

Abstract
Experimental allergic encephalomyelitis (EAE) is an autoimmune disease of the central nervous system (CNS), and the most commonly used experimental model for multiple sclerosis. It is mediated by autoreactive T cell clones exhibiting a T helper cell (Th) 1 cytokine profile. Nonencephalitogenic T lymphocytes specific for self or exogenous antigens have been found to suppress encephalitogenic T cell responses and to protect against autoimmune disease. The mechanisms by which exogenous antigens modulate autoimmunity are not fully understood. In this study, we tested the hypothesis that a Th2-type immune response against an exogenous, nonself antigen, keyhole limpet hemocyanin (KLH), by releasing IL-4 in the microenvironment, could shift the cytokine profile of encephalitogenic T cells from an inflammatory Th1 to a protective Th2 type. SJL/J mice were preimmunized with the KLH in incomplete Freund's adjuvant to induce a population of Th2 memory cells that would be expected to release Th2 cytokines when activated by the specific antigen at the time of EAE induction. Four weeks later, mice received an encephalitogenic challenge containing guinea pig myelin in complete Freund's adjuvant with or without KLH. All KLH primed animals not receiving the exogenous antigen at the time of EAE induction developed a severe clinical disease indistinguishable from control mice not KLH primed. In contrast, animals preimmunized and challenged with the encephalitogenic inoculum containing KLH showed either no, or markedly reduced, clinical signs. Enzyme-linked immunospot analysis demonstrated that KLH-specific T cells in the primed mice were producing IL-4 characteristic of Th2 cells. In the KLH-primed and restimulated mice, the cytokine profile of the autoreactive, myelin basic protein–specific T cells was shifted from an inflammatory Th1 towards a protective Th2 type. We infer that the presence of IL-4 secreted by KLH-specific memory Th2 cells in the lymphoid system microenvironment in which the autoreactive T cells were engaged by the encephalitogenic stimulus were able to bias their cytokine profile towards a protective Th2 phenotype. This interpretation is supported by the observation that the protective effect of preimmunization with KLH was overcome by rm– IL-12, which inhibited the production of IL-4 by the Th1 cells and biased the autoimmune response to a predominantly Th1 type. Since IL-4 mRNA could not be detected by reverse transcriptase PCR in the CNS, the protective effect was inferred to be mediated by Th2 cells in the lymphoid system, and not the target organ. We conclude that exogenous, nonself antigens that can induce Th2 responses, can modify the cytokine environment sufficiently to alter the cytokine phenotype of inflammatory, autoreactive T cell clones, and ultimately, to provide significant protection against EAE and possibly other T cell–mediated autoimmune diseases.
Experimental allergic encephalomyelitis (EAE)1 is an autoimmune disease of the central nervous system (CNS) mediated by CD4+ T lymphocytes specific for autoantigens of the myelin sheath, including myelin basic protein (MBP) and proteolipid protein (1, 2). Encephalitogenic T cell clones have a classical Th1 cytokine profile, secreting the cytokines IFN-γ and IL-2, and induce local macrophage activation, inflammation, and demyelination (3, 4). Th2 cell clones specific for encephalitogenic peptides are unable to induce the disease and can inhibit Th1 autoimmune clones, presumably by secreting IL-4, IL-10, and TGF-β (5–8). In a transgenic mouse model, the presence of lymphocytes expressing a transgenic TCR specific for MBP was not sufficient to induce the autoimmune disease. Rather, these transgenic mice developed spontaneous EAE only when the RAG-1 gene was deleted and the transgenic, MBPspecific T cells were present in a context in which no other lymphocytes expressing different T cell receptors existed (9, 10). This observation supports the view that the pathogenicity of predetermined autoimmune T cells can be effectively modulated by lymphocytes of other specificities.
Infectious agents have often been implicated in the etiology of autoimmune diseases (11), and considerable evidence has accumulated indicating that immune responses to foreign antigens are able to modulate the activation of autoimmune T cell clones. For example, it has been shown that the immune response against bacterial antigens can attenuate the outcome of experimental autoimmune diseases such as adjuvant arthritis, insulin-dependent diabetes mellitus in NOD mice, and EAE in mice and rats (12–18). In several studies, administration of mycobacterial proteins such as heat-shock cognate proteins of 65 and 70 kD, a 12-kD protein, or tuberculin PPD in IFA, induced protection against EAE (19–21). Understanding the mechanisms involved in the modulation of experimental autoimmune diseases could provide insights into immunologic strategies for developing useful interventions in human autoimmune diseases.
At present, the mechanisms responsible for protection against autoimmune diseases induced by microbial antigens remain unclear. One hypothesis to explain how an immune response against an exogenous antigen may interfere with autoimmunity holds that the bacterial proteins may induce a state of tolerance, as suggested by a downregulation of the CD4 coreceptor detected at the time of onset of the autoimmune disease in mice protected by treatment with mycobacterial hsp60 (19). In this paper we test an alternative hypothesis, namely, that the immunization against nonself antigens can inhibit the development of EAE by modifying the cytokine environment in which autoreactive T cell clones develop and expand upon autosensitization, such that their cytokine profile is shifted from an inflammatory Th1 towards a protective Th2 type.
Materials and Methods
Mice.
Female SJL/J mice (8–12-wk old) were purchased from the Jackson Laboratory (Bar Harbor, ME). Animals were housed in the animal facility of the Albert Einstein College of Medicine, maintained on standard laboratory chow and water ad libitum, and were free of 12 murine pathogens. The experiments were done in accordance to the animal use guidelines of the National Institutes of Health.
Antigens.
The guinea pig myelin was purified from guinea pig spinal cords (Pel-Freeze, Rockland, PA) according to the method of Norton and Poduslo (22). KLH of Megathura crenulata was purchased from Calbiochem Corp. (San Diego, CA) and bovine MBP from Sigma Chemical Co. (St. Louis, MO).
Preimmunization with KLH.
SJL/J mice were injected intraperitoneally 4 wk before the induction of EAE with 50 μg of KLH emulsified in IFA (Difco Labs., Detroit, MI). Control mice received PBS at the same time.
Induction of EAE.
SJL/J mice were injected in four different sites in the back with 0.1 ml of inoculum containing 700 μg of guinea pig myelin in CFA (700 μg/ml of Mycobacterium tuberculosis H37Ra strain in IFA). One group of mice received again the KLH antigen (50 μg) with the encephalitogenic inoculum. All groups of mice received 100 ng of pertussigen/mouse intravenously on days 0, 2, and 7.
Clinical Evaluation.
Mice were monitored daily and a clinical score was assigned using the following scale (23): 0, no clinical sign; 1, a limp tail; 2, hind limb weakness; 3, complete hind limb paralysis; 4, tetraplegia; 5, death.
Recombinant Murine IL-12.
The recombinant murine (rm) IL-12 (Genetics Institute, Boston, MA) was administered intraperitoneally at 0.3 μg/mouse on days 0, 1, and 2 p.i. to a group of SJL/J mice pretreated with the KLH antigen that received a second KLH immunization at the time of EAE induction (24).
Establishment of KLH- and MBP-specific T Cell Lines.
Axillar and inguinal lymph nodes were removed 10 d p.i., homogenized, pooled, and resuspended (4 × 106/ml) in RPMI 1640 (GIBCO BRL, Gaithersburg, MD) supplemented with 10% FCS, 100 U/ml penicillin/streptomycin, 2 mM glutamine, 15 mM Hepes, 100 μM nonessential amino acids, 1 mM Na-pyruvate, and 50 μM 2-mercaptoethanol. Cells were stimulated with KLH or MBP (50 μg/ ml) for 6 d at 37°C in 5% CO2.
Determination of Cytokine Profiles of Specific T Cell Lines by Enzyme-linked Immunospot.
An adaptation of the enzyme-linked immunospot (ELISPOT) assay (25) was used to enumerate IFN-γ and IL-4 secreting T cells among the KLH- and MBP-specific short-term lines. In brief, 96-well nitrocellulose based plates (Millititer HA; Millipore Corp., Bedford, MA) were coated overnight with mAb anti-murine IL-4 or IFN-γ mAbs (10 μg/ml; PharMingen, San Diego, CA). The plates were washed with PBS, and the T cell lines were purified on ficoll-hypaque and added to individual wells at serial twofold dilutions (2 × 105–6 × 103). Splenocytes from donor SJL/J mice were negatively selected for CD3+ T cells using a rat anti–murine CD3 mAb (GIBCO BRL) and magnetic beads coated with antibodies to rat IgG (DYNAL, Inc., Great Neck, NY), irradiated, and added to the plate (5 × 104/well) together wth Con A (2.5 μg/ml). After 20 h, the plates were washed with PBS containing 0.05% Tween and incubated overnight with biotinylated mAbs anti–IL-4 or anti–IFN-γ (4 μg/ml). The plates were then washed with PBS–Tween and incubated with avidin-peroxidase (2.5 μg/ml). Spots representing single IL-4– or IFN-γ–secreting T cells were developed with the substrate 3-amino-9-ethylcarbazole in 0.1 M sodium acetate buffer + H2O2. The number of spots was enumerated using a dissecting microscope.
Determination of Cytokine Profiles in the CNS by Quantitative Reverse Transcriptase–PCR.
The CNS of KLH-protected and control EAE mice were removed 18 d p.i., and the total RNA was extracted using TRIzol Reagent (GIBCO BRL). RNA samples were reverse transcribed using random hexamers (GIBCO BRL) as primers and Superscript reverse transcriptase (GIBCO BRL). The cDNA was then amplified with the following specific primers: hypoxanthine phosphoribosyl transferase (HPRT), 5′-GTTGGATACAGGCCAGACTTTGTTG, 3′-GAGGGTAGGCTGGCCTATGGCT; IFN-γ, 5′-TGCATCTTGGCTTTGCAGCTCTTCCTCATGGC, 3′-TGGACCTGTGGGTTGTTGACCTCAAACTTGGC; IL-4, 5′-CATCGGCATTTTGAACGAGGTCA, 3′-CTTATCGATGAATCCAGGCATCG. To normalize the amount of cDNA, we performed a quantitative PCR analysis of HPRT mRNA levels using serial dilution of the pQRS polycompetitor (provided by Dr. D.B. Corry, University of California at San Francisco, San Francisco, CA; 26). The amount of IFN-γ–specific RNA in the two cDNA normalized samples was measured by quantitative PCR using serial dilutions of the mouse IFN-γ PCR MIMIC (Clontech, Palo Alto, CA). Reactions were performed in a programmable thermal controller (Perkin-Elmer Cetus Instrs., Norwalk, CT) for 35 cycles. Each cycle consisted of 94°C for 40 s, 60°C for 20 s, and 72°C for 40 s. After amplification, 10 μl of PCR product was separated by electrophoresis on 1% agarose gels and visualized by ethidium bromide staining.
Results
Priming and Challenge of Mice with an Exogenous Antigen, KLH, in IFA Induces Protection from EAE.
To test the hypothesis that T cells specific for a foreign antigen could alter the course of EAE in SJL/J mice, two groups of mice were primed intraperitoneally with KLH in IFA. A control group received PBS at the same time. After 4 wk all mice received an encephalitogenic inoculum containing guinea pig myelin in CFA, and one of the two groups of mice previously primed with KLH received the KLH antigen at this time. EAE was fully developed after 10–14 d in the animals that were not primed, or that received the KLH in IFA 4 wk before but were not given the foreign antigen at the time of the encephalitogenic immunization. In contrast, when the KLH-specific T cells were reactivated at the time of EAE induction by a second antigenic challenge, the KLHprimed mice were almost completely protected. In this group of mice, both the incidence and the mean clinical score were significantly reduced compared to controls (Fig. 1). These results indicated that stimulating primed mice with the nonself antigen at the time of activation of autoimmune T cell clones could alter the course of the autoimmune disease.
Figure 1.
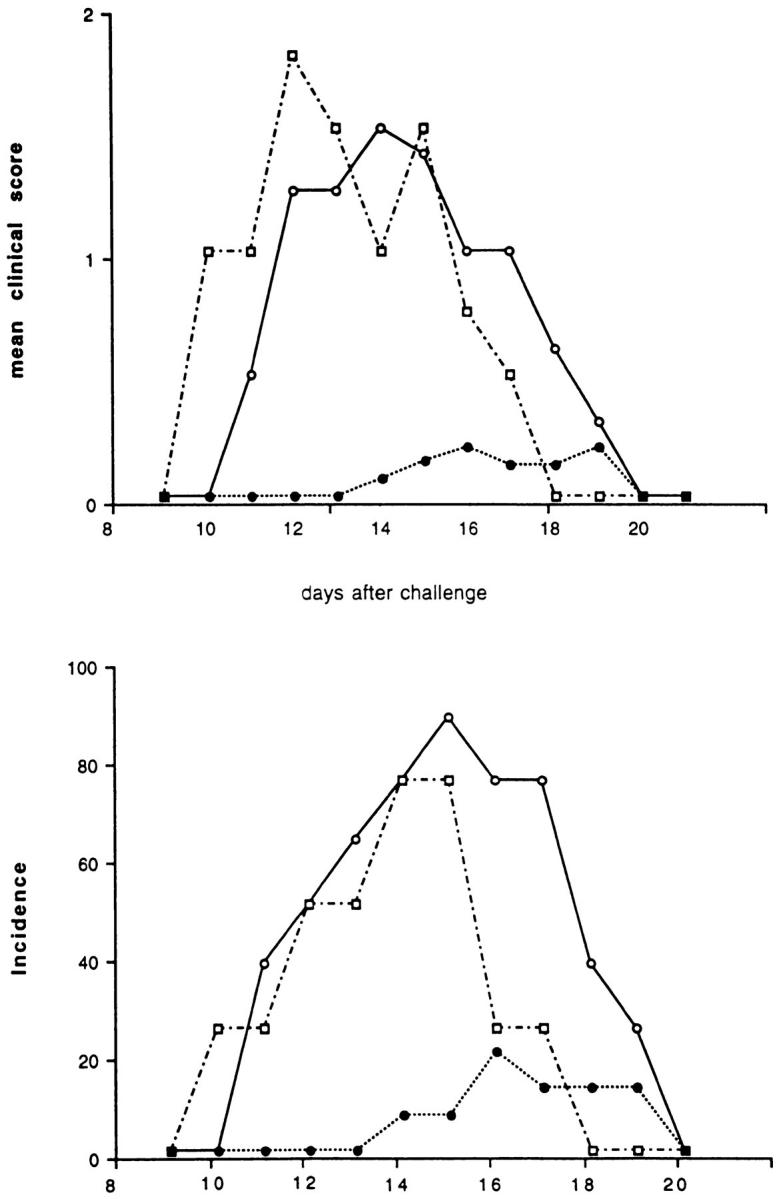
Effect of the KLH preimmunization and challenge at the time of EAE induction on the incidence and clinical score of the disease. Two groups of SJL/J mice were injected intraperitoneally with an emulsion of KLH in IFA. One control group was injected with PBS. After 4 wk, EAE was induced by immunizing the animals with guinea pig myelin in CFA. One of the two groups of mice pretreated with KLH received the foreign antigen with the encephalitogenic inoculum. This group of mice was almost completely protected (filled circles) against EAE. The control group that was not preimmunized with KLH (open circles) or the group that was primed, but not challenged a second time with KLH (open squares) both showed high incidences and clinical scores for the disease.
Protection Against EAE Is Abolished when the KLH-primed Mice Are Treated with rmIL-12.
To test the hypothesis that the protection induced by the KLH was due to a shift in the cytokine profile of the encephalitogenic clones from a Th1 to a Th2 type, we attempted to reverse the KLH effect by modifying the cytokine profiles of the KLH- and MBPspecific T cells in the KLH protected mice. Since previous studies established that IL-12, the major Th1 “priming” cytokine, is able to inhibit KLH-specific memory Th2 cells in primed mice (27), we treated the KLH-primed and challenged mice with rmIL-12. SJL/J mice were preimmunized with KLH as before. After 4 wk the exogenous antigen was given again with encephalitogenic inoculum and, at the same time, a portion of the mice were treated for three consecutive days with rmIL-12. In these mice, the KLHinduced protection was completely abolished, and, in fact, the mice receiving IL-12 showed even greater mean clinical score than the control, non-KLH primed mice (Fig. 2).
Figure 2.

rmIL-12 at the time of immunization overcomes protection engendered by pretreatment and reticulation with KLH. One group of mice that was pretreated with KLH in IFA received the antigen again with the encephalitogenic inoculum together with rmIL-12 (0.3 mg/ mouse), given on days 0, 1, and 2 after immunization (filled squares). The mice receiving the minimal treatment with rmIL-12 were not protected against EAE and showed a significantly higher mean clinical score compared to the KLH-protected mice (filled circles).
KLH-specific T Cells in KLH-preimmunized Mice Have a Th2 Cytokine Profile.
To verify our hypothesis that KLH administered in IFA induces the Th2 response that alters the nature of autoimmune T cells, we examined the cytokine profiles of both KLH- and MBP-specific T cell lines in these mice. T cells were isolated from lymph nodes from the three groups of animals at 10 d p.i. and stimulated in vitro with KLH or MBP, the predominant autoantigen in EAE. After 6 d, the cytokine profiles of these short-term lines directly obtained from the animals were determined by ELISPOT. As expected, the KLH-specific T cells from animals preimmunized intraperitoneally with KLH in IFA showed a predominantly Th2 phenotype. In contrast, when the KLH-primed mice were treated with rmIL-12, both IFN-γ– and IL-4–producing cells were found among the KLH-specific T cells, but the number of IL-4 secreting T cells was consistently decreased (Fig. 3 a). In control mice not primed with KLH, the KLH-specific T cells receiving the first antigenic stimulus in vitro, expressed a Th1 profile. This indicated that primary antigenic stimulation of naive T cells with KLH in vitro leads to a predominantly Th1 phenotype.
Figure 3.


Determination of cytokine profiles of KLH (a) and MBP (b) specific short-term T cell lines. Two mice of each group were killed at day 10 p.i. and lymph node cells stimulated in vitro with KLH or MBP for 6 d. The number of IL-4 (crosshatched bars) and IFN-γ (solid bars) was determined by ELISPOT analysis. The KLH-specific lymph node T cells of mice immunized with the antigen in IFA showed a Th2 cytokine profile. When the mice were treated with rmIL-12, the number of IL-4–producing cells was decreased. In control mice receiving only PBS, the primary antigenic stimulation in vitro with KLH induced a Th1 phenotype. The MBP-specific T cells showed a Th1 inflammatory profile in nonprotected controls (not pretreated with, or not restimulated with KLH) while in KLH-protected mice, the cytokine profile was shifted to a Th2 type. Treatment with rmIL-12 for only 3 d was sufficient to modify the phenotype of the MBP-specific T cells in the KLH/IFA-treated mice from Th2 from Th1.
In the Protected, KLH-primed Mice, the Cytokine Profile of the Encephalitogenic T Cells Is Shifted Towards a Th2 Type.
Among the MBP-specific T cell lines isolated from lymph nodes, the number of IL-4–secreting T cells was significantly higher in the KLH-protected mice compared to the controls (Fig. 3 b). The number of IFN-γ–producing, inflammatory Th1 cells in these mice is very low, a result consistent with the protection against EAE that we observed. When the KLHprimed and challenged mice were treated for 3 d with rmIL-12, the cytokine profile of the MBP-specific T cells was reversed to a predominantly inflammatory Th1 type (Fig. 3 b).
Th2 Autoreactive Cells Are Not Found in the CNS.
We sought to address the question whether the protective effect on EAE induced by KLH-specific Th2 cells was mediated centrally or in the CNS target organ. Histopathological analysis of the brain and spinal cords of KLH-protected and control mice confirmed a lower degree of lymphocyte infiltration in the CNS of the protected mice, consistent with the reduced clinical score (data not shown). To determine the cytokine profile phenotype of the T cells found in the CNS, we performed a reverse transcriptase (RT)-PCR analysis on the total RNA samples extracted from brains and spinal cords of both groups of mice. After normalization for HRPT DNA, we evaluated IL-4 and IFN-γ mRNA expression in the two samples. No IL-4 RNA expression was found in either group of mice. The amount of IFN-γ expression was measured by quantitative PCR. As expected, the attomoles per microgram of IFN-γ mRNA was significantly lower in the protected mice (5 × 10−2 attomoles) than in the control (1.2 × 10−1 attomoles). Although the failure to detect the IL-4 mRNA expression in the brain of the protected mice does not allow us formally to exclude the possibility that Th2 cells enter the CNS, the data support the interpretation that the downregulation of Th1 inflammatory clones takes place in the lymph nodes at the time of autoantigenic stimulation, so that a lower number of Th1 encephalitogenic clones develop and reach the CNS.
Discussion
Autoreactive T lymphocytes are present in the T cell repertoire of healthy individuals and the mechanisms that modulate their activation in T cell–mediated autoimmune diseases remain unclear. Multiple lines of evidence suggest that simultaneous T cell immune responses against nonself antigens, for example, during infections, may modulate the activity of autoreactive T cell clones. Epidemiological studies have clearly shown that environmental factors play a role in the pathogenesis of human autoimmune diseases (28, 29). Experimental studies on transgenic mice expressing the MBP T cell receptor indicate that the ability of the encephalitogenic clones to induce EAE is modulated by lymphocytes with different specificity, presumably specific for antigens present in the environment (9, 10). Most of these studies have stressed the ability of immune responses to environmental antigens to activate the autoreactive T cells (30–32) and induce disease, but in a number of cases, it has been observed that responses against exogenous antigens can downregulate the encephalitogenic T cell clones (17–21). Several laboratories have reported that immunization with mycobacterial proteins such as PPD, hsp65, and hsp70 can confer protection against EAE, but the mechanisms responsible for this protection have not been elucidated. Another example of downmodulation of autoimmune T cell clones by an exogenous antigen is the protection against EAE reported after oral administration of ovalbumin (33). Although a “bystander suppression” mechanism mediated by Th2 cytokines has been proposed, a shift in the cytokine profile of the autoreactive T cells was not clearly demonstrated, and other immunosuppressive cytokines such as TGF-β and IL-10 were implicated in the protection induced.
Several attempts have been made to shift the cytokine profile of autoimmune T cell clones from a Th1 to a Th2 type, for example, by oral administration of the autoantigen (5), or targeting of the encephalitogenic peptide of MBP on B cells (34), and recently by modifying of the amino acid sequence of the encephalitogenic peptide (35). In most such studies, protection against EAE and clinical disease was observed, but a shift in cytokine profiles of the autoreactive T clones from an inflammatory Th1 to a protective Th2, IL-4–producing phenotype has not been clearly shown. At the present time, the mechanisms that regulate the Th1 or Th2 differentiation remain to be clarified, and currently only the presence of “priming” cytokines such as IL-12 or IL-4, in the microenvironment where the naive T cells receive their primary antigenic stimulus, appears to be determinative (36). In other systems it has been demonstrated that Th1 and Th2 subsets with different antigen specificities can interact in vivo. In particular, Th2-type immune responses against parasitic agents can alter Th1 responses to mycobacterial antigens and viruses by an IL-4–dependent mechanism (37–40).
Previous studies have shown that the administration of an antigen in IFA biases the response towards a Th2 phenotype, while CFA more likely induces a Th1 cytokine profile in T helper cells (41). In our model, we find that the production of Th2 cells, defined by the ability to produce IL-4, induced by an exogenous antigen by administration in IFA was sufficient to prime for a cytokine microenvironment in which the autoimmune stimulus was modulated to an attenuated or protective one. This finding indicates that the exogeneous antigen need not be a cognate hsp or cross-reactive with autoantigens to supress EAE. When the effect of IL-4 was countered by the administration of IL-12, the protection provided by the KLH-specific, Th2 cells was abrogated. However, our findings suggest that the modulation of the response mediated by exogenous antigen occurs in the lymphoid system rather than the target organ, since by RT-PCR we were unable to detect IL-4 mRNA in the CNS, although IFN-γ mRNA was readily detectable in the control group that developed EAE. This observation suggests that Th2 autoreactive cells either do not reach, or are not retained in the CNS. Our interpretation is that MBP-specific Th2 cells do not induce inflammation in the CNS and, at the same time, inhibit the Th1 autoreactive clones at the level of the peripheral lymph nodes such that fewer are developed or migrate to the CNS. This is indicated by the lower amount of IFN-γ mRNA we found in the CNS of the KLH-protected mice.
In summary, our data suggest a mechanism for modulation of disease-causing autoimmune T cell clones development by Th2 cells specific for nonself antigens. We believe that a Th2-type immune response against an exogenous antigen modulates the autoimmune disease by modifying the cytokine environment at the time of primary autoantigenic stimulation of the autoreactive T cell clones, shifting their cytokine profile from an inflammatory Th1 to a protective Th2 type. These findings may serve to clarify the role of Th2 immune responses against exogenous antigens, such as those found to occur during parasitic infections, in modulating the pathogenesis of Th1-mediated autoimmune diseases. At the same time, because of obvious risks in immunizing individuals with specific autoantigens to prevent autoimmunity, they provide further support for the possibility of modulating the cytokine profile of encephalitogenic T cell clones by means of exogenous antigens to induce protection in experimental autoimmune diseases of CNS, and encourage the view that it may ultimately be possible to modify the immune responses of autoreactive T cell clones therapeutically to prevent or attenuate human T cell–mediated autoimmune diseases.
Figure 4.

IFN-γ and IL-4 mRNA in the CNS of KLHprotected and control mice sensitized to myelin. The RT-PCR was performed on RNA samples extracted from brain and spinal cord of the mice at day 16 after immunization using specific primers for IFN-γ and IL-4. The mRNAs for IFN-γ and IL-4 were measured by quantitative PCR using a competitor DNA fragment for each, and the quantities expressed as attomoles of cDNA per microgram of total RNA.
Acknowledgments
We thank Celia Brosnan, Robert Modlin, and Marco Salvetti for review of the manuscript.
Footnotes
Dr. M. Falcone was supported by a Fellowship from the Cenci-Bolognetti Foundation, Pasteur Institute of Rome, Italy. This work was supported by the National Multiple Sclerosis Society, National Institutes of Health AI 07118, and the Howard Hughes Medical Institute.
1 Abbreviations used in this paper: CNS, central nervous system; EAE, experimental allergic encephalomyelitis; ELISPOT, enzyme-linked immunospot; HPRT, hypoxanthine phosphoribosyl transferase; MBP, myelin basic protein; rm, recombinant murine; RT, reverse transcriptase.
References
- 1.Ben-Nun A, Wekerle H, Cohen IR. The rapid isolation of clonable antigen-specific T lymphocytes lines capable of mediating autoimmune encephalomyelitis. Eur J Immunol. 1981;11:195–199. doi: 10.1002/eji.1830110307. [DOI] [PubMed] [Google Scholar]
- 2.Pettinelli CB, Fritz RB, Chau CHJ, McFarlin DE. Encephalitogenic activity of guinea pig myelin basic protein in the SJL/J mouse. J Immunol. 1982;129:1209–1211. [PubMed] [Google Scholar]
- 3.Liblau RS, Singer SM, McDevitt HO. Th1 and Th2 CD4+T cells in the pathogenesis of organ-specific autoimmune diseases. Immunol Today. 1995;1:34–38. doi: 10.1016/0167-5699(95)80068-9. [DOI] [PubMed] [Google Scholar]
- 4.Miller SD, Karpus WJ. The immunopathogenesis and regulation of T-cell–mediated demyelinating diseases. Immunol Today. 1994;8:356–361. doi: 10.1016/0167-5699(94)90173-2. [DOI] [PubMed] [Google Scholar]
- 5.Chen Y, Kuchroo VK, Inobe J, Hafler DA, Weiner HL. Regulatory T cell clones induced by oral tolerance: suppression of autoimmune encephalomyelitis. Science (Wash DC) 1994;265:1237–1240. doi: 10.1126/science.7520605. [DOI] [PubMed] [Google Scholar]
- 6.Racke MK, Bonomo A, Scott DE, Cannella B, Levine A, Raine CS, Shevach EM, Rocken M. Cytokine-induced immune deviation as a therapy for inflammatory autoimmune disease. J Exp Med. 1994;180:1961–1966. doi: 10.1084/jem.180.5.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van der Veen RC, Stohlman SA. Encephalitogenic Th1 cells are inhibited by Th2 cells with related peptide specificity: relative roles of interleukin (IL)-4 and IL-10. J Neuroimmunol. 1993;48:213–220. doi: 10.1016/0165-5728(93)90194-4. [DOI] [PubMed] [Google Scholar]
- 8.Miller A, Al-Sabbagh A, Santos LMB, Prabhu-Das M, Weiner HL. Epitopes of myelin basic protein that trigger TGF-b release after oral tolerization are distinct from encephalitogenic epitopes and mediate epitope–driven bystander suppression. J Immunol. 1993;151:7307–7315. [PubMed] [Google Scholar]
- 9.Lafaille JJ, Nagashima K, Katsuki M, Tonegawa S. High incidence of spontaneous autoimmune encephalomyelitis in immunodeficient anti-myelin basic protein T cell receptor transgenic mice. Cell. 1994;78:399–408. doi: 10.1016/0092-8674(94)90419-7. [DOI] [PubMed] [Google Scholar]
- 10.Governman J, Woods A, Larson L, Weiner LP, Hood L, Zaller DM. Transgenic mice that express a myelin basic protein–specific T cell receptor develop spontaneous autoimmunity. Cell. 1993;72:551–560. doi: 10.1016/0092-8674(93)90074-z. [DOI] [PubMed] [Google Scholar]
- 11.Oldstone MBA. Overview: Infectious agents as etiologic triggers of autoimmune disease. Curr Top Microbiol Immunol. 1989;145:1–3. doi: 10.1007/978-3-642-74594-2_1. [DOI] [PubMed] [Google Scholar]
- 12.Yang X-D, Feige U. Heat shock proteins in autoimmune diseases. From causative antigen to specific therapy? . Experientia (Basel) 1992;48:650–656. doi: 10.1007/BF02118311. [DOI] [PubMed] [Google Scholar]
- 13.van den Broek MF, Hogervorst EJM, van Bruggen MCJ, van Eden W, van der Zee R, van den Berg WB. Protection against streptococcal cell wall–induced arthritis by pretreatment with the 65-kD mycobacterial heat shock proteins. J Exp Med. 1989;170:449–466. doi: 10.1084/jem.170.2.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Billingham MEJ, Carney S, Butler R, Colston MJ. A mycobacterial heat shock protein induces antigenspecific suppression of adjuvant arthritis, but is not itself arthritogenic. J Exp Med. 1990;171:339–344. doi: 10.1084/jem.171.1.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elias D, Markovits D, Reshef T, van der Zee R, Cohen IR. Induction and therapy of autoimmune diabetes in the non-obese diabetic (NOD/Lt) mouse by a 65-kDa heat shock protein. Proc Natl Acad Sci USA. 1990;87:1576–1580. doi: 10.1073/pnas.87.4.1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elias D, Reshef T, Birk OS, van der Zee R, Walker MD, Cohen IR. Vaccination against autoimmune mouse diabetes with a T cell epitope of the human 65kDa heat-shock protein. Proc Natl Acad Sci USA. 1991;88:3088–3091. doi: 10.1073/pnas.88.8.3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hempel K, Freitag A, Freitag B, Endres B, Mai B, Liebaldt G. Unresponsiveness to experimental allergic encephalomyelitis in Lewis rats pretreated with complete Freund's adjuvant. Int Arch Allerg Appl Immunol. 1985;76:193–199. doi: 10.1159/000233691. [DOI] [PubMed] [Google Scholar]
- 18.Lehmann D, Ben-Nun A. Bacterial agents protect against autoimmune disease. I. Mice pre-exposed to Bordetella pertussis or Mycobacterium tuberculosisare highly refractory to induction of experimental autoimmune encephalomyelitis. J Autoimmun. 1992;5:675–690. doi: 10.1016/0896-8411(92)90185-s. [DOI] [PubMed] [Google Scholar]
- 19.Fiori, P., G. Ristori, C. Buttinelli, M. Falcone, A. Cacciani, S. Di Giovanni, C. Pozzilli, and M. Salvetti. 1996. Downregulation of cell-surface CD4 coreceptor expression and modulation of experimental allergic encephalomyelitis. Int. Immunol. In press. [DOI] [PubMed]
- 20.Ben-Nun A, Yossefi S, Lehmann D. Protection against autoimmune disease by bacterial agents. II. PPD and pertussis toxin as proteins active in protecting mice against experimental autoimmune encephalomyelitis. Eur J Immunol. 1993;23:689–696. doi: 10.1002/eji.1830230318. [DOI] [PubMed] [Google Scholar]
- 21.Ben-Nun A, Mendel I, Sappler G, Kerlero de Rosbo N. A 12-kDa protein of Mycobacterium tuberculosisprotects mice against experimental autoimmune encephalomyelitis. J Immunol. 1995;154:2939–2948. [PubMed] [Google Scholar]
- 22.Norton WT, Poduslo SE. Myelin in rat brain: method of myelin isolation. J Neurochem. 1973;21:749–757. doi: 10.1111/j.1471-4159.1973.tb07519.x. [DOI] [PubMed] [Google Scholar]
- 23.Merrill JE, Kono DH, Clayton J, Ando DG, Hinton DR, Hofman FM. Inflammatory leukocytes and cytokines in the peptide-induced disease of experimental allergic encephalomyelitis in SJL and B10.PL mice. Proc Natl Acad Sci USA. 1992;89:574–578. doi: 10.1073/pnas.89.2.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leonard JP, Waldburger KE, Goldman SJ. Prevention of experimental autoimmune encephalomyelitis by antibodies against interleukin-12. J Exp Med. 1995;181:381–386. doi: 10.1084/jem.181.1.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taguchi T, McGhee JR, Coffman RL, Beagley KW, Eldridge JH, Takatsu K, Kiyono H. Detection of individual mouse splenic T cells producing IFN-γ and IL-5 using the enzyme-linked immunospot (ELISPOT) assay. J Immunol Methods. 1990;128:65–73. doi: 10.1016/0022-1759(90)90464-7. [DOI] [PubMed] [Google Scholar]
- 26.Reiner SL, Zheng S, Corry DB, Locksley RM. Constructing polycompetitor cDNA for quantitative PCR. J Immunol Methods. 1993;165:37–46. doi: 10.1016/0022-1759(93)90104-f. [DOI] [PubMed] [Google Scholar]
- 27.deKruyff RH, Fang Y, Wolf SF, Umetsu DT. IL-12 inhibits IL-4 synthesis in keyhole limpet hemocyaninprimed CD4+ T cells through an effect on antigen-presenting cells. J Immunol. 1995;154:2578–2587. [PubMed] [Google Scholar]
- 28.Kurtzke, J.F. 1983. Epidemiology of MS. In Multiple Sclerosis. J.F. Hallpike, C.W.M. Adams, and W.E. Tourtellote, editors. Williams and Wilkins, Baltimore, MD. 49–95.
- 29.Ebers GC, Bulman DE, Sadovnick AD, Paty DW, Warren S, Hader W, Murray TJ, Seland TP, Duquette P, Grey T, et al. A population based study of MS in twins. N Engl J Med. 1986;315:1638–1642. doi: 10.1056/NEJM198612253152603. [DOI] [PubMed] [Google Scholar]
- 30.Oldstone MBA. Molecular mimicry and autoimmune disease. Cell. 1987;50:819–820. doi: 10.1016/0092-8674(87)90507-1. [DOI] [PubMed] [Google Scholar]
- 31.Wucherpfennig KW, Strominger JL. Molecular mimicry in T cell–mediated autoimmunity: viral peptides activate human T cell clones specific for myelin basic protein. Cell. 1995;80:695–705. doi: 10.1016/0092-8674(95)90348-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brocke S, Veromaa T, Weissman I, Gijbels K, Steinman L. Infections and multiple sclerosis: A possible role for superantigens? . Trends Microbiol. 1994;2:250–254. doi: 10.1016/0966-842x(94)90630-0. [DOI] [PubMed] [Google Scholar]
- 33.Miller A, Lider O, Weiner HL. Antigen-driven bystander suppression after oral administration of antigens. J Exp Med. 1991;174:791–798. doi: 10.1084/jem.174.4.791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saoudi A, Simmonds S, Huitinga I, Mason D. Prevention of experimental allergic encephalomyelitis in rats by targeting autoantigen to B cells: evidence that the protective mechanism depends on changes in the cytokine response and migratory properties of the autoantigen-specific T cells. J Exp Med. 1995;182:335–344. doi: 10.1084/jem.182.2.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Windhagen A, Scholz C, Hollsberg P, Fukaura H, Sette A, Hafler DA. Modulation of cytokine patterns of human autoreactive T cell clones by a single amino acid substitution of their peptide ligand. Immunity. 1995;2:373–380. doi: 10.1016/1074-7613(95)90145-0. [DOI] [PubMed] [Google Scholar]
- 36.Seder RA, Paul WE, Davis MM, Fazekas de St B, Groth The presence of interleukin-4 during in vitro priming determines the lymphokine-producing potential of CD4+T cells from T cell receptor transgenic mice. J Exp Med. 1992;176:1091–1098. doi: 10.1084/jem.176.4.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pearlman E, Kazura JW, Hazlett FE, Boom WH. Modulation of murine cytokine responses to mycobacterial antigens by helminth-induced T helper 2 cell responses. J Immunol. 1993;151:4857–4864. [PubMed] [Google Scholar]
- 38.Kullberg MC, Pearce EJ, Hieny SE, Sher A, Berzofsky JA. Infection with Schistosoma mansonialters Th1/Th2 cytokine responses to a non-parasite antigen. J Immunol. 1992;148:3264–3270. [PubMed] [Google Scholar]
- 39.Actor JK, Shirai M, Kullberg MC, Buller RML, Sher A, Berzofsky JA. Helminth infection results in decreased virus-specific CD8+cytotoxic T-cell and Th1 cytokine responses as well as delayed virus clearance. Proc Natl Acad Sci USA. 1993;90:948–952. doi: 10.1073/pnas.90.3.948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Curry AJ, Else KJ, Jones F, Bancroft A, Grencis RK, Dunne DW. Evidence that cytokine-mediated immune interactions induced by Schistosoma mansoni alter disease outcome in mice concurrently infected with Trichuris muris. . J Exp Med. 1995;181:769–774. doi: 10.1084/jem.181.2.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Forsthuber T, Yip HC, Lehmann PV. Induction of Th1 and Th2 immunity in neonatal mice. Science (Wash DC) 1996;271:1728–1730. doi: 10.1126/science.271.5256.1728. [DOI] [PubMed] [Google Scholar]