

Abstract
Human immunodeficiency virus-1 (HIV-1) expression in monocyte-derived macrophages (MDM) infected in vitro is known to be inhibited by lipopolysaccharide (LPS). However, the mechanisms are incompletely understood. We show here that HIV-1 suppression is mediated by soluble factors released by MDM stimulated with physiologically significant concentrations of LPS. LPS-conditioned supernatants from MDM inhibited HIV-1 replication in both MDM and T cells. Depletion of C–C chemokines (RANTES, MIP-1α, and MIP-1β) neutralized the ability of LPS-conditioned supernatants to inhibit HIV-1 replication in MDM. A combination of recombinant C–C chemokines blocked HIV-1 infection as effectively as LPS. Here, we report an inhibitory effect of C–C chemokines on HIV replication in primary macrophages. Our results raise the possibility that monocytes may play a dual role in HIV infection: while representing a reservoir for the virus, they may contribute to the containment of the infection by releasing factors that suppress HIV replication not only in monocytes but also in T lymphocytes.
Monocyte/macrophages are key players in the pathogenesis of HIV-1 infection. Macrophages are major reservoirs for HIV-1 during all stages of infection (1, 2) and may be among the first cells to be infected by HIV-1 in patients (3, 4). Unlike T cells, HIV-infected monocytes show little or no virus-induced cytopathic effects in vitro (5, 6). HIV-infected macrophages therefore may persist in tissues for long periods of time and represent a vector for the spread of the infection to different tissues both within the patients and between individuals. In addition to this Trojan horse– like role, it has been recognized that monocytes may play a regulatory role during HIV infection by controlling the pace of disease progression through the release of soluble products (reviewed in 5, 7, 8).
Monocyte/macrophages are critically involved in the immune response to bacterial infections. LPS/endotoxin, the major constituent of the cell wall in gram-negative bacteria, has been shown to activate monocyte/macrophages by interacting with a specific receptor, CD14 (9), a glycosylphosphatidylinositol-linked glycoprotein expressed on the monocytic lineage at high density and less intensely on neutrophils (10, 11). CD14 plays a pivotal role in LPS-induced monokine release during infections and toxic shock (9, 12). More recently, LPS/CD14 interactions have been shown to result in the induction of HIV expression in monocytoid tumor cell lines (13, 14), but to protect primary macrophages from productive infection by HIV-1 in vitro (15, 16). Notably, the concentrations of LPS that affect HIV-1 replication in vitro can be easily reached in vivo and may thus affect viral replication in patients with HIV superinfected with bacteria. The mechanisms underlying the complex effects of LPS on HIV-1 expression in monocytic cells have not been elucidated so far.
We have studied the effects of LPS on HIV-1 expression in cultures of monocyte-derived macrophages (MDM)1 and T cells isolated from normal donors, and infected with different strains of HIV-1. Our results show that LPS-dependent inhibition of HIV infection affected T lymphocytes, as well as MDM, and involved the release of suppressive factors, most notably, the C–C chemokines RANTES (regulated upon activation, normal T expressed and secreted), macrophage inflammatory protein (MIP)-1α, and MIP-1β.
Materials and Methods
Reagents.
PE-conjugated anti-CD14 mAb P9 (anti-Leu-M3, IgG2b) and an isotype control were purchased from Becton Dickinson (Mountain View, CA). A neutralizing rat anti–human IL-10 mAb (J53-19F1, IgG2a) was a gift from Dr. J. Abrams (DNAX Research Institute, Palo Alto, CA). rTNF-α, recombinant C–C chemokines (RANTES, MIP-1α, and MIP-1β), and neutralizing goat polyclonal antibodies against IL-1 receptor antagonist (IL-1Ra: neutralizing dose, ND50 = 5–10 μg/ml), MIP-1α (ND50 = 10 μg/ml), MIP-1β (ND50 = 40 μg/ml), and RANTES (ND50 = 100–200 μg/ml) were obtained from R&D Systems (Minneapolis, MN). The mAbs used in the ELISA assay for soluble TNF receptor 1, and in the immunofluorescence analysis of membrane TNF-α expression were provided by Dr. A. Corti (Department of Biological and Technological Research, San Raffaele Scientific Institute). Concentrations of TNF-α, IL-6, MIP-1α, MIP-1β, and RANTES in culture supernatants were determined by ELISA (Quantikine, R&D Systems).
LPS from Salmonella minnesota and purified goat IgG were purchased from Sigma Chemical Co. (St. Louis, MO). The endotoxin content of all cell culture reagents was assessed by the Limulus amebocyte lysate assay (Whittaker Bioproducts, Walkersville, MD), and was always <0.125 EU/ml. Polymixin B sulfate was purchased from Calbiochem Novabiochem (La Jolla, CA).
Isolation of MDM and HIV-1 Infection.
PBMC were isolated by Ficoll–HyPaque (Pharmacia, Uppsala, Sweden) density gradient centrifugation from buffy-coat preparations obtained from healthy donors. The cells were then resuspended in RPMI-1640 (GIBCO BRL, Gaithersburg, MD) supplemented with 10% AB+ serum (Sigma), 20% FCS (Biological Industries, Israel), 2 mM glutamine, 50 μg/ml streptomycin, and 100 U/ml penicillin, and cultured at a concentration of 1 × 106 cells/cm2 for 5 d at 37°C in 6-well tissue culture plates (Nunc, Roskilde, Denmark), in a 3 ml volume. Non-adherent cells were then removed by extensive washing with medium. The MDM preparations contained ⩾90% CD14+ cells, as assessed by immunofluorescence.
Cells were infected with the monocytotropic HIV-1Ba-L viral strain (tissue culture infectious dose50, TCID50: 110/106 MDM) grown in primary MDM and never previously passaged in continuous cell lines, or with HIV-1IIIB grown in PHA-activated PBMC (TCID50: 45/106 MDM). Furthermore, cells were infected with primary viral isolates (HIV-15088 and HIV-1181) with the biological characteristics of non syncitium-inducing (NSI) strains.
For infection, MDM were incubated with the viral strains at a concentration of 500 pg/ml of p24 Ag in RPMI-1640, 20% FCS, in a total volume of 2 ml of cell-free viral supernatant. After overnight incubation, unbound virus was removed by extensive washing, fresh medium (3 ml) was added, and the cultures were further incubated at 37°C. Supernatants were harvested every day for p24 Ag detection and reverse transcriptase (RT) determination. Culture medium was fully replaced every 3–4 d, without washing.
Isolation of Lymphocytes and HIV-1 Infection.
Normal peripheral blood lymphocytes depleted of monocytes by two cycles of adherence to plastic were activated by a 3-d incubation with PHA (1.5 μg/ml; Sigma). The resulting PHA blasts were collected, resuspended at 2.5 × 106 cells/ml in medium containing 10% FCS, and supplemented with polybrene (2 mg/ml; Sigma) and IL-2 (10 U/ml: Amersham, Buckinghamshire, UK), and incubated overnight in the presence of HIV-1Ba-L. Subsequently, free virus was removed by washing twice in RPMI-1640, and the cells (1.5 × 106/ml) were cultured in 6-well plates in the presence of IL-2. Culture supernatants were harvested every 3–4 d, and tested for the presence of HIV-1 p24 Ag by ELISA.
Preparation of LPS-conditioned and Monokine-depleted Supernatants.
LPS-conditioned supernatants were prepared by incubating cultures of normal uninfected MDM in the presence or absence of LPS (1 μg/ml). 2 d later, supernatants were harvested, centrifuged, and stored at −20°C until used. To deplete LPS-conditioned supernatants of chemokines (MIP-1α, MIP-1β, RANTES), petri dishes were coated for 2 h at room temperature with neutralizing antibodies in PBS, at concentrations (10–30 μg/ml) expected to neutralize the amounts of cytokines found in culture supernatants. Control plates were coated with normal goat IgG (55 μg/ml). LPS-conditioned supernatants were incubated in the sensitized dishes overnight at 37°C, then collected and used immediately.
HIV-1 Detection.
HIV-1 p24 Ag concentrations in the culture supernatants were determined by ELISA (17). In brief, p24 Ag from a detergent lysate of virions was captured by an immobilized anti-p24 Ag polyclonal antibody (D7320; Aalto Bio Reagents, Dublin, Ireland). Bound p24 Ag was then detected using an alkaline phosphatase–conjugated anti-p24 Ag monoclonal antibody (BC 1071-AP; Aalto Bio Reagents) and the AMPAK ELISA amplification system (DAKO A/S, Glostrup, Denmark).
RT activity in the supernatants of HIV-infected MDM was assayed as described in reference 18. In brief, 10 μl of cell-free culture supernatants were added to 50 μl of a mixture containing poly(A), oligo (dT) (Pharmacia), MgCl2, and 32P-labeled deoxythymidine 5′-triphosphate (dTTP) (Amersham) in a 96-well V-bottomed microtiter plate, and incubated 1.5 h at 37°C. 5 μl of the RT reaction mixture were then dotted onto DE81 paper (Whatman, Maidstone, England), dried, washed, and subsequently counted on a microplate scintillation counter (Packard Instrument Co., Meriden, CT).
Immunofluorescence.
Expression of CD14 was detected by direct immunofluorescence, as previously described (19). Cultured monocytes and/or MDM in staining buffer (RPMI-1640, 10% AB+ serum, containing 0.01% sodium azide) were incubated with fluorochrome-conjugated P9 mAb or isotype control for 40 min at 4°C. The cells were then extensively washed and fixed in 2% paraformaldehyde. Percentages of positive cells and mean fluorescence intensity (MFI) were analyzed by a FACScan® (Becton Dickinson) gating on the monocyte population, as defined by forward and side light scatter.
Competitive PCR Amplification.
This procedure was described in detail elsewhere (20). In brief, total RNA was extracted according to the guanidine thiocyanate procedure (21), and treated with RNase-free DNase I (Boehringer, Mannheim, Germany) to remove traces of contaminating DNA. First-strand cDNA synthesis was obtained by priming with random hexamers and reverse transcription in 20 μl of RT mix containing 75 mM KCl, 50 mM Tris–HCl (pH 8.3), 3 mM MgCl2, 0.4 mM each dNTP (Pharmacia), 400 U Moloney murine leukemia virus (MMLV)–RT (Promega, Madison, WI), 20 U RNasin (Promega). RNA was preheated at 65°C for 5 min and incubated with the reaction mix at 37°C. After 1 h, the reaction was stopped by incubation at 95°C for 5 min and samples were cooled on ice. Amplification of CC–CKR-5 cDNA was performed using primers CKR-9 (5′-CATCATCCTCCTGACAATCG) and CKR-10 (5′-ATGGTGAAGATAAGCCTCACAG). Quantification of CC–CKR-5 mRNA levels in MDM was carried out by a competitive PCR procedure using a competitor DNA fragment carrying the primer recognition sites for β-actin (BA1 and BA4 [22]) and for CKR-5 (primers CKR-9 and CKR-10). β-actin is used as a standard to monitor the efficiency of total DNA extraction. A schematic representation of this competitor is shown in Fig. 6 A and its construction is described in the legend to Fig. 6.
Figure 6.

MDM express CC–CKR-5 mRNA. Total RNA was extracted from untreated MDM. RNA samples were treated with DNase I to remove traces of contaminating DNA and reverse transcribed using random hexameric primers. The cDNA products were mixed to scalar amounts of a synthetic competitor DNA fragment containing primer recognition sites for both β-actin and CC–CKR-5 amplification, and amplified with the respective primer pairs. (A) Schematic representation of the competitor DNA fragment used for the quantification of CC–CKR-5 and β-actin cDNA. The fragment contains a core sequence derived from the human β-actin cDNA, carrying a 20-bp insertion in the middle (closed box). Amplification with the β-actin-specific primer set BA1–BA4 detects a 226-bp product on human cDNA, and a 246-bp product from the competitor DNA. To this core sequence, the primer recognition sites for human CC–CKR-5 amplification were added at the two ends (indicated by gray boxes) by reamplification with composite primers corresponding to the CKR-9+BA1 sequence at one end and CKR-10+BA4 at the other end. Amplification with CKR-9 and CKR-10 generates a 288-bp fragment from the competitor template and a 368-bp fragment from the CC–CKR-5 cDNA. (B) Competitive PCR for the quantification of CC– CKR-5 and β-actin mRNAs. cDNA samples from untreated MDM were mixed with tenfold dilution of the competitor DNA fragment as indicated, and amplified with primer sets CKR-9/CKR-10 and BA1/BA4 for CC–CKR-5 and β-actin mRNA quantification. Amplification products were resolved by polyacrylamide gel electrophoresis, stained with ethidium bromide, and quantified by densitometric scanning. According to the principles of competitive PCR, quantification of the target molecules in the samples was obtained by estimation of the ratio between the amplification products, as reported at the bottom of each gel. Furthermore, since the same competitor DNA fragment acts as a competitor for quantification of both CC–CKR-5 and β-actin, standardization for mRNA input is obtained by estimating the ratio between the two measurements, as indicated at the bottom of the figure. M, molecular weight markers.
Competitive PCR amplifications were carried out by adding to the sample increasing concentrations of the competitive templates, in 100 μl of PCR buffer (50 mM KCl, 10 mM Tris–HCl, 2 mM MgCl2) containing the two primers (100 pmol each), the four dNTPs (200 μM each), and 2.5 U of Taq DNA polymerase (Perkin Elmer, Emeryville, CA). Samples were submitted to 50 cycles of amplification with the following cycle profiles: denaturation at 95°C for 30 s, annealing at 60°C (primer sets CC–CKR-5 and β-actin) for 30 s, extension at 72°C for 30 s. After amplification, 10 μl of each PCR reaction were resolved on a 8% nondenaturing polyacrylamide gel, visualized under UV light after ethidium bromide staining and photographed. Quantification of the amplification products was obtained by densitometric scanning.
Determination of Viral DNA Load by Semiquantitative PCR.
High molecular weight DNA was extracted from MDM cultures exposed to HIV-HH1Ba-L for 14 h by overnight incubation at 37°C in lysis buffer (10 mM Tris–HCl, pH 8.3, 1 mM EDTA, 0.5% Nonidet P-40, 0.5% Tween 20, 0.3 mg/ml proteinase K) followed by extraction with a phenol/chloroform/isoamyl alcohol mixture (25:24:1) and ethanol precipitation. Samples were amplified using a sets of nested primers specific for the pol gene (outer primer set, JA79/82; inner primer set, JA 80/81), as previously described (23). Viral DNA load was determined by progressively diluting the samples, and testing each dilution in five parallel reactions. As a positive control, HIV-1-infected cells were diluted in uninfected cells so as to contain 10 and 1 HIV-1 viral DNA copies. The number of viral DNA copies per 106 MDM was calculated according to the Poisson distribution formula. The sensitivity of the PCR assay was shown to be one copy of HIV-1 per 105 cells (24).
Results
LPS Suppresses HIV-1 Replication in MDM Cultures Infected in Vitro.
To characterize the effects of LPS on the replication of HIV-1 in monocytic cells, MDM from normal donors were infected in vitro with the monocytotropic HIV-1Ba-L strain, in the presence or absence of LPS (1 μg/ml). Fig. 1 A shows that p24 Ag secretion in untreated MDM cultures rapidly reached high levels, which were maintained for over 10 d. In contrast, p24 Ag secretion by LPStreated MDM remained extremely low throughout the culture time. RT activity in the same cultures showed a similar pattern (data not shown). Fig. 1 B shows that LPSdependent inhibition of p24 Ag secretion was also observed in MDM cultures infected in vitro with HIV-15088, a primary isolate from an asymptomatic HIV-1-infected patient with the biological characteristics of an NSI isolate. LPS had a potent inhibitory effect on the replication of both HIV-1Ba-L and HIV-15088. Fig. 1 C shows that p24 Ag secretion was inhibited by >70% using LPS at a concentration of 1 ng/ml. Notably, inhibition was still apparent when LPS was added at 10 pg/ml, a physiologically significant concentration (13). Interestingly, LPS addition did not inhibit HIV-1 expression in MDM cultures infected with the SI laboratory strain, HIV-1IIIB (Fig. 1 D). The surprisingly high levels of replication of our HIV-1IIIB in MDM are likely to result from multiple passages of the viral stock in human primary PBMC. Addition of LPS did not result in significant cell death, nor in apoptosis, as assessed by Trypan blue or propidium iodide staining (data not shown).
Figure 1.

LPS suppresses HIV-1 replication in MDM cultures infected in vitro. MDM from healthy donors were infected with HIV-1Ba-L (A), the primary NSI isolate HIV-15088 (B), or HIV-1IIIB (D), all at 500 pg/ml, in the presence or absence of LPS (1 μg/ml). MDM were washed 1 d later and further cultured, adding LPS every 3 d. Culture supernatants were harvested daily, and tested for p24 Ag secretion by ELISA. The data are representative of 10 (A), 3 (B), and 2 (D) separate experiments. In (C) MDM were infected with HIV-1Ba-L or HIV-15088 in the presence of decreasing concentrations of LPS. p24 Ag secretion was assessed 5 d after infection.
LPS-induced inhibition of HIV-1 replication was dependent on the time of addition of LPS to the culture. Fig. 2 shows that HIV-1 expression was completely blocked when LPS was added at the time of infection or 1 d later, but was progressively less affected when LPS was added 2 or 3 d after infection with HIV-1. Notably, viral replication was completely inhibited by pretreating MDM with LPS for 48 h before infection. However, the inhibitory effect of LPS pretreatment was abolished if the cells were subsequently washed before virus addition (data not shown). These data suggest that LPS interferes with early events in HIV-1 infection.
Figure 2.
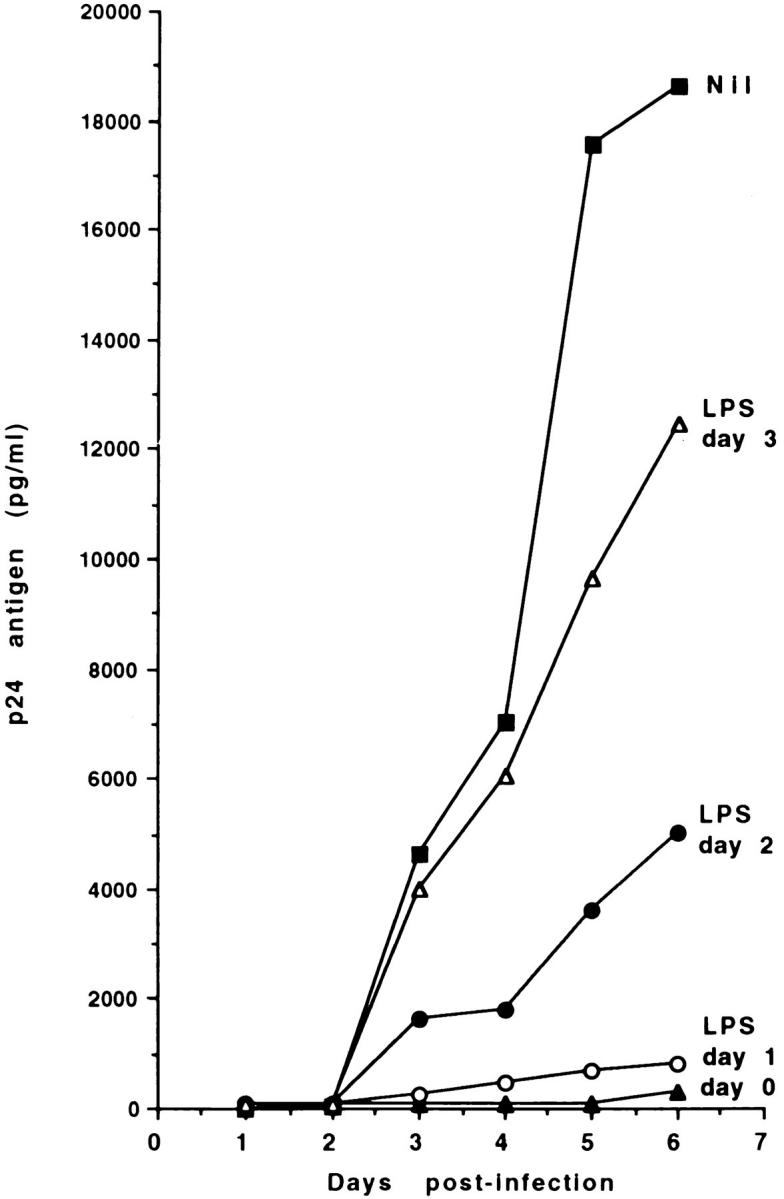
LPS-induced inhibition of HIV-1 expression in MDM cultures is dependent on the time of addition of LPS. MDM were infected with HIV-1Ba-L (500 pg/ml), and stimulated with LPS (1 μg/ml) at different times from the initiation of the culture. Culture supernatants were harvested daily, and tested for p24 Ag secretion by ELISA. The data represent the mean of two separate experiments.
The Expression of CD14, the LPS Receptor, Is Upregulated in LPS-treated, HIV-1-infected MDM.
LPS has been shown to upregulate the expression of its own receptor, CD14, in whole blood (25). Therefore, we asked whether a modulation of CD14 expression may contribute to the effects of LPS on HIV-1 replication in MDM. Immunofluorescence analysis of MDM cultures 2 d after HIV-1 infection showed that CD14 expression was upregulated not only in uninfected, LPS-treated MDM, but also in in vitro HIV-1-infected, LPS-untreated cells (Fig. 3). Interestingly, LPS and HIV-1 synergized in upregulating CD14 expression. These data suggest that the combined effects that HIV-1 infection and LPS stimulation have on CD14 expression may amplify the LPS-induced, CD14-mediated suppression of HIV-1 replication.
Figure 3.

LPS and HIV-1 synergize in upregulating CD14 expression in MDM. MDM were infected with HIV-1Ba-L (500 pg/ml) in the presence or absence of LPS (1 μg/ml). After 2 d of culture, CD14 expression was assessed by direct immunofluorescence, using PE-conjugated mAb P9 and an unrelated isotype control. The data are representative of three separate experiments.
LPS-induced HIV-1 Suppression Is Not Mediated by an Effect on the Secretion of IL-6 and TNF-α.
A number of cytokines have been described that regulate HIV-1 expression. In particular, TNF-α and IL-6 enhance HIV-1 replication in acutely infected MDM. The HIV-1–inducing effect of TNF-α is mainly, if not exclusively, mediated by the activation of NF-κB, which activates LTR-driven viral RNA transcription (26). IL-6 induces expression of viral proteins and RT activity to levels comparable to those induced by TNF-α, but unlike TNF-α, does not increase significantly the levels of steady-state viral mRNA (27). Therefore, we investigated whether a decrease in the production of these HIV-1 stimulatory cytokines may underlie LPS-dependent inhibition of HIV-1 replication in MDM. Fig. 4 shows that LPS-induced IL-6 secretion was vigorous and comparable in both uninfected and HIV-1–infected MDM cultures. In contrast, infected cultures treated with LPS showed an impairment in their ability to sustain TNF-α secretion over time. However, stimulation with LPS released high and comparable levels of TNF-α (>40 ng/ml) from uninfected and infected cells at the initiation of the culture, before removal of unbound virus. The decrease in TNF-α detected after ⩾2 d of culture did not result from masking by shed soluble TNF receptors, nor from a selective upregulation of membrane TNF-α (data not shown). Addition of rTNF-α (10 and 100 U/ml) did not restore HIV-1 expression, as detected by p24 Ag (data not shown). Thus, the decrease in TNF-α was not responsible for the inhibitory effect of LPS on HIV-1 replication. Loss of sensitivity of HIV-1-infected MDM to TNF-α–mediated upregulation of HIV expression, rather than decreased levels of TNF-α, may be involved in LPS-induced inhibition of HIV infection. The mechanisms involved in TNF-α suppression are currently under investigation.
Figure 4.

Effects of LPS stimulation and/or HIV-1 infection on IL-6 and TNF-α secretion by MDM. Uninfected or HIV-1Ba-Linfected MDM were cultured in the presence or absence of LPS (1 μg/ml). LPS was added to the cultures every 3 d. IL-6 and TNF-α concentrations in the supernatants were measured by ELISA. The data are representative of four separate experiments.
LPS-induced Inhibition of HIV Replication Is Mediated by Soluble Factors Active on Both MDM and T Lymphocytes.
The finding that pretreatment with LPS inhibited HIV-1 infection only if the cells were not washed before adding the virus prompted us to investigate whether the effects of LPS are mediated by soluble factors. To this purpose, LPSconditioned supernatants were obtained from MDM cultures stimulated with LPS for 24 h, and LPS was neutralized by the addition of polymixin B (15 μg/ml). Normal MDM were then infected with HIV-1 and cultured either with LPS, or with these supernatants (100% vol/vol) in the absence of LPS. Table 1 shows that the supernatants from LPS-treated MDM inhibited HIV-1 replication as actively as LPS itself, even in the presence of polymixin B. Interestingly, the effect of the soluble inhibitory factor(s) was not MDM specific. Indeed, Table 2 shows that the same LPSconditioned supernatants also suppressed viral expression in T lymphocytes infected with the NSI strains HIV-1Ba-L and HIV-1181, a primary isolate. The inhibitory effect of LPSconditioned MDM supernatants on HIV replication in T cells was particularly remarkable, because LPS per se had no effect when added directly to purified infected T cells. However, LPS-conditioned supernatants failed to suppress the replication of an SI primary isolate, HIV-15233, in T cells. These results suggest that suppressive monokines released by MDM upon stimulation with LPS are responsible for the observed inhibition of HIV replication.
Table 1.
LPS-induced Inhibition of HIV Replication in MDM Is Mediated by the Release of Soluble Factors
| Culture | Supernatant added | Polymixin | HIV-1 p24 Ag release | |||||
|---|---|---|---|---|---|---|---|---|
| Day 4 | Day 7 | |||||||
| pg/ml | ||||||||
| MDM+HIV-1 | Nil | − | 3,209 | 12,616 | ||||
| MDM+HIV-1 | Nil | + | 3,620 | 13,917 | ||||
| MDM+HIV-1+ 1LPS | Nil | − | 108 | 992 | ||||
| MDM+HIV-1+ 1LPS | Nil | + | 2,953 | 12,408 | ||||
| MDM+HIV-1 | Untreated MØ | + | 3,048 | 13,726 | ||||
| MDM+HIV-1 | LPS-treated MØ | + | 100 | 300 | ||||
MDM from healthy donors were infected in vitro with HIV-1, in the presence of LPS (1 μg/m), LPS-conditioned supernatants (100% vol/ vol), or polymixin B sulfate (15 μg/ml). Supernatants from infected cultures were harvested at different timepoints, and assayed by ELISA for p24 Ag secretion.
Table 2.
Soluble Factors Released by LPS-treated MDM Inhibit the Replication of NSI HIV-1 Strains in T Lymphocytes
| Culture | Supernatant added | Polymixin | HIV-1 p24 Ag release | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ba-L | 181 | 5233 | ||||||||
| pg/ml | ||||||||||
| T cells+HIV-1 | − | − | 2,155 | 8,755 | 7,057 | |||||
| T cells+HIV-1 | − | + | 2,355 | 6,390 | 7,592 | |||||
| T cells+HIV-1+LPS | − | − | 2,344 | 7,795 | 6,793 | |||||
| T cells+HIV-1+LPS | − | + | 2,086 | 7,885 | 7,738 | |||||
| T cells+HIV-1 | Untreated MØ | + | 2,225 | 8,927 | 7,462 | |||||
| T cells+HIV-1 | LPS-treated MØ | + | 47 | 281 | 7,198 | |||||
Lymphocytes from healthy donors were infected in vitro with two NSI HIV-1 strains, HIV-1Ba-L and HIV-1181, or with an SI strain, HIV-15233, in the presence or absence of LPS (1 μg/m), LPS-conditioned supernatants (100% vol/vol) or polymixin B, sulfate (15 μg/ml). Supernatants from infected cultures were harvested 5 d after infection, and assayed by ELISA for p24 Ag secretion.
C–C Chemokines Released by LPS-stimulated MDM Mediate the Suppression of HIV Replication.
Several monokines have been reported to suppress HIV-1 replication. Among them, IL-10 blocks HIV replication by inhibiting the secretion of endogenous TNF-α and IL-6 (28), cytokines that upregulate HIV expression. IL-1Ra, on the other hand, has been described to be produced by HIV-infected MDM in excess relative to IL-1α and IL-β, and thus effectively counteracts IL-1-mediated induction of HIV expression (29). We tested whether the release of these monokines was responsible for the LPS-induced inhibition of HIV-1 expression in MDM. To this purpose, neutralizing anti-IL-10 or antiIL-1Ra antibodies were added to MDM cultures infected with HIV-1 and stimulated with LPS. Fig. 5 shows that addition of neither antibody reversed the suppression of HIV-1 replication caused by LPS, thus ruling out a role of IL-10 and IL-1Ra in HIV-1 suppression.
Figure 5.

Effects of neutralizing antibodies against HIV-1-inhibitory cytokines. MDM were infected with HIV-1Ba-L and stimulated with LPS (1 μg/ml), in presence or absence of neutralizing anti-IL-1Ra or anti-IL-10 antibodies (10 μg/ml). Culture supernatants were harvested daily, and tested for p24 Ag secretion by ELISA. The data represent the mean of two separate experiments. Control antibodies had no effect on p24 Ag secretion.
CD8+ T lymphocytes release soluble factors that inhibit HIV-1 replication in CD4+ T cells in a manner not restricted by the major histocompatibility complex (30). HIV-1 inhibition was recently shown to depend on the presence of the C–C chemokines RANTES, MIP-1α, and MIP-1β (31), the natural ligands of CC–CKR-5, the second receptor for primary NSI strains (32, 33, 34). Therefore, in preliminary experiments, we assessed whether CC–CKR-5 is expressed in MDM, and whether stimulation with LPS induces the release of these chemokines. Competitive PCR experiments were carried out to quantitatively determine the levels of CC–CKR-5 mRNA in total cDNA isolated from MDM. Quantification was achieved by using a DNA fragment that acts as a dual competitor for PCR amplification of both β-actin (as an internal standard) and CC– CKR-5 cDNA (Fig. 6 A). Fig. 6 B shows that high levels of CC–CKR-5 mRNA were expressed by MDM at the time of infection. Stimulation with LPS did not upregulate the expression of CC–CKR-5 in infected MDM (data not shown).
Next, we investigated whether stimulation with LPS induces MDM to release C-C chemokines. Table 3 shows that addition of LPS resulted in vigorous production of these C–C chemokines by MDM, both uninfected and infected in vitro with HIV. Then, we investigated whether the C–C chemokines released in LPS-conditioned supernatants played a role in the inhibition of HIV-1 replication. The simultaneous neutralization of RANTES, MIP-1α, and MIP-1β has been shown to be required to abrogate the HIV suppressive effects of CD8+ T cell supernatants. Thus, high concentrations of antibodies are necessary to achieve neutralization (31). Because monocytes and MDM express all types of Fcγ receptors (CD64, CD32, and CD16), the engagement of which is known to modulate HIV expression (35), supernatants from LPS-stimulated MDM cultures were simultaneously depleted of RANTES, MIP-1α, and MIP-1β by adsorption on specific antibodies immobilized on plastic. After polymixin B was added to neutralize LPS, the chemokine-depleted supernatants were added to HIV1-infected MDM from different donors. In the representative experiment shown in Table 4, LPS-conditioned supernatants completely inhibited p24 Ag secretion. Depletion of C–C chemokines neutralized the inhibitory activity of the supernatants. In contrast, supernatants adsorbed on control goat IgG were almost as inhibitory as the undepleted ones. Our data suggest that the LPS-dependent release of HIV-1 suppressive chemokines plays a major role in the inhibition of HIV-1 replication in MDM.
Table 3.
C–C Chemokine Secretion in MDM Cultures
| Day 2 | Day 5 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MIP-1α | MIP-1β | RANTES | MIP-1α | MIP-1β | RANTES | |||||||
| pg/ml | pg/ml | |||||||||||
| Experiment 1 | ||||||||||||
| Nil | 1,710 | 591 | 198 | 900 | 610 | 190 | ||||||
| LPS | 39,297 | 17,164 | 12,740 | 2,014 | 2,302 | 2,494 | ||||||
| HIV | 2,040 | 627 | 281 | 1,227 | 1,612 | 542 | ||||||
| HIP+LPS | 34,452 | 20,253 | 8,380 | 2,266 | 3,719 | 818 | ||||||
| Experiment 2 | ||||||||||||
| Nil | 380 | 50 | 38 | 1,200 | 1,300 | 67 | ||||||
| LPS | 22,680 | 37,590 | 12,080 | 25,480 | 47,220 | 2,279 | ||||||
| HIV | 1,750 | 2,330 | 47 | 2,960 | 8,200 | 75 | ||||||
| HIV+LPS | 21,710 | 20,110 | 8,890 | 7,440 | 13,960 | 332 | ||||||
Uninfected or HIV-1Ba-L-infected MDM were cultured in the presence or absence of LPS (1 μg/ml). LPS was added to the cultures every 3 d. Supernatants were harvested after 2 and 5 d of culture. The concentrations of C–C chemokines in the supernatants were measured by ELISA.
Table 4.
Antibody-mediated Depletion of C–C Chemokines Neutralizes the HIV Suppressive Activity of LPS-conditioned Supernatants
| Culture | Supernatant added | Polymixin | Depletion | HIV-1 p24 Ag | ||||
|---|---|---|---|---|---|---|---|---|
| pg/ml | ||||||||
| MDM+HIV-1 | − | − | − | 4,516 | ||||
| MDM+HIV-1 | − | + | − | 4,426 | ||||
| MDM+HIV-1+LPS | − | − | − | 597 | ||||
| MDM+HIV-1+LPS | − | + | − | 4,500 | ||||
| MDM+HIV-1 | Untreated MØ | + | − | 5,739 | ||||
| MDM+HIV-1 | LPS-treated MØ | + | − | 176 | ||||
| MDM+HIV-1 | LPS-treated MØ | + | Anti-chemokines | 3,597 | ||||
| MDM+HIV-1 | LPS-treated MØ | + | Normal goat IgG | 806 |
MDM from healthy donors were infected in vitro with HIV-1Ba-L, in the presence of LPS-conditioned supernatants (100% vol/vol), undepleted or depleted of monokines by adsorption on specific neutralizing antibodies or control IgG immobilized on plastic. Polymixin B sulfate was added at a concentration of 15 μg/ml. Supernatants from infected cultures were harvested after 4 d of culture, and assayed by ELISA for p24 Ag secretion. The table shows the results of a representative experiment.
Competitive inhibition of HIV-1 coreceptor utilization by released chemokines is expected to result in the inhibition of HIV entry into MDM (32–34). Therefore, we tested the effects of LPS and LPS-conditioned supernatants on the early stages of the HIV-1 replication cycle by assessing the levels of proviral DNA in MDM incubated with HIV-1Ba-L for 14 h, in the presence or absence of LPS and LPS-conditioned supernatants. Viral DNA load was determined by a semiquantitative nested PCR procedure, using two primer sets specific for the pol gene (23, 24). In a representative experiment, 5,860 viral DNA copies were detected in 106 infected MDM 14 h after infection. Addition of LPS or LPS-conditioned supernatants reduced the number of viral DNA copies to 407 and 585 per 106 MDM, respectively, thus decreasing viral load by 93 and 90%. The finding that LPS treatment suppressed the rate of proviral DNA formation at an early time after MDM infection is consistent with the reported ability of C–C chemokines to interfere with HIV-1 entry.
Recombinant C–C Chemokines Inhibit HIV-1 Replication in Human MDM.
To assess whether C–C chemokines are sufficient to inhibit HIV-1 replication in MDM, recombinant RANTES, MIP-1α, and MIP-1β were added to HIVinfected MDM, alone or in combination. Fig. 7 (left) shows that a combination of the three chemokines, each at a concentration of 50 ng/ml, inhibited the replication of HIV-1Ba-L in infected MDM by 76%. In the same experiments, addition of LPS reduced p24 Ag release by 75%. Among the three chemokines, RANTES was the most effective one, because it inhibited HIV-1Ba-L infection as efficiently as LPS when used at a concentration of 250 ng/ml. Notably, the inhibitory effect of C–C chemokines on HIV-1 replication was even more pronounced in MDM cultures infected with NSI primary viral isolates. Indeed, Fig. 7 (right) shows that RANTES, MIP-1α, and MIP-1β blocked the replication of HIV-15088 by >75% even when used individually at a concentration as low as 10 ng/ml. The combination of the three chemokines suppressed HIV-15088 by over 90%. The concentrations of recombinant chemokines used in our experiments were physiologically significant. Indeed, the assessment of the concentrations of endogenous chemokines released by MDM during the overnight incubation with virus and LPS before washing (data not shown) demonstrated that at the time of in vitro infection, HIV is exposed to similar amounts of chemokines. These results show that recombinant chemokines are sufficient to inhibit HIV infection in human MDM.
Figure 7.

Recombinant C–C chemokines inhibit HIV-1 replication in human MDM. MDM from healthy donors were infected in vitro with HIV-1Ba-L (left) or with the NSI primary viral isolate HIV-15088 (right), in the presence or absence of LPS (1 μg/ml) and recombinant chemokines. Chemokines were added to HIV-1Ba-L-infected cultures at a concentration of 250 ng/ml when used individually, and 50 ng/ml each when used in combination. For HIV-15088infected cultures, chemokines were used at 10 ng/ml, individually and in combination. Supernatants from infected cultures were harvested at different timepoints, and assayed by ELISA for p24 Ag secretion.
Discussion
For several years, it has been known that stimulation with bacterial LPS protects macrophages from productive infection by HIV-1 in vitro (15, 16). Despite the potential implications of this finding for the pathogenesis and treatment of HIV infection, the mechanisms responsible for the HIV suppressive effect of LPS have remained unknown. Our present results indicate that LPS stimulates human MDM to release soluble factors, the C–C chemokines RANTES, MIP-1α, and MIP-1β, that strongly inhibit HIV replication, not only in macrophages but also in T lymphocytes.
These data may help redefine our current understanding of the role played by monocyte/macrophages in the pathogenesis of HIV infection. Macrophages have been viewed mostly negatively, as major targets for infection (3, 4), reservoirs for the virus (1, 2), triggers for T cell apoptosis (36, 37), and last but not least, as a source of soluble factors (TNF-α, IL-1, IL-6) that sustain viral replication (27, 38, 39). The potential for a defensive role of macrophages only became clear after C–C chemokines have been shown to exert a potent inhibitory effect on HIV replication (31). These chemoattractants are vigorously secreted not only by CD8+ T lymphocytes, the cells traditionally implicated in HIV-1 suppression, but also by activated monocyte/macrophages (40). Important indications about the possible mechanism of action of these HIV-suppressive chemokines have emerged from a series of recent reports, which demonstrated that selected chemokine receptors act as critical surface membrane cofactors for HIV infection (32–34, 41–43). These include CC–CKR-5, a RANTES, MIP-1α, and MIP-1β receptor that is used by most primary NSI strains, and LESTR/fusin, the receptor for the lymphocyte chemoattractant SDF-1 (44, 45), used by cell line–adapted SI strains, as well as CC– CKR-2b and CC–CKR-3, which may also be used by a limited number of isolates. C–C chemokines block membrane fusion and HIV-1 entry, either by competing for the HIV-1 binding site on CC–CKR-5, and/or through the downregulation of surface receptor expression (32, 33, 34).
Within this context, our data suggest that a receptor for RANTES, MIP-1α, and MIP-1β is a major cofactor for HIV-1 entry in macrophages, as well as T cells. This hypothesis is supported by a number of findings presented in this paper: (a) at the time of infection, MDM expressed mRNA for CC–CKR-5, the β-chemokine receptor; (b) stimulation with LPS induced the release of endogenous C–C chemokines, and reduced viral DNA load in infected MDM by >90% as early as 14 h after infection; (c) depletion of C–C chemokines strongly reduced the HIV-suppressive capacity of LPS-conditioned MDM supernatants; (d) recombinant chemokines at physiologically significant concentrations inhibited the replication of HIV-1 NSI strains in MDM, and (e) activation of MDM with LPS inhibited the replication of NSI primary isolates, but not of HIV-1IIIB, an SI laboratory strain. This pattern is consistent with the observation that C–C chemokines do not block the replication of HIV-1IIIB (31), because this SI strain, unlike HIV-1Ba-L and the NSI strains, requires LESTR/fusin, rather than CC–CKR-5, as a coreceptor for entry and fusion (41).
The suppressive effect of C–C chemokines on MDM infection by NSI HIV-1 strains is supported by the observation that recombinant RANTES, MIP-1α, and MIP-1β potently inhibited fusion between primary macrophages and HIV-1Ba-L Env-expressing cells (34). Furthermore, it has been recently shown that both macrophages and CD4+ T cells from multiply exposed uninfected individuals (46) resist infection by primary NSI isolates of HIV-1, while remaining susceptible to infection by SI strains (47). It is not yet clear whether resistance results from a defect in second receptor usage secondary to C–C chemokine hyperproduction (46) and/or from mutations in the CC–CKR-5 gene that generate a nonfunctional receptor unable to support cell fusion and infection by NSI HIV-1 strains (48). However, it seems likely that susceptibility to HIV-1 infection of both macrophages and T cells is genetically determined, and critically regulated by the interactions between C–C chemokines and their receptors.
The issue of C–C chemokine-induced inhibition of HIV-1 replication in macrophages is still somewhat controversial. Dragic et al. (33) recently reported that entry of NSI HIV-1 strains into primary macrophages was relatively insensitive to C–C chemokines. This discrepancy with our results is likely to be caused by differences in the experimental conditions. The readout in our experiments was p24 Ag secretion in MDM cultures infected with several HIV-1 strains, including primary isolates from HIV-1-infected patients. In contrast, Dragic et al. assessed virus entry by a single-cycle infection with an env-deficient virus, which also carries the luciferase reporter gene, complemented by envelope glycoprotein expressed in trans (33). This assay, although elegant, is necessarily artificial, and may not capture the full complexity of virus–host cell interactions in macrophages infected with naturally occurring HIV-1 strains. On the other hand, a number of differences in culture conditions (i.e., MDM propagation and stimulation and/or virus source and/or activity of recombinant chemokines) may have determined the lack of HIV suppression observed by Schmidtmayerova et al. (49). In our hands, different batches of recombinant C–C chemokines have reproducibly suppressed infection by different HIV-1 NSI strains over several months.
While the results obtained with recombinant chemokines clearly show that these chemoattractants are sufficient to suppress HIV replication in MDM, it is possible that LPS-conditioned supernatants contain additional factor(s) with HIV suppressive effects. Preliminary experiments in our laboratory indicate that the replication of some HIV-1 strains is insensitive to C–C chemokine-mediated inhibition, but is blocked by LPS-conditioned supernatants (Verani, A., G. Scarlatti, and D. Vercelli, manuscript in preparation). The nature of other potential HIV-suppressive factor(s) contained in the LPS-conditioned MDM supernatants, and their function vis-a-vis chemokines, is currently under investigation. IFN (α and/or β) is a good candidate, because it is known to be released by LPS-stimulated MDM (50), and to block HIV replication (51).
Finally, the observation that LPS-stimulated macrophages release soluble factors that effectively inhibit HIV replication in both macrophages and T cells may prompt a reinterpretation of the role played by bacterial superinfections in the pathogenesis and progression of HIV infection. It has been recently shown that CD14 is not just the receptor for LPS of gram-negative bacteria (9) but is a multipurpose receptor for foreign lipoglycans of gram-positive bacteria and mycobacteria (52, 53). Thus, a vast array of exogenous stimuli derived from microbial pathogens may conceivably trigger intense chemokine release. In this perspective, the effect of bacterial superinfections in patients with HIV-1 immunodeficiency may be complex and somewhat counterintuitive. The chemokine response triggered by the infectious agent upon interaction with the macrophages of the host in fact may contribute to the containment of HIV-1 infection in the main targets of the virus, T cells, and mononuclear phagocytes.
Acknowledgments
This work was supported by AIDS Project, Istituto Superiore di Sanitá, Italy (grant 9306-39 to D. Vercelli, 9306-20 to G. Scarlatti, 9402-11 to M. Giacca, 9304-78 to P. Lusso, and 9405-04 to A.G. Siccardi). A. Verani was the recipient of a fellowship from Istituto Superiore di Sanitá.
Footnotes
1 Abbreviations used in this paper: MDM, monocyte-derived macrophages; MIP, macrophage inflammatory protein; NSI, nonsyncitium-inducing; RANTES, regulated upon activation, normal T expressed and secreted; RT, reverse transcriptase.
References
- 1.Stoler MH, Eskin TA, Benn S, Angerer RC, Angerer LM. Human T-cell lymphotrophic virus type III infection of the central nervous system. Preliminary in situ analysis. JAMA. 1986;256:2360–2364. [PubMed] [Google Scholar]
- 2.Koenig S, Gendelman HE, Orenstein JM, Canto MCD, Pezeshkpour GM, Yungbluth M, Janotta F, Aksamit A, Martin MA, Fauci AS. Detection of AIDS virus in macrophages in brain tissue from AIDS patients with encephalopathy. Science (Wash DC) 1986;233:1089–1093. doi: 10.1126/science.3016903. [DOI] [PubMed] [Google Scholar]
- 3.Gendelman HE, Orenstein JM, Martin MA, Ferrua C, Mitra R, Phipps T, Wahl LA, Lane HC, Fauci AS, Burke DS, et al. Efficient isolation and propagation of human immunodeficiency virus on recombinant colony-stimulating factor 1–treated monocytes. J Exp Med. 1988;167:1428–1441. doi: 10.1084/jem.167.4.1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weiss SH, Goedert JJ, Gartner S, Popovic M, Waters D, Markham P, Di Marzo F, Veronese, Gail MH, Barkley WE, Gibbons J, et al. Risk of human immunodeficiency virus infection among laboratory workers. Science (Wash DC) 1988;239:68–71. doi: 10.1126/science.3336776. [DOI] [PubMed] [Google Scholar]
- 5.Ho DD, Rota TR, Hirsh MS. Infection of monocytes/macrophages by human T lymphotropic virus type III. J Clin Invest. 1986;77:1712–1715. doi: 10.1172/JCI112491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Salahuddin SZ, Rose RM, Groopman JE, Markham PD, Gallo RC. Human T lymphotropic virus type III infection in human alveolar macrophages. Blood. 1986;68:281–284. [PubMed] [Google Scholar]
- 7.Ho DD, Roger MD, Pomerantz RJ, Kaplan JC. Pathogenesis of infection with human immunodeficiency virus. N Engl J Med. 1987;317:278–286. doi: 10.1056/NEJM198707303170505. [DOI] [PubMed] [Google Scholar]
- 8.Meltzer MS, Skillman DR, Gomatos PJ, Kalter DC, Gendelman HE. Role of mononuclear phagocytes in the pathogenesis of human immunodeficiency virus infection. Annu Rev Immunol. 1990;8:169–194. doi: 10.1146/annurev.iy.08.040190.001125. [DOI] [PubMed] [Google Scholar]
- 9.Wright SD, Ramos RA, Tobias PS, Ulevitch RJ, Mathison JC. CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science (Wash DC) 1990;249:1431–1433. doi: 10.1126/science.1698311. [DOI] [PubMed] [Google Scholar]
- 10.Haziot A, Chen S, Ferrero E, Low MG, Silber R, Goyert SM. The monocyte differentiation antigen, CD14, is anchored to the cell membrane by a phosphatidylinositol linkage. J Immunol. 1988;141:547–552. [PubMed] [Google Scholar]
- 11.Simmons DL, Tan S, Tenen DG, Nicholson-Weller A, Seed B. Monocyte antigen CD14 is a phospholipid anchored membrane protein. Blood. 1989;73:284–289. [PubMed] [Google Scholar]
- 12.Dentener MA, Bazil V, Von Asmuth EJU, Ceska M, Buurman WA. Involvement of CD14 in lipopolysaccharide-induced tumor necrosis factor-α, IL-6 and IL-8 release by human monocytes and alveolar macrophages. J Immunol. 1993;150:2885–2891. [PubMed] [Google Scholar]
- 13.Pomerantz RJ, Feinberg MB, Trono D, Baltimore D. Lipopolysaccharide is a potent monocyte/macrophagespecific stimulator of human immunodeficiency virus type 1 expression. J Exp Med. 1990;172:253–261. doi: 10.1084/jem.172.1.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bagasra O, Wright SD, Seshamma T, Oakes JW, Pomerantz RJ. CD14 is involved in control of human immunodeficiency virus type 1 expression in latently infected cells by lipopolysaccharide. Proc Natl Acad Sci USA. 1992;89:6285–6289. doi: 10.1073/pnas.89.14.6285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kornbluth RS, Oh PS, Munis JR, Cleveland PH, Richman DD. Interferons and bacterial lipopolysaccharide protect macrophages from productive infection by human immunodeficiency virus in vitro. J Exp Med. 1989;169:1137–1151. doi: 10.1084/jem.169.3.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bernstein MS, Tong-Starksen SE, Locksley RM. Activation of human monocyte-derived macrophages with lipopolysaccharide decreases human immunodeficiency virus replication in vitro at the level of gene expression. J Clin Invest. 1991;88:540–545. doi: 10.1172/JCI115337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moore JP, McKeating JA, Huan YX, Ashkenazi A, Ho DD. Virions of primary human immunodeficiency virus type 1 isolates resistant to soluble CD4 (sCD4) neutralization differ in sCD4 binding and glycoprotein gp120 retention from sCD4-sensitive isolates. J Virol. 1992;66:235–243. doi: 10.1128/jvi.66.1.235-243.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Willey RL, Smith DH, Lasky LA, Theodore TS, Earl PL, Moss B, Copon DJ, Martin MA. In vitromutagenesis identifies a region within the envelope gene of the human immunodeficiency virus that is critical for infectivity. J Virol. 1988;62:139–147. doi: 10.1128/jvi.62.1.139-147.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cosentino G, Soprana E, Thienes CP, Siccardi AG, Viale G, Vercelli D. IL-13 downregulates CD14 expression and TNF-α secretion in human monocytes. J Immunol. 1995;155:3145–3151. [PubMed] [Google Scholar]
- 20.Comar M, Marzio G, Dagaro P, Giacca M. Quantitative dynamics of HIV type 1 expression. AIDS Res Hum Retroviruses. 1996;12:117–126. doi: 10.1089/aid.1996.12.117. [DOI] [PubMed] [Google Scholar]
- 21.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate–phenol– chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 22.Grassi G, Zentilin L, Tafuro S, Diviacco S, Ventura A, Falaschi A, Giacca M. A rapid procedure for the quantitation of low abundance RNAs by competitive reverse transcription–polymerase chain reaction. Nucleic Acids Res. 1994;22:4547–4549. doi: 10.1093/nar/22.21.4547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Albert J, Fenyo EM. Simple, sensitive, and specific detection of human immunodeficiency virus type 1 in clinical specimens by polymerase chain reaction with nested primers. J Clin Microbiol. 1990;28:1560–1564. doi: 10.1128/jcm.28.7.1560-1564.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brinchmann JE, Albert J, Vartdal F. Few infected CD4+ T cells but a high proportion of replicationcompetent provirus copies in asymptomatic human immunodeficiency virus type 1 infection. J Virol. 1991;65:2019–2023. doi: 10.1128/jvi.65.4.2019-2023.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marchant A, Duchow J, Delville J-P, Goldman M. Lipopolysaccharide induces up-regulation of CD14 molecule on monocytes in human whole blood. Eur J Immunol. 1992;22:1663–1665. doi: 10.1002/eji.1830220650. [DOI] [PubMed] [Google Scholar]
- 26.Cullen BR, Green WC. Regulatory pathways governing HIV-1 replication. Cell. 1989;58:423–426. doi: 10.1016/0092-8674(89)90420-0. [DOI] [PubMed] [Google Scholar]
- 27.Poli G, Bressler P, Kinter A, Duh E, Timmer WC, Rabson A, Justement JS, Stanley S, Fauci AS. Interleukin 6 induces human immunodeficiency virus expression in infected monocytic cells alone and in synergy with tumor necrosis factor α by transcriptional and post-transcriptional mechanisms. J Exp Med. 1990;172:151–158. doi: 10.1084/jem.172.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weissman D, Poli G, Fauci AS. Interleukin 10 blocks HIV replication in macrophages by inhibiting the autocrine loop of tumor necrosis factor α and interleukin 6 induction of virus. AIDS Res Hum Retroviruses. 1994;10:1199–1206. doi: 10.1089/aid.1994.10.1199. [DOI] [PubMed] [Google Scholar]
- 29.Zavala F, Rimaniol A-C, Boussin F, Dormont D, Bach J-F, Descamps-Latscha B. HIV predominantly induces IL-1 receptor antagonist over IL-1 synthesis in human primary monocytes. J Immunol. 1995;155:2784–2793. [PubMed] [Google Scholar]
- 30.Walker CM, Moody DJ, Stites DP, Levy JA. CD8+ lymphocytes can control HIV infection in vitro by suppressing virus replication. Science (Wash DC) 1986;234:1563–1566. doi: 10.1126/science.2431484. [DOI] [PubMed] [Google Scholar]
- 31.Cocchi F, DeVico AL, Garzino-Demo A, Arya SK, Gallo RC, Lusso P. Identification of RANTES, MIP-1α and MIP-1β as the major suppressive factors produced by CD8+ T cells. Science (Wash DC) 1995;270:1811–1815. doi: 10.1126/science.270.5243.1811. [DOI] [PubMed] [Google Scholar]
- 32.Deng H, Liu R, Elimeier W, Choe S, Unutmaz D, Burkhart M, Di Marzio P, Marmon S, Sutton RE, Hill CM, et al. Identification of a major co-receptor for primary isolates of HIV-1. Nature (Lond) 1996;381:661–666. doi: 10.1038/381661a0. [DOI] [PubMed] [Google Scholar]
- 33.Dragic T, Litwin V, Allaway GP, Martin SR, Huang Y, Nagashima KA, Cayanan C, Maddon PJ, Koup RA, Moore JP, et al. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC–CKR-5. Nature (Lond) 1996;381:667–673. doi: 10.1038/381667a0. [DOI] [PubMed] [Google Scholar]
- 34.Alkhatib G, Combardiere C, Broder CC, Feng Y, Kennedy PE, Murphy PM, Berger EA. CC CKR5: A RANTES, MIP-1α, MIP-1β receptor as a fusion cofactor for macrophage-tropic HIV-1. Science (Wash DC) 1996;272:1955–1958. doi: 10.1126/science.272.5270.1955. [DOI] [PubMed] [Google Scholar]
- 35.Tsitsikov EN, Fuleihan R, McIntosh K, Scholl PR, Geha RS. Cross-linking of Fcγ receptors activates HIV-1 long terminal repeat–driven transcription in human monocytes. Int Immunol. 1995;7:1665–1670. doi: 10.1093/intimm/7.10.1665. [DOI] [PubMed] [Google Scholar]
- 36.Finkel TH, Tudor-Williams G, Banda NK, Cotton MF, Curiel T, Monks C, Baba TW, Ruprecht RM, Kupfer A. Apoptosis occurs predominantly in bystander cells and not in productively infected cells of HIV- and SIVinfected lymph nodes. Nature Med. 1995;1:129–134. doi: 10.1038/nm0295-129. [DOI] [PubMed] [Google Scholar]
- 37.Wu MX, Daley JF, Rasmussen RA, Schlossman SF. Monocytes are required to prime peripheral blood T cells to undergo apoptosis. Proc Natl Acad Sci USA. 1995;92:1525–1529. doi: 10.1073/pnas.92.5.1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Folks TM, Clouse KA, Justement J, Rabson A, Duh E, Kehrl JH, Fauci AS. Tumor necrosis factor alpha induces expression of human immunodeficiency virus in a chronically infected T cell clone. Proc Natl Acad Sci USA. 1989;86:2365–2368. doi: 10.1073/pnas.86.7.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Poli G, Kinter AL, Fauci AS. Interleukin 1 induces expression of the human immunodeficiency virus alone and in sinergy with interleukin 6 in chronically infected U1 cells: inhibition of inductive effects by the interleukin 1 receptor antagonist. Proc Natl Acad Sci USA. 1994;91:108–112. doi: 10.1073/pnas.91.1.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Murphy PM. The molecular biology of leukocyte chemoattractant receptors. Annu Rev Immunol. 1994;12:593–633. doi: 10.1146/annurev.iy.12.040194.003113. [DOI] [PubMed] [Google Scholar]
- 41.Feng Y, Broder CC, Kennedy PE, Berger EA. HIV-1 entry cofactor: functional cDNA cloning of a seven transmembrane, G-protein coupled receptor. Science (Wash DC) 1996;272:872–877. doi: 10.1126/science.272.5263.872. [DOI] [PubMed] [Google Scholar]
- 42.Doranz BJ, Rucker J, Yi Y, Smyth RJ, Samson M, Peiper SC, Parmentier M, Collman RG, Doms RW. A dual-tropic primary HIV-1 isolate that uses fusin and the β-chemokine receptors CKR-5, CKR-3, and CKR-2b as fusion cofactors. Cell. 1996;85:1149–1158. doi: 10.1016/s0092-8674(00)81314-8. [DOI] [PubMed] [Google Scholar]
- 43.Choe H, Farzan M, Sun Y, Sullivan N, Rollins B, Ponath PD, Wu L, Mackay CR, LaRosa G, Newman W, et al. The β-chemokine receptor CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell. 1996;85:1135–1148. doi: 10.1016/s0092-8674(00)81313-6. [DOI] [PubMed] [Google Scholar]
- 44.Bleul CC, Farzan M, Choe H, Parolin C, Clark-Lewis I, Sodroski J, Springer TA. The lymphocyte chemoattractant SDF-1 is a ligand for LESTR/fusin and blocks HIV-1 entry. Nature (Lond) 1996;382:829–832. doi: 10.1038/382829a0. [DOI] [PubMed] [Google Scholar]
- 45.Oberlin E, Amara A, Bachelerie F, Bessia C, Virelizier J-L, Arenzana-Seisdedos F, Schwartz O, Heard J-M, Clark-Lewis I, Legler DF, et al. The CXC chemokine SDF-1 is the ligand for LESTR/fusin and prevents infection by T-cell-line-adapted HIV-1. Nature (Lond) 1996;382:833–835. doi: 10.1038/382833a0. [DOI] [PubMed] [Google Scholar]
- 46.Paxton WA, Martin SR, Tse D, O'Brian TR, Skurnick J, Van Devanter NL, Padian N, Braun JF, Kotler DP, Wolinsky SM, et al. Relative resistance to HIV-1 infection of CD4 lymphocytes from persons who remain uninfected despite multiple high-risk sexual exposures. Nature Med. 1996;2:412–417. doi: 10.1038/nm0496-412. [DOI] [PubMed] [Google Scholar]
- 47.Paxton WJ, Dragic T, Koup RA, Moore JP. The β chemokines, HIV type 1 second receptors, and exposed uninfected persons. AIDS Res Hum Retroviruses. 1996;12:1203–1207. doi: 10.1089/aid.1996.12.1203. [DOI] [PubMed] [Google Scholar]
- 48.Samson M, Libert F, Doranz BJ, Rucher J, Liesnard C, Farber C-M, Saragosti S, Lapouméroulie C, Cognaux J, Forceille C, et al. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature (Lond) 1996;382:722–725. doi: 10.1038/382722a0. [DOI] [PubMed] [Google Scholar]
- 49.Schmidtmayerova H, Sherry B, Bukrinsky M. Chemokines and HIV replication. Nature (Lond) 1996;382:767. doi: 10.1038/382767a0. [DOI] [PubMed] [Google Scholar]
- 50.Gessani S, Testa U, Varano B, Marzio PD, Borghi P, Conti L, Barberi T, Tritarelli E, Martucci R, Seripa D. Enhanced production of LPS-induced cytokines during differentiation of human monocytes to macrophages. Role of LPS receptors. J Immunol. 1993;151:3758–3766. [PubMed] [Google Scholar]
- 51.Gendelman HE, Baca LM, Turpin J, Kalter DC, Hansen B, Orenstein JM, Dieffenbach CW, Friedman RM, Meltzer MS. Regulation of HIV replication in infected monocytes by IFN-α. Mechanisms for viral restriction. J Immunol. 1990;145:2669–2676. [PubMed] [Google Scholar]
- 52.Pugin J, Heumann D, Tomasz A, Kravchenko VV, Akamatsu Y, Nishijima M, Glauser MP, Tobias PS, Ulevitch RJ. CD14 is a pattern recognition receptor. Immunity. 1994;1:509–516. doi: 10.1016/1074-7613(94)90093-0. [DOI] [PubMed] [Google Scholar]
- 53.Kusunoki T, Hailman E, Juan TS-C, Lichenstein HS, Wright SD. Molecules from Staphylococcus aureus that bind CD14 and stimulate innate immune responses. J Exp Med. 1995;182:1673–1682. doi: 10.1084/jem.182.6.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]