifugation through oil gradients. The cellassociated radioactivity of wildtype ligand was defined as 100% and those of the variant ligands are expressed in percent. The mean values and standard deviations were calculated from triplicates.
For quantification, the autoradiograms were evaluated by phospho-imaging. The reading for the wild-type ligand was defined as 100% and the readings for the other ligands expressed relative to this value (Fig. 2 B). To assess whether TCR photoaffinity labeling truthfully reflects the actual TCR–ligand binding, S14 cells were incubated likewise with the same ligands and the cell-associated ligand was separated from free ligand by spinning the cells through oil gradients. As shown in Fig. 2 C, essentially the same binding pattern was observed, indicating that for a given clone TCR photoaffinity labeling is proportional to the TCR– ligand binding. While the TCR photoaffinity labeling assay had no detectable background, the background in the direct binding assay was high (∼30%) (Fig. 2, B and C). For this reason the detection limit in the TCR photoaffinity labeling assay was considerably lower (∼1% versus 30%).
Recognition of Six IASA-YIPSAEK(ABA)I Variants by Seven CTL Clones.
We tested the first six variants of IASA-YIPSAEK(ABA)I (Table 1) for recognition on seven IASA-YIPSAEK(ABA)I-specific CTL clones. As shown in Fig. 3 A, the normalized relative antigenic activities of the variants I254A, P255A, and S256A were in the range of 0.1 to 6 (e.g., were recognized between 10-fold less to 6-fold more efficiently than IASA-YIPSAEK(ABA)I), indicating that none of these substitutions dramatically affected the antigen recognition by these clones. Conversely, the variant E258A was efficiently recognized only by clones S1, S4, S15, and S18, but only inefficiently by clone S14 and not detectably by clones S17 and T1. Of the K259(ABA) variants, K259(ASA) was efficiently recognized by all clones, except by clone S15, which recognized this variant only partially (e.g., the specific lysis never exceeded 30%). An even lower plateau of lysis was observed for the variant IASA-YIPSAEK(IASA)I (data not shown). Conversely, the variant K259(BA) was efficiently recognized only by clones S4, S17, S18, inefficiently by clones S1, S15, and T1 clones, and not detectably by clone S14. More drastic modifications of the K(ABA) side chain, such as shortening by one methylene group, deletion of the ABA group, or alanine substitution obliterated antigen recognition and TCR–ligand binding (reference 29; data not shown).
Figure 3.


Antigen recognition and TCR–ligand binding of IASA-YIPSAEK(ABA)I by seven CTL clones. Antigen recognition, as expressed in normalized relative antigenic activities were assessed as described for Fig. 1 and TCR–ligand binding, expressed as normalized TCR photoaffinity labeling, as described for Fig. 2. The TCR binding of Kd-“125 IASA”-YIPSAEK(BA) was assessed as described in Materials and Methods. Shown in gray shaded bars are cases in which TCR-ligand binding was ⩾fivefold more efficient than antigen recognition; in vertical-striped bars cases in which the antigen recognition was ⩾fivefold more efficient than TCR ligand binding; in diagonal-striped bars cases in which TCR antagonists and in checker-striped bars a partial agonist exhibiting low plateau (lp) of lysis. (A) Six IASA-YIPSAEK(ABA)I variants were examined, containing either single alanine substitutions of PbCS residues or modifications of the K(ABA) side chain. (B) Six additional conjugate variants containing L, S, N, D, K, or H in place of PbCS P255 likewise were tested. The indicated mean values and standard deviations were calculated from at least three independent experiments.
TCR Photoaffinity Labeling by Six Ligand Variants on Seven CTL Clones.
We next assessed TCR–ligand binding for the same peptide derivative variants by TCR photoaffinity labeling as described for Fig. 3. Because the variants I254A and K259(ASA) were efficiently recognized by all clones, except for K259(ASA) by S15 CTL, only this case was tested. As shown in Fig. 3 A, the normalized TCR photoaffinity labeling and the normalized antigenic relative antigenic activities were less than fivefold different, except for six cases. For the variants, P255A on clone T1 and S256A on clone S17 antigen recognition was more efficient than TCR–ligand binding (eight and fivefold different, respectively). The opposite situation was observed for the variant E258A on clones S4, S14, and S15, for which the normalized antigenic activity was five- to sevenfold lower than the TCR photoaffinity labeling. Finally, the variant K259(BA) was not recognized by S14 CTL, but as assessed by inhibition of S14 TCR photoaffinity labeling (see Materials and Methods), its Kd complex detectably bound to S14 TCR.
Correlation Between Antigen Recognition and TCR–Ligand Binding for PbCS P-255 Substituted Peptide Derivative Variants.
From structural analysis of TCR–ligand interactions, evidence was obtained indicating that P-255 of IASA-YIPSAEK(ABA)I is a secondary TCR contact residue for most of the clones (unpublished data). Therefore, we tested likewise six variants containing different amino acids in this position (Table 1). As shown in Fig. 3 B, these substitutions dramatically affected the antigen recognition by at least some of the seven CTL clones. The least dramatic effects were observed for P255S and the most dramatic ones for P255H, which was significantly recognized only by S17 CTL.
Testing of the variants in TCR photoaffinity labeling experiments showed that in most cases (36 of 42) TCR– ligand binding and antigen recognition differed by less than fivefold. As shown in Fig. 3 B, only in six cases (P255L on CTL S4, and S14, variant P255S on CTL S17, variant P255K on CTL S4, and variant P255D on CTL S14) the two parameters differed by more than fivefold. In all cases the TCR–ligand binding was more efficient than the antigen recognition.
Variant K259(ASA) on S15 CTL Activates Fas, but Not Perforin-mediated Cytotoxicity.
To find out whether the partial recognition of variant K259(ASA) by S15 CTL (Fig. 3 A) was accounted for by Fas-mediated cytotoxicity, antigen recognition experiments were performed by using as target cells either A20 B lymphoma cells or an A20 variant that lacked functional Fas (A20 Fas−). As shown for a representative experiment in Fig. 4, A20 cells were efficiently lysed by S15 CTL in the presence of the wild-type peptide derivative, with half-maximal lysis reached at ∼2 × 10−12 M of IASA-YIPSAEK(ABA)I. Conversely, the variant K259 (ASA) was recognized only partially, with half-maximal lysis of only 13% observed at about × 10−10 M. Similar results were obtained on P815 target cells (see Figs. 3 A and 5 C). However, A20 Fas− target cells were not detectably lysed in the presence of this variant, although they were efficiently killed in the presence of the wild-type epitope (half-maximal lysis at about 2 × 10−11 M) (Fig. 4 B). These results strongly suggest that the low lysis of K259(ASA) sensitized A20 cells was accounted for essentially by Fas-mediated cytotoxicity, similarly as has been described in other systems (18, 19).
Figure 4.

S15 CTL recognize variant K259(ASA) only by Fas-mediated cytotoxicity. Cloned S15 CTL were incubated with 51Cr-labeled A20 (A) or A20 Fas− cells (B) in the presence of graded concentrations of IASA-YIPSAEK(ABA)I (closed circles) or IASA-YIPSAEK(ASA)I (open circles) at an E/T ratio of 3:1 for 8 h.
Figure 5.
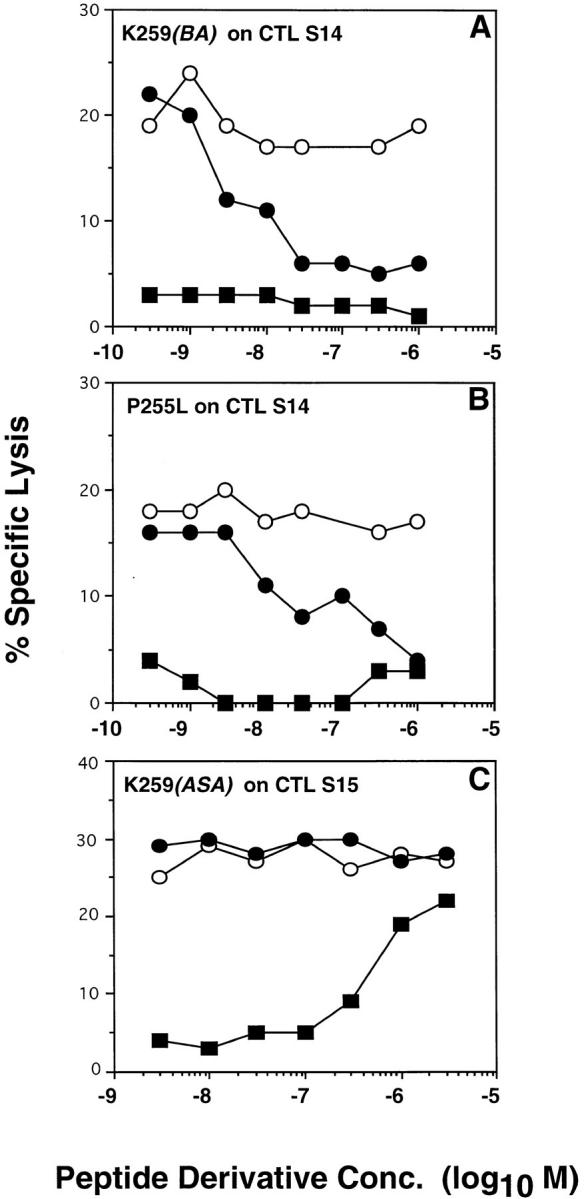
Recognition of target cells expressing covalent Kd-“IASA”- YIPSAEK(ABA)I complexes by S14 CTL is inhibited by variants P255L and K259(BA). Kd molecules on 51Cr-labeled P815 target cells were photocross-linked with a suboptimal concentration of IASA-YIPSAEK(ABA)I. After extensive washing the target cells were incubated with CTL S14 (A and B) or S15 (C) in the presence of the indicated concentrations of peptide PbCS 252-260 (open circles), or peptide variants K259(BA) (closed circle in A), or P255L (closed circle in B) or K259(ASA) (closed circle in C). The recognition of the respective conjugate variants on normal 51Cr-labeled target cells is shown (closed squares). After 4 h, of incubation 51Cr-release was measured and the specific lysis calculated as described for Fig. 1.
Variants P255L and K259(BA) Are TCR Antagonists for S14 CTL.
The conjugate variants P255L and K259(BA) were not recognized by S14 CTL, although their Kd complexes significantly bound to S14 TCR (see Fig. 3). Therefore, we examined their ability to inhibit recognition of IASA-YIPSAEK(ABA)I. To rule out competition of K259 (BA) with the wild-type epitope for Kd binding, we photocross-linked IASA-YIPSAEK(ABA)I to target cell–associated Kd molecules. As shown for a representative experiment in Fig. 5 A, thus sensitized P815 cells were lysed by S14 CTL with ∼22% specific lysis. This lysis was gradually inhibited in the presence of increasing amounts of IASAYIPSAEK(BA)I to ∼5% specific lysis at >5 × 10−8 M of IASA-YIPSAEK(BA)I. No significant inhibition was observed when the parental PbCS peptide was added, demonstrating that this inhibition was accounted for by antagonism and not by competition on the level of Kd–peptide binding.
Likewise similar findings were obtained when the variant P255L was tested, except that a ∼10-fold higher concentration of P255L was required for maximal inhibition (Fig. 5 B). In contrast, when the variant K259(ASA) was tested the same way on S15 CTL, no inhibition was observed (Fig. 5 C). All experiments were performed with target cells expressing different densities of Kd–“IASA”-YIPSAEK(ABA)I complexes, which showed that the inhibition decreased with increasing degrees of sensitization and vanished above 60% specific lysis (data not shown). The same observation has been made in another system (9). These results indicate that the variants K259(BA) and P255L were antagonists for the S14 CTL clone, but that the partial recognition of K259 (ASA) by the S15 clone (see Fig. 4 C) was not accounted for by antagonism.
Aberrant TCR Signaling and CD8 Dependence.
From Fig. 3, it emerges that epitope modifications affect antigen recognition and TCR–ligand binding in a clone-specific manner. To find out whether this is accounted for by CD8, we assessed the CD8 dependence of the clones, as well as the avidity of their TCR–ligand binding. As shown in Fig. 6, the recognition of IASA-YIPSAEK(ABA)I by the different clones was inhibited by the anti-CD8β mAb H35-17 in a diverse manner. The recognition by the S1, S17, and T1 clones was barely affected, but significantly impaired for clones S4 and S15 and abolished for clones S14 and S18. Because anti-CD8 mAb can have diverse effects on CTL, we repeated these experiments using the anti-CD8α mAb 53.6.72, which binds the other chain of CD8 as well as Fab′ fragments of this mAb. Essentially the same findings were obtained (Fig. 6; data not shown), indicating that clones S14, S17, and T1 are CD8 independent, clones S14, S15, and S18 are CD8 dependent, and clone S4 is intermediate. Interestingly, TCR antagonism was observed mainly among the CD8-dependent clones (see Figs. 3 and 6).
Figure 6.

CD8 dependence and avidities of TCR–ligand binding. The recognition of IASA-YIPSAEK(ABA)I by the different CTL clones was assessed in the presence or absence of the antiCD8β mAb H35-17 or the antiCD8α mAb 53.6.72 as described for Fig. 1. Alternatively, and as shown in the inserts, the cloned CTL were incubated in the absence (closed bars) or presence (open bars) of anti-Kd mAb 208-4S with Kd-“125 IASA”-YIPSAEK(ABA)I and the cell-associated radioactivity was measured as described for Fig. 2 C. 100% refers to the highest degree of binding, as observed on CTL S4.
The TCR–ligand binding avidity of the different CTL clones was assessed by the TCR–ligand binding assay described for Fig. 2 C. As shown in the inserts in Fig. 6, the highest binding was observed for S4 CTL and was defined as 100%. The second highest TCR–ligand binding was observed on T1 CTL (80%), followed by clones S14 (40%) and S17 (30%). Intermediate binding was recorded on clones S14 and S17, and for clones S1 and S15, the specific binding was barely above the background. According to TCR photoaffinity labeling, the weakest binding was observed for S15 CTL (7%), followed by CTL S1 (22%) and S18 (25%). The bindings avidities correlated poorly with the observed CD8 dependence, but rather well with the ability of the CTL clones to recognize the different epitope modifications (see Figs. 3 and 6). This is probably explained by that low avidity TCR–ligand interactions are more likely to be reduced below a critical threshold required for T cell activation.
Discussion
The availability of CD8+ CTL clones that permit direct assessment of TCR–ligand binding by TCR photoaffinity labeling, provided an unique opportunity to study in a systematic manner the correlation between CTL function and TCR–ligand binding. Whereas correlations between T cell responses to altered peptide ligands and TCR–ligand binding have been studied previously with purified recombinant TCR and ligands (13, 25, 26), the present study is novel in that TCR–ligand binding was measured on living cells, which takes into account CD8 contributions (27). In addition, since the study was performed on a family of seven CTL clones of the same origin and the same specificity (29), it also provides insights into clone-specific features of antigen recognition. By testing 12 variants of the PbCS peptide derivative IASA-YIPSAEK(ABA)I for antigen recognition and TCR photoaffinity labeling, we found that generally there exists a good, though rarely strict, correlation between cytotoxicity and TCR–ligand binding (Fig. 3) and in only 12 of these 2 parameters diverged by fivefold or more. Whereas similar findings have been obtained in a previous study, in which TCR–ligand interactions were measured with purified TCR and ligand by plasmon resonance (25), another report contends that some 40% of peptide variants were antagonists, e.g., antagonized lysis by three OVA-specific CTL clones (6). In contrast, we only found two TCR antagonists (2,4%) (Figs. 3 and 5). This discrepancy may be explained, at least in part, by that in our system, but not in the other, competition of the peptide variant (antagonist) and the stimulatory peptide (agonist) for MHC binding was strictly ruled out (Fig. 5). Since at least some MHC class I–peptide complexes have limited stability on living cells under physiological conditions, such competition may give exaggerated positive readings in antagonist assays, in which peptide variants usually are used in large excess to the wild-type peptide (Fig. 5; references 6, 9).
If one disregards the cases in which the efficiency of TCR–ligand binding and antigen recognition differed by less than 5-fold, as well as the two TCR antagonists, 10 situations were observed, in which these two parameters diverge by up to 10-fold (Fig. 3). These can be grouped in two categories: (a) the antigen recognition was more efficient than TCR–ligand binding (2 cases) and (b) conversely, it was less efficient than TCR–ligand binding (8 cases). The first and larger group we call partial agonists, though it may contain partial TCR antagonists, e.g., variants that partially antagonize antigen recognition. The second group we call heteroclitic agonists, because they activate CTL more efficiently than expected from their TCR–ligand binding. A similar case has been observed when measuring TCR–ligand binding by plasmon resonance (25). Because in most previous studies on heteroclitic T cell responses TCR–ligand binding was not directly assessed, it is conceivable that at least some of the reported heteroclitic T cell responses actually belong to this category. The only case observed in this study, in which epitope modification increased both the efficiency of antigen recognition and TCR–ligand binding relative to the wild-type epitope, was P255A on S14 CTL (Fig. 3). This effect, however, was only twofold.
Our data clearly show that there exists no correlation between the avidity of TCR–ligand binding and the relative efficiency of antigen recognition. For example, the peptide-derivative variants K259(BA), P255L, P255D, and E258A bind to S14 TCR with comparable avidities, yet the former two were antagonists, whereas the latter two were partial agonists (Fig. 3). Also K259(BA) was a more potent antagonist for S14 CTL than P255L (Fig. 5), yet its Kd complex bound to S14 TCR less efficiently than the one containing P255L (Fig. 3). Moreover, other variants on other clones exhibited similarly reduced TCR–ligand binding, but were recognized more efficiently than the corresponding TCR–ligand binding (i.e., P255S on T1, and P255H on S17). Even more strikingly, the variant S256A was recognized sixfold more efficiently by S17 CTL than the wild-type epitope, whereas the variant P255S was recognized fivefold less efficiently, yet the TCR–ligand binding was the same for both variants (Fig. 3).
It has recently been shown that TCR–ligand complexes with antagonists dissociate faster than those of TCR agonists, but that the corresponding association rates are comparable (13). The finding that our two antagonists and most of the partial agonists displayed significantly reduced TCR– ligand binding (Fig. 3) is consistent with this. However, other data are difficult to explain this way. For example, the finding that the TCR–ligand binding avidity can be the same for TCR antagonists and partial agonists (see above) according to this concept would imply that the corresponding TCR–ligand association rates are significantly different. Moreover, for clones S15 and S17 partial agonists were found (E258A and P255S, respectively), which exhibited TCR–ligand binding equal or better than the wild-type ligand (Fig. 3). Also, it has been reported that certain viral CTL epitope mutants can antagonize CTL responses at very low concentrations, suggesting that these variants too avidily interact with TCR (15, 16). Preliminary results surprisingly indicated that ligands containing partial agonists (i.e., variant P255S on CTL S17 or E258A on CTL S4) dissociate considerably slower from TCR than the wild-type ligand, whereas those containing heteroclitic agonist (i.e., variant S256A on CTL S17 or P255A on CTL T1) dissociate faster (our unpublished results). While these studies need to be extended, they suggest that the half-life of TCR–ligand complexes, rather than TCR–ligand binding avidities, are critical for recognition of epitope variant but that the efficiency of recognition is actually inversely related to the TCR– ligand dissociation rates. This observation seems best explained by serial TCR–ligand engagement during CTL–target cell interaction (28), in that ligands that rapidly dissociate from TCR interact with TCR more frequently, thus providing more TCR signaling. However, this relationship is applicable only as long as TCR–ligand interactions produce normal TCR signaling and one may expect that if TCR–ligand dissociations exceed a certain threshold, aberrant TCR signaling will result, as described by Lyonis et al. (13).
However, other factors may also play a role in aberrant CTL activation. An interesting situation is encountered where epitope variants can induce only Fas, but not perforin mediated cytotoxicity (Fig. 4; references 18, 19). As both lytic mechanisms are activated by TCR ligation, yet involve different signaling pathways (20, 21) and TCR antagonism, at least in our system, is ruled out (Fig. 5 C), the question arises how TCR ligand variants can selectively activate the Fas pathway. An interesting observation is that the anti-CD8β mAb H35-17, but not the anti-CD8α mAb 53.6.72, impaired the recognition of IASA-YIPSAEK(ABA)I by S15 CTL in a manner similar to the epitope modification K259(ASA) (Figs. 4 and 6), suggesting that anti-CD8β mAb may also selectively inhibit perforin mediated cytotoxicity. The role of the coreceptor in aberrant T cell activation is presently poorly understood. It has been shown that blocking of CD4 by anti-CD4 mAb can convert a TCR agonist in an antagonists (34, 35). We made similar observations in the here-described CD8 systems (our unpublished results). We have previously shown that CD8 considerably strengthens TCR–ligand binding, which, at least in part, was explained by CD8-mediated decrease of TCR–ligand dissociation (27). According to the kinetic proofreading concept (11–13), these observations thus may be explained by accelerated dissociation of TCR–ligand complexes.
An interesting finding of the present study is that all antagonists and partial agonist, except one, were observed on CD8-dependent clones (Figs. 3 and 6), suggesting that CD8 may interfere with CTL activation. The poor correlation between CD8 dependence and avidity of TCR– ligand binding (Fig. 6), suggests that CD8 dependence is explained by a requirement of CD8 signaling for CTL activation, rather than by CD8-mediated increase of TCR– ligand binding avidity. The same conclusion has been reached in a different system (33). Studies by Mescher et al. have clearly demonstrated that CD8 signaling requires CD8 activation, which is induced by TCR/CD3 ligation (36, 37). Moreover, there is ample evidence indicating that TCR ligation initiates various signaling cascades, and that TCR ligand modifications can affect these in a diverse manner (4, 5, 21). Thus, it seems conceivable, if not likely, that ligand modifications can interfere with CD8 signaling, which is predicted to adversely affect the function of CD8dependent, but not of independent CTL clones.
The present study indicates that generally epitope modifications affect antigen recognition by cloned CD8+ CTL in accordance with changes in TCR–ligand binding. However, exceptions exist, and from these we learn that factors other than TCR–ligand binding avidities account for aberrant CTL activation. Our data suggest that CD8 can interfere with CTL activation. Because CD8 modulates the kinetics of TCR–ligand interactions (27) and by association with the tyrosine kinase p56lck and the phosphatase CD45 participates in critical phosphorylation/dephosphorylation events (1, 2, 4, 5, 9, 10, 38), it clearly has the potential to do so. As CD8 functions are induced by TCR–ligand interactions (36, 37), the interesting possibility is suggested that epitopes modifications can perturb CTL function via CD8. Further investigations are clearly needed to elucidate the role of CD8 and other factors in aberrant CTL function. The systems described here should greatly facilitate such future studies as they permit direct assessment of TCR–ligand binding and kinetics on living cells, as well as vigorous control of critical experimental parameters.
Acknowledgments
We thank C. Horvath and C. Servis for excellent technical assistance, Drs J. Tschopp and M. Schroeter for A20 and A20 Fas− cells, and Drs P. Romero and E. Palmer for helpful discussions and are grateful to A. Zoppi for preparing the manuscript.
Footnotes
1 Abbreviations used in this paper: ABA, 4-azidobenzoic acid; ASA, azidosalicylic; IASA, iodo-4-azidosalicylic acid.
References
- 1.Janeway CA., Jr The T cell receptor as a multicomponent signaling machine: CD4/CD8 coreceptors and CD45 in T cell activation. Annu Rev Immunol. 1992;10:645–674. doi: 10.1146/annurev.iy.10.040192.003241. [DOI] [PubMed] [Google Scholar]
- 2.Weiss A, Littman DR. Signal transduction by lymphocyte antigen receptors. Cell. 1994;76:263–274. doi: 10.1016/0092-8674(94)90334-4. [DOI] [PubMed] [Google Scholar]
- 3.Evavold BD, Allen PM. Separation of IL-4 production from Th cell proliferation by an altered T cell receptor ligand. Science (Wash DC) 1991;252:1308–1310. doi: 10.1126/science.1833816. [DOI] [PubMed] [Google Scholar]
- 4.Sloan-Lancaster J, Allen PM. Altered peptide ligand–induced partial T cell activation: Molecular mechanisms and role in T cell biology. Annu Rev Immunol. 1996;14:1–27. doi: 10.1146/annurev.immunol.14.1.1. [DOI] [PubMed] [Google Scholar]
- 5.Kersh GJ, Allen PM. Essential flexibility in the T-cell recognition of antigen. Nature (Lond) 1996;380:495–498. doi: 10.1038/380495a0. [DOI] [PubMed] [Google Scholar]
- 6.Jameson SC, Carbone FR, Bevan MJ. Clonespecific T cell receptor antagonists of major histocompatibility complex class I–restricted cytotoxic T cells. J Exp Med. 1993;177:1541–1550. doi: 10.1084/jem.177.6.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Racioppi L, Ronchese F, Matis LA, Germain RN. Peptide–major histocompatibility complex class II complexes with mixed agonist/antagonist properties provide evidence for ligand-related differences in T cell receptor–dependent intracellular signaling. J Exp Med. 1993;177:1047–1060. doi: 10.1084/jem.177.4.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoon ST, Dianzani U, Bottomly K, Janeway CJ. Both high and low avidity antibodies to the T cell receptor can have agonist or antagonist activity. Immunity. 1994;1:563–569. doi: 10.1016/1074-7613(94)90046-9. [DOI] [PubMed] [Google Scholar]
- 9.Reis e Sousa C, Levine EH, Germain RN. Partial signaling by CD8+T cells in response to antagonist ligands. J Exp Med. 1996;184:149–157. doi: 10.1084/jem.184.1.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Madrenas J, Wange RL, Wang JL, Isakov N, Samelson LE, Germain RN. ζ phosphorylation without ZAP-70 activation induced by TCR antagonists or partial agonists. Science (Wash DC) 1995;267:515–518. doi: 10.1126/science.7824949. [DOI] [PubMed] [Google Scholar]
- 11.McKeithan TW. Kinetic proofreading in T-cell receptor signal transduction. Proc Natl Acad Sci USA. 1995;92:5042–5046. doi: 10.1073/pnas.92.11.5042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rabinowitz JD, Beeson C, Lyons DS, Davis MM, McConnell HM. Kinetic discimination in T-cell activation. Proc Natl Acad Sci USA. 1996;93:1401–1405. doi: 10.1073/pnas.93.4.1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lyons DS, Lieberman SA, Hampl J, Boniface JJ, Chien Y-H, Berg LJ, Davis MM. A TCR binds to antagonist ligands with lower affinities and faster dissociation rates than to agonists. Immunity. 1996;5:53–61. doi: 10.1016/s1074-7613(00)80309-x. [DOI] [PubMed] [Google Scholar]
- 14.Janeway CJ. Ligands for the T-cell receptor: hard times for avidity models. Immunol Today. 1995;16:223–225. doi: 10.1016/0167-5699(95)80163-4. [DOI] [PubMed] [Google Scholar]
- 15.Bertoletti A, Sette A, Chisari FV, Penna A, Levrero M, De Carli M, Fiaccadori F, Ferrari C. Natural variants of cytotoxic epotopes are T-cell receptor antagonists for anticiral cytotoxic T cells. Nature (Lond) 1994;369:407–410. doi: 10.1038/369407a0. [DOI] [PubMed] [Google Scholar]
- 16.Klenerman P, Rowland-Jones S, McAdam S, Edwards J, Daenke S, Lalloo D, Köppe B, Rosenberg W, Boyd D, Edwards A, et al. Cytotoxic T-cell activity antagonized by naturally occurring HIV-1 Gag variants. Nature (Lond) 1994;369:403–406. doi: 10.1038/369403a0. [DOI] [PubMed] [Google Scholar]
- 17.England RD, Kullberg MC, Cornette JL, Berzofsky JA. Molecular analysis of a heteroclitic T cell response to the immunodominant epitope of sperm whale myoglobin. Implications for peptide partial agonists. J Immunol. 1995;155:4295–4306. [PubMed] [Google Scholar]
- 18.Cao W, Tykodi SS, Esser MT, Braciale VL, Braciale TJ. Partial activation of CD8+T cells by a selfderived peptide. Nature (Lond) 1995;378:295–298. doi: 10.1038/378295a0. [DOI] [PubMed] [Google Scholar]
- 19.Brossart P, Bevan MJ. Selective activation of Fas/Fas ligand–mediated cytotoxicity by a self peptide. J Exp Med. 1996;183:2449–2458. doi: 10.1084/jem.183.6.2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anel A, Simon AK, Auphan N, Buferne M, Boyer C, Golstein P, Schmitt-Verhulst AM. Two signaling pathways can lead to Fas ligand expression in CD8+cytotoxic T lymphocyte clones. Eur J Immunol. 1995;25:3381–3387. doi: 10.1002/eji.1830251227. [DOI] [PubMed] [Google Scholar]
- 21.Esser MT, Krishnamurthy B, Braciale VL. Distinct T cell receptor signaling requirements for perforin- or Fas L-mediated cytotoxicity. J Exp Med. 1996;183:1697–1706. doi: 10.1084/jem.183.4.1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van der Burg SH, Visseren MJW, Brandt RMP, Kast WM, Melief CJM. Immunogenicity of peptides bound to MHC class I molecules depends on the MHC–peptide complex stability. J Immunol. 1996;156:3308–3314. [PubMed] [Google Scholar]
- 23.Luescher IF, Cerottini JC, Romero P. Photoaffinity labeling of the T cell receptor on cloned cytotoxic T lymphocytes by covalent photoreactive ligand. J Biol Chem. 1994;269:5574–5582. [PubMed] [Google Scholar]
- 24.Sykulev Y, Brunmark A, Jackson M, Cohen RJ, Peterson PA, Eisen HN. Kinetics and affinity of reactions between an antigen-specific T cell receptor and peptide–MHC complexes. Immunity. 1994;1:15–22. doi: 10.1016/1074-7613(94)90005-1. [DOI] [PubMed] [Google Scholar]
- 25.Al-Ramadi BK, Jelonek MT, Boyd LF, Margulies DH, Bothwell AL. Lack of strict correlation of functional sensitization with the apparent affinity of MHC/peptide complexes for the TCR. J Immunol. 1995;155:662–673. [PubMed] [Google Scholar]
- 26.Munir Alam, S., P.J. Travers, J.L. Wung, W. Nasholds, S. Redpath, S.C. Jameson, and N.R.J. Gascoigne. T-cellreceptor affinity and thymocyte positive selection. Nature. 1996;381:616–620. doi: 10.1038/381616a0. [DOI] [PubMed] [Google Scholar]
- 27.Luescher IF, Vivier E, Layer A, Mahiou J, Godeau F, Malissen B, Romero P. CD8 modulation of T-cell antigen receptor–ligand interactions on living cytotoxic T lymphocytes. Nature (Lond) 1995;373:353–356. doi: 10.1038/373353a0. [DOI] [PubMed] [Google Scholar]
- 28.Valitutti S, Muller S, Cella M, Padovan E, Lanzavecchia A. Serial triggering on many T-cell receptors by a few peptide–MHC complexes. Nature (Lond) 1995;375:148–151. doi: 10.1038/375148a0. [DOI] [PubMed] [Google Scholar]
- 29.Luescher IF, Anjuere F, Peitsch MC, Jongeneel CV, Cerottini JC, Romero P. Structural analysis of TCR–ligand interactions studied on H-2Kd-restricted cloned CTL specific for a photoreactive peptide derivative. Immunity. 1995;3:51–63. doi: 10.1016/1074-7613(95)90158-2. [DOI] [PubMed] [Google Scholar]
- 30.Anjuere F, Layer A, Cerottini JC, Servis C, Luescher IF. Synthesis of a radioiodinated photoreactive MAGE-1 peptide derivative and photoaffinity labeling of cell-associated human leukocyte antigen-A1 molecules. Anal Biochem. 1995;229:61–67. doi: 10.1006/abio.1995.1379. [DOI] [PubMed] [Google Scholar]
- 31.Hahne M, Rimoldi D, Schröter M, Schreyer M, Romero P, Schneider P, Bornand T, French LE, Fontana A, Carrel S, et al. Melanomas express Fas(Apo-1/ CD95) ligand and constitute a rejection-resistant immuneprivileged tissue. Science (Wash DC) 1996;274:1364–1366. [Google Scholar]
- 32.Romero P, Eberl G, Casanova JL, Cordey AS, Widmann C, Luescher IF, Corradin G, Maryanski JL. Immunization with synthetic peptides containing a defined malaria epitope induces a highly diverse cytotoxic T lymphocyte response. Evidence that two peptide residues are buried in the MHC molecule. J Immunol. 1992;148:1871–1878. [PubMed] [Google Scholar]
- 33.Kwan-Lim G-E, Ong T, Aosai F, Stauss H, Zamoyska R. Is CD8 dependence a true reflection of TCR affinity for antigen? . Int Immunol. 1993;5:1219–1228. doi: 10.1093/intimm/5.10.1219. [DOI] [PubMed] [Google Scholar]
- 34.Mannie MD, Rosser JM, White GA. Autologous rat myelin basic protein is a partial agonist that is converted into a full antagonist upon blockade of CD4. Evidence for the integration of efficacious and nonefficacious signals during T cell antigen recognition. J Immunol. 1995;154:2642–2654. [PubMed] [Google Scholar]
- 35.Vidal K, Hsu BL, Williams CB, M AP. Endogenous altered peptide ligands can affect peripheral T cell responses. J Exp Med. 1996;183:1311–1321. doi: 10.1084/jem.183.4.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O'Rourke AM, Rogers J, Mescher MF. Activated CD8 binding to class I protein mediated by the T-cell receptor results in signaling. Nature (Lond) 1990;346:187–255. doi: 10.1038/346187a0. [DOI] [PubMed] [Google Scholar]
- 37.Kane KP, Mescher MF. Activation of CD8dependent cytotoxic T lymphocyte adhesion and degranulation by peptide class I antigen complexes. J Immunol. 1993;150:4788–4797. [PubMed] [Google Scholar]
- 38.Mittler RS, Rankin BM, Kiener PA. Physical associations between CD45 and CD4 or CD8 occur as late activation events in antigen receptor–stimulated human T cells. J Immunol. 1991;147:3434–3440. [PubMed] [Google Scholar]