

Abstract
Proteins cleaved by interleukin-1β converting enzyme family proteases during apoptosis are common targets for autoantibody production in patients with systemic lupus erythematosus (SLE). We have tested the possibility that proteins phosphorylated in cells undergoing apoptosis are also targets for autoantibody production in patients with autoimmune disease. Sera from 9/12 patients containing antinuclear antibodies (10/12 meeting diagnostic criteria for SLE or a lupus overlap syndrome), precipitated new phosphoproteins from lysates derived from Jurkat T cells treated with apoptotic stimuli (i.e., Fas-ligation, gamma irradiation, ultraviolet irradiation), but not with an activation (i.e., CD3-ligation) stimulus. Sera derived from individual patients precipitated different combinations of seven distinct serine-phosphorylated proteins. None of these phosphoproteins were included in precipitates prepared using sera from patients with diseases that are not associated with autoantibody production or using serum from rheumatoid arthritis patients. Protein phosphorylation precedes, or is coincident with, the induction of DNA fragmentation, and is not observed when apoptosis is inhibited by overexpression of bcl-2. Serum from four patients precipitated a serine/threonine kinase from apoptotic cell lysates that phosphorylates proteins of 23-, 34-, and 46-kD in in vitro kinase assays. Our results suggest that proteins phosphorylated during apoptosis may be preferred targets for autoantibody production in patients with SLE.
Acommon feature of autoimmune diseases such as systemic lupus erythematosus (SLE)1, systemic sclerosis, Sjögren's disease (SD), and mixed connective tissue disease is the breakdown of tolerance to self antigens. A consequence of this immune dysfunction is the production of antibodies reactive with multiple self proteins (1). Remarkably, the self proteins recognized by these antibodies are culled from a relatively small subset of total cellular proteins. Protein targets for autoantibody production can be grouped into distinct classes sharing structural and/or functional properties. One such class is the ribonucleoprotein (RNP) particles involved in the regulation of RNA metabolism. Autoantigens belonging to this class include heterogeneous nuclear RNPs, small nuclear RNPs, the Th/To RNP complex, and the Ro complex (1–5). It is not known why tolerance to RNP particles is commonly circumvented in patients with autoimmune disease.
It was recently reported that substrates for IL-1β converting enzyme (ICE) family proteases that are cleaved during apoptosis comprise a second class of proteins that are commonly recognized by antibodies found in the serum of patients with autoimmune disease. Autoantigens belonging to this class include poly (A) ribose polymerase, U1 70-kD snRNP, DNA-dependent protein kinase (DNA-PK), nuclear mitotic apparatus protein, and lamin B (6–10). ICE family proteases function in the effector phase of apoptotic cell death. Their substrates are commonly proteins involved in cellular repair processes, suggesting that they may function to ensure the irreversibility of the programmed cell death program. Although proteolysis has the potential to create novel epitopes in protein substrates, most autoantibodies recognize both native and processed substrates (1). Moreover, only a small subset of the over 100 autoantigens that have been described are known to undergo proteolysis during apoptosis, suggesting that other mechanisms contribute to the immunogenicity of these proteins (7, 9). Interestingly, several proteins, including the U1 70-kD protein, have been shown to translocate from the nucleus to large “apoptotic blebs” at the surface of cultured keratinocytes after UV irradiation (8, 11). This and other observations raise the intriguing possibility that cells undergoing apoptosis are uniquely suited to present modified self proteins to the immune system in such a way that overcomes normal mechanisms of peripheral tolerance (12, 13).
The possibility that cells undergoing apoptosis might be reservoirs of autoantigens led us to examine the possibility that proteins selectively phosphorylated during apoptosis might also be commonly recognized by autoantisera derived from patients with autoimmune disease. Recent results have established that inflammatory cytokines (e.g., TNF-α, Fasligand) and environmental stress (e.g., heat shock, UV light, and x irradiation) are potent triggers of apoptotic cell death (14–20). Stress-induced apoptosis requires the activation of a cascade of stress-activated protein (SAP) kinases that phosphorylate their specific substrates on serine or threonine residues (14–20). Although the relevent substrates for these kinases are largely unknown, and neither the kinases nor their substrates have been implicated in the etiology of autoimmune disease, we found that autoimmune sera from patients with SLE and lupus overlap syndromes commonly recognize proteins that are phosphorylated during apoptosis. In addition, serum from four of five patients with SLE or a lupus overlap syndrome were found to selectively precipitate a serine/threonine kinase from apoptotic cell extracts. Our results implicate a SAP kinase in phosphorylation of autoantigens during apoptosis and link a common protein modification to autoantibody production in patients with SLE.
Materials and Methods
Cell Culture.
Jurkat cells were grown in 5% CO2 at 37°C using RPMI 1640 (BioWhittaker, Inc., Walkersville, MD) supplemented with 10% heat-inactivated FCS (HI-FCS; Tissue Culture Biologicals, Tulare, CA) and penicillin and streptomycin (Mediatech, Inc., Herndon, VA). Cells were grown and harvested at mid-log phase. Jurkat T cells overexpressing bcl-2 (or empty vector), a gift from John Reed (The La Jolla Cancer Research Foundation, La Jolla, CA), were grown in RPMI medium as described above, and supplemented with G418 (GIBCO BRL, Gaithersburg, MD) at a final concentration of 500 μg/ml. Protein overexpression was confirmed by Western blotting before each experiment.
Metabolic Labeling.
Jurkat cells were incubated at a density of 2 × 106 cells/ml in labeling medium containing the following: 45% RPMI 1640, 45% RPMI 1640 lacking either phosphate (GIBCO BRL) or methionine and cysteine (GIBCO BRL), 2 mM glutamine (Mediatech, Inc.), 5% HI-FCS, and 5% HI-FCS that had been dialyzed to equilibrium against 10 mM Hepes buffer (Sigma Chemical Co., St. Louis, MO). 32P-labeled orthophosphate or 35S-labeled methionine and cysteine (Dupont–New England Nuclear, Boston, MA) was added at a concentration of 0.1 mCi/ml. Cells were incubated at 37°C for 10–12 h to allow the cells to reach a steady state before each treatment, unless otherwise indicated.
Cell Lysis.
Lysis of cells was performed using nonidet P40 (NP40) (Sigma Chemical Co.) lysis buffer (1% NP40, 150 mM NaCl, 50 mM Tris, pH 7.8, 1 mM EDTA). NP40 lysis buffer was supplemented immediately before use with 1 mM sodium vanadate (Sigma Chemical Co.) and a 1:100 dilution of a 100× protease inhibitor cocktail prepared by dissolving 10 mg chymostatin, 1.5 mg leupeptin, 7 mg pepstatin A, 850 mg phenylmethylsulfonyl fluoride, 500 mg benzamidine, and 5 mg aprotonin in 50 ml of ethanol by stirring overnight. The solution was sterilized by filtration and stored at room temperature (21). All chemicals were purchased from Sigma Chemical Co. After addition of 1 ml lysis buffer, the lysate was incubated on ice for 30 min, centrifuged in a refrigerated microfuge (5402; Eppendorf Inc., Hamburg, Germany) at 14,000 rpm for 15 min, and the supernatant was immediately used for each experiment.
UV Irradiation.
Labeled Jurkat cells were placed on 100 × 15 mm polystyrene petri dishes (Nunc, Thousand Oaks, CA) at a concentration of 2 × 106 cells/ml and irradiated (Stratalinker 2400; Stratagene Corp., La Jolla, CA) at a distance of 9 cm for 12 s. After irradiation, cells were incubated at 37°C for the indicated times before harvesting.
Gamma Irradiation.
Labeled cells were placed in a 50-ml conical tube and irradiated at a dose of 3,300 rad from a Cesium 137 source using an irradiator (Gammacell 1000; Nordion International, Kanata, Ontario, Canada). After irradiation, cells were placed in culture dishes at 37°C and incubated for the indicated times before harvesting.
Cellular Activation.
Labeled Jurkat cells were treated with the following antibodies: anti-Fas antibody 7C11 (provided by Michael Robertson, Indiana University, Bloomington, IN) from hybridoma supernatant at a final dilution of 1:500, and anti-CD3 antibody (Coulter Immunology, Hialeah, FL) at a concentration of 5 μg/ml followed by goat anti–mouse antibody (Jackson ImmunoResearch Labs., West Grove, PA) at the same concentration. Cells were incubated at 37°C for the indicated times before harvesting.
Immunoprecipitation and Western Blot Analysis.
Lysates were precleared once with 25 μl of a 50% solution of protein A–Sepharose (Pharmacia, Uppsala, Sweden) in PBS and 5 μg rabbit anti–mouse (RAM) IgG (Jackson ImmunoResearch Labs., West Grove, PA) for 1 h, followed by two preclears with protein A–Sepharose overnight. Mouse monoclonal antibodies (5 μg) and 5 μg RAM, or 3.5–5 μl patient serum alone were used in precipitation experiments. Serum from all Brigham and Women's Hospital Arthritis Center (Boston, MA) patients who had a serum sample submitted to the Brigham and Women's Hospital Clinical Immunology Laboratory over an 8-mo-period was collected and stored at −20°C until used. Serum from healthy control patients was a gift from P. Fraser (Brigham and Women's Hospital). Diagnoses and serum characterization were confirmed by chart review by P.J. Utz. Immunoprecipitations were performed after addition of 1% BSA (Intergen Co., Purchase, NY) in PBS to a total volume of 500 μl, and rotation in a 4°C cold room for 2–24 h. Comparison of precipitates showed no difference between incubation times for periods of up to 72 h. Precipitates were harvested by centrifuging for 15 s at 14,000 rpm in a refrigerated Eppendorf microfuge, washing three times with NP40 lysis buffer supplemented with protease inhibitor cocktail, resuspending in SDS loading buffer with 9% 2-mercaptoethanol, boiling for 5 min, and electrophoresing on SDS–polyacrylamide gels as described (22). Proteins were transferred to nitrocellulose (Schleicher & Scheull, Keene, NH) for Western blotting experiments or to polyvinylidene difluoride (PVDF), (Dupont–New England Nuclear) for phosphoaminoacid analysis, and either exposed for autoradiography or subjected to Western blot analysis as indicated (23). The mouse monoclonal antibody 4D7, anti-bcl-2 (PharMingen, San Diego, CA) was used for blotting studies at a dilution of 1:1,000. Nitrocellulose blots were blocked with 3% BSA in PBS overnight at 4°C. Bands were visualized using RAM conjugated to horse radish peroxidase (Amersham Corp., Arlington Heights, IL) at a dilution of 1:7,500 in 1% BSA in PBS, and developed using enhanced chemiluminescence performed according to the manufacturer's instructions (Amersham Corp.).
Phosphoaminoacid Analysis.
Immunoprecipitates that had been electrophoresed and transferred to PVDF were rinsed thoroughly with water, exposed for radiography, and appropriate bands excised with a razor blade. The radiolabeled bands were than subjected to acid hydrolysis as described (24), with the exception that two-dimensional electrophoresis was performed at 14°C rather than at 4°C.
DNA Fragmentation.
Unlabeled Jurkat cells were induced to undergo apoptosis using the above triggers in parallel experiments to those using radiolabeled cells. Cells were collected at the indicated times and centrifuged for 5 min at 1,000 rpm. The cell pellet was lysed by adding 500 μl DNA lysis buffer (20 mM Tris, pH 7.4, 5 mM EDTA, and 0.4% Triton X-100) and incubating on ice for 15 min, mixing several times. After centrifuging at 4°C at 14,000 rpm for 5 min, supernatants were extracted with a 25 phenol:24 chloroform:1-isoamyl alcohol mixture (GIBCO BRL). Next, 100 μl 5 M NaCl and 500 μl isopropanol were added to each tube before incubating overnight at −70°C. Samples were thawed and centrifuged at 14,000 rpm for 5 min, washed once with 70% ethanol, and dried in a Speed-Vac. Pellets were resuspended in 30 μl of Tris-EDTA buffer containing 0.1 mg/ml RNase A (Sigma Chemical Co.) and incubated at 37°C for 30 min. After the addition of 10 μl loading buffer, 10 μl of each sample, corresponding to 1 million cells per lane, was separated on 0.8% agarose gels and visualized by ethidium bromide staining under UV light.
In Vitro Kinase Assays.
Individual immunoprecipitates were washed three times in NP40 lysis buffer, then once with TBS (150 mM NaCl, 20 mM Tris, pH 7.6) before resuspending in 30 μl kinase buffer (20 mM Tris, pH 7.6, 10 mM MgCl2, 2 mM MnCl2, and 20 μCi [32P]-gamma ATP [Dupont–New England Nuclear] 150 mCi/ml) for 30 min at 30°C. The reactions were terminated by addition of sample buffer and boiling for 5 min. Proteins were separated on an SDS-PAGE gel before transfer to PVDF and autoradiography for 2–5 min (25).
Results
Autoimmune Sera Recognize Proteins Phosphorylated during Stress-Induced Apoptosis.
Serum from 12 random patients with positive tests for antinuclear antibodies (ANA; defined as ⩾1:20 titer on immunofluorescence staining using Hep2 cells as a substrate), as well as serum from 10 healthy control patients, 5 rheumatoid arthritis patients, and 15 patients with diseases considered to be unassociated with autoantibodies (including fibrositis, tendonitis, bursitis, chronic fatigue syndrome, carpal tunnel syndrome, and osteoarthritis), were chosen from the sera collected as described in Materials and Methods. All patients with a positive ANA test were further screened by ELISA for antibodies against DNA, the Smith complex, Ro, La, and RNP, and the patients' charts were reviewed to obtain clinical data sufficient to establish a diagnosis (1). As summarized in Table 1, most patients (10/12) met published criteria for either SLE or lupus in association with a second inflammatory condition (referred to as SLE overlap syndrome; 26). Other conditions were also represented, including SD (patient 6), and undifferentiated connective tissue disease (patient 9). One patient with rheumatoid arthritis (RA; patient 13) and a patient with fibrositis (patient 14) are also presented for comparison.
Table 1.
Characterization of Autoimmune Sera
| Patient | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ANA | 1:2,560 | + | 1:640 | 1:640 | 1:640 | 1:20 | + | 1:640 | 1:640 | 1:40 | 1:2560 | 1:160 | ND | − | ||||||||||||||
| Pattern | D/C | S/D | P/D | P/D | S/D | ND | S/D | D | S | D/N | Nu | S/D | ND | − | ||||||||||||||
| RF | ND | ND | ND | ND | ND | − | ND | ND | − | − | − | ND | ND | ND | ||||||||||||||
| Ro | + | − | − | − | + | + | + | + | − | − | + | − | ND | ND | ||||||||||||||
| La | − | − | − | − | − | − | − | − | − | − | + | − | ND | ND | ||||||||||||||
| Sm | − | − | − | − | − | − | − | − | − | − | − | − | ND | ND | ||||||||||||||
| dsDNA | + | + | − | − | − | − | + | + | − | − | − | − | ND | ND | ||||||||||||||
| ssDNA | + | − | − | + | + | + | + | + | − | − | + | + | ND | ND | ||||||||||||||
| RNP | + | − | − | − | − | − | − | − | − | − | + | − | ND | ND | ||||||||||||||
| APLA | ND | + | + | ND | − | ND | − | − | − | ND | ND | ND | ND | ND | ||||||||||||||
| Comp | ↓ | Nl | ↓ | ↓ | Nl | ↑ | Nl | Nl | ND | Nl | Nl | Nl | ND | ND | ||||||||||||||
| Disease | SLE | SLE | SLE | SLE | Over | SD | Over | Over | UCTD | SLE | SLE | SLE | RA | Fib | ||||||||||||||
| Bands | pp200 | pp54 | pp54 | pp54 | pp17 | pp46 | pp54 | pp54 | pp54 | |||||||||||||||||||
| pp54 | pp42 | pp17 | pp42 | pp42 | pp42 | |||||||||||||||||||||||
| pp17 | pp34 | pp34 | pp34 | pp34 | ||||||||||||||||||||||||
| pp23 | pp23 | pp23 | ||||||||||||||||||||||||||
| pp17 | pp17 |
Individual patient sera are identified by the numbers above each column. ANA, antinuclear antibody titer; Pattern, immunoflourescence staining pattern using Hep 2 cells as substrate (P, peripheral; D, diffuse or homogeneous; C, cytoplasmic; N, homogeneous nuclear; S, speckled; Nu, nucleolar); RF, rheumatoid factor; Ro, RNA binding protein Ro; La, RNA binding protein La; Sm, Smith antigen; dsDNA, double-stranded DNA; ssDNA, single-stranded DNA; APLA, antiphospholipid antibody, determined by anticardiolipin ELISA assay; Comp, complement determined by CH 50 assay. Test results are labeled as positive (+); negative (−); normal (NI); not done (ND); increased ( ↑ ); or decreased ( ↓ ). Over, SLE overlap syndrome; UCTD, undifferentiated connective tissue disease; Fib, fibrositis. The relative migration of phosphoproteins precipitated using sera derived from individual patients (derived from Fig. 1 A) are as indicated.
Jurkat cells metabolically labeled with 32P-orthophosphate were cultured for 2.5 h in the absence or presence of a monoclonal antibody reactive with Fas (anti-7C11), solubilized in NP40 lysis buffer, and immunoprecipitated using the indicated autoimmune or control sera. Immunoprecipitates were separated on a 12% SDS–polyacrylamide gel, transferred to nitrocellulose, and subjected to autoradiograpy. Fig. 1 A shows that 9/12 ANA+ autoimmune sera, representing 9/10 SLE or SLE overlap patients, precipitated at least one new phosphoprotein from cells undergoing Fas-mediated apoptosis compared to untreated cells. The phosphorylation of these proteins did not result from a nonspecific, general increase in kinase activity after Fas engagement, as 32P-labeled,whole cell extracts prepared from untreated and apoptotic cells were identical when compared on SDS-PAGE gels (data not shown). The individual phosphoproteins precipitated by several of the patient sera is strikingly similar in profile, but variable in intensity of phosphorylation. For example, serum from patients 1, 2, 3, 4, 8, 11, and 12 precipitates a protein of ∼54 kD (pp54) that is weakly phosphorylated in untreated cell lysates and strongly phosphorylated in lysates from apoptotic cells. Similarly, a 34-kD protein (pp34) was precipitated using serum derived from patients 3, 8, 11, and 12; and a doublet of ∼42 kD (pp42) was precipitated using serum derived from patients 3, 8, 11, and 12. None of these phosphoproteins were precipitated using ANA (−) sera derived from patients 13 or 14, nor using sera derived from 12 healthy control patients or 4 additional patients with RA (data not shown). The level of phosphorylation of pp42, pp34, and pp17 differed significantly between patients (patients 3, 8, 11, and 12) and was independent of the ANA titer as detected by immunfluorescence (Table 1), suggesting that these phosphoproteins may be novel and independent of the major proteins responsible for the immunfluorescence detectable as an ANA. In addition to the phosphoproteins described above, three other new phosphoproteins can be seen as bands migrating at the following positions: 17 kD doublet (pp17; patients 1, 4, 5, 8, and 11), 23 kD (pp23; patients 3, 8, and 11), and 46 kD (pp46; patient 7). A seventh protein migrating between 96 and 200 kD (pp200) was observed for patient 1 (Fig. 2, A–C).
Figure 1.


Human autoimmune sera precipitate phosphoproteins from apoptotic Jurkat cell lysates. (A) Jurkat cells were labeled with 32P-orthophosphate, treated with the anti-Fas monoclonal antibody 7C11, and lysed either before (odd numbered lanes) or 2.5 h after (even numbered lanes) the addition of antibodies. Proteins were then precipitated using the indicated autoimmune serum, separated on a 12% SDS–polyacrylamide gel, transferred to nitrocellulose, and exposed for autoradiography. Arrows point to new phosphoproteins in the anti-Fas–treated lanes. (B) The identical experiment with 35S-labeled cells. Patient numbers are located above each figure and correspond to those in Table 1. Lane numbers appear beneath the corresponding lane. The relative migration of molecular size markers in kilodaltons are indicated on the left side of the gel.
Figure 2.




Phosphorylation of autoantigens in response to apoptotic or mitogenic stimuli. Jurkat cells were labeled with 32Porthophosphate, triggered with apoptotic or mitogenic stimuli, and solubilized using NP40 lysis buffer at the indicated times before immunoprecipitation using sera derived from the indicated patient. Immunoprecipitates were separated on a 12% SDS–polyacrylamide gel, transferred to nitrocellulose, and subjected to autoradiographic analysis. (A) Anti-Fas treatment; (B) gamma irradiation; (C) UV irradiation; (D) CD3 cross-linking. Arrows point to new phosphoproteins. The patient number is indicated above each time course. The time, in hours, from initial exposure to each stimulus is indicated at the top of each gel. Lane numbers appear beneath the corresponding lane. The relative migration of molecular size markers in kilodaltons is indicated on the left side of each panel.
The preferential inclusion of phosphoproteins in precipitates prepared from apoptotic versus nonapoptotic lysates could result from de novo phosphorylation of autoantigens, increased extractability of the phosphoproteins during the detergent lysis, or recruitment of preexisting or new phosphoproteins to the autoantigen complex during apoptosis. To differentiate between these three possibilities, the experiment shown in Fig. 1 B was performed using cells that were metabolically labeled with 35S-methionine and cysteine in a manner identical to the experiment depicted in Fig. 1 A, which used cells labeled with 32P-orthophosphate. In most cases, immunoprecipitates prepared from apoptotic and nonapoptotic lysates contained similar 35S-labeled proteins. Two exceptions were observed. A 60-kD protein and a >200-kD protein were included in immunoprecipitates prepared from apoptotic, but not nonapoptotic lysates using sera derived from patients 10 (Fig. 1 B, lane 20), and 11 (Fig. 1 B, lane 22), respectively (indicated with arrows on the right side of the panel). Although neither of these proteins clearly corresponded to the phosphoproteins identified in Fig. 1 A, 35S-labeled proteins (Fig. 1 B) migrating similarly to the phosphoproteins identified in Fig. 1 A were observed in all cases. Taken together, these results are most consistent with de novo phosphorylation of autoantigens during apoptosis.
Phosphorylation of Autoantigens Accompanies Apoptosis, but Not T Cell Receptor Stimulation.
The results shown in Fig. 1 indicate that autoimmune sera preferentially precipitate proteins phosphorylated in response to Fas ligation. To determine whether these proteins are also phosphorylated during apoptosis triggered by stimuli other than Fas ligation, selected patient sera were used to precipitate 32P-labeled Jurkat lysates prepared from cells subjected to apoptotic stimuli or an activation stimulus for various times. This kinetic analysis reveals that phosphorylation of autoantigens is induced between 1 and 2.5 h after Fas ligation (Fig. 2 A), between 2.5 and 4.5 h after gamma irradiation (Fig. 2 B), and between 1 and 2.5 h after UV irradiation (Fig. 2 C). Individual autoantisera precipitate a similar cadre of phosphoproteins regardless of the apoptotic trigger. In contrast, ligation of the T cell receptor complex using a monoclonal antibody reactive with CD3, a stimulus that induces IL-2 production and enhances proliferation in these cells (data not shown), induced neither new protein phosphorylation, nor DNA fragmentation over the course of this experiment (Figs. 2 D and 3 D). Control sera derived from an individual without autoimmune disease did not precipitate phosphoproteins from apoptotic lysates, nor from lysates prepared from CD3-stimulated cells (Fig. 2, A–D, right). The kinetics of DNA fragmentation induced by apoptotic or activation stimuli was also determined. As shown in Fig. 3, A–D, the onset of DNA fragmentation is approximately coincident with the phosphorylation of autoantigens regardless of the apoptotic stimulus.
Figure 3.
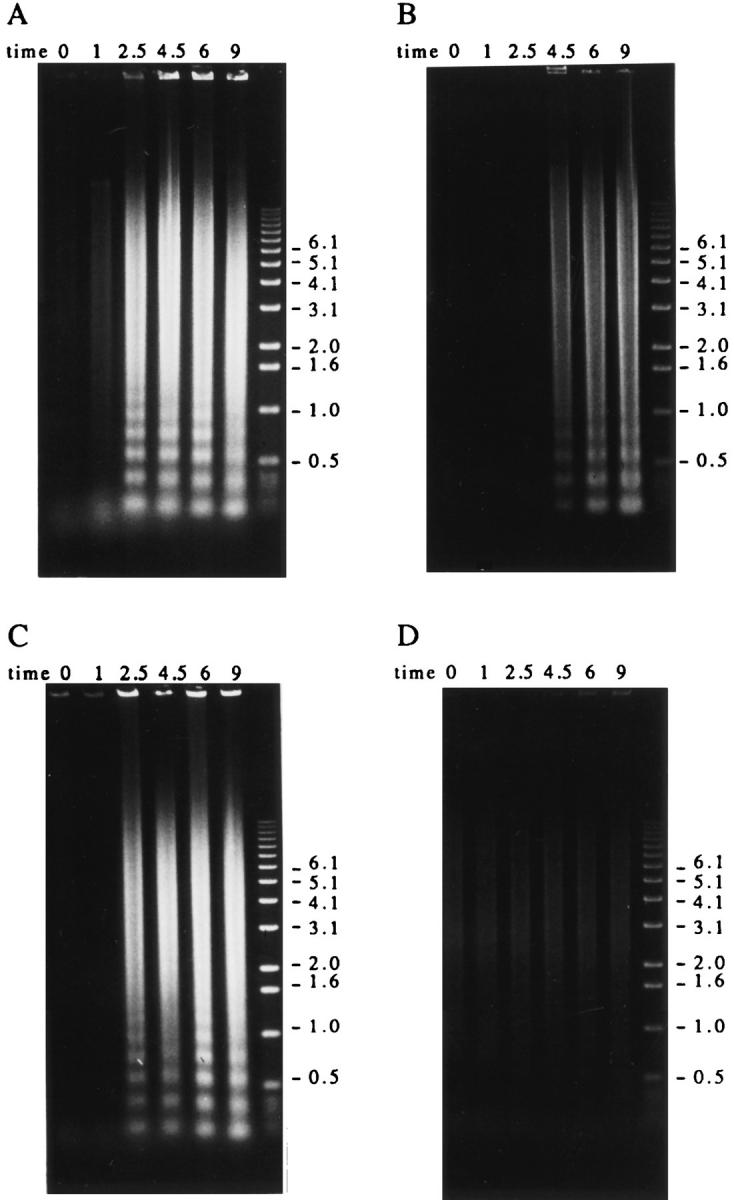
Autoantigen phosphorylation coincides with or precedes the onset of DNA fragmentation in apoptotic Jurkat cells. Jurkat cells were triggered to undergo apoptosis and harvested at the indicated times. Each time point represents a total of 1 million cells. The DNA was prepared as described in the Materials and Methods, separated on a 0.8% agarose gel and visualized by staining with ethidium bromide before UV exposure. (A) Anti-Fas treatment; (B) gamma irradiation; (C) UV irradiation; (D) anti-CD3 treatment. The time, in hours, from initial exposure to each stimulus is indicated at the top of each lane. The relative migration of molecular size markers in kilobases is indicated on the right side of each panel.
In addition to the phosphorylation of autoantigens during apoptosis, selected phosphoproteins appear to be rapidly dephosphorylated, and then rephosphorylated in a reproducible manner over the course of the kinetic assay (pp17 and pp23; Fig. 2 B, lanes 1–4). The level of basal phosphorylation of several autoantigens, particularly pp34, pp23, and pp17 was somewhat variable in each experiment (e.g., patient 1, Fig. 2, A–D), and appeared to be related to the initial density of the cells at the time of labeling, with less dense (and presumably more active) cells labeling more uniformly (our unpublished observations).
Phosphoaminoacid Analysis of Autoantigens.
Since both tyrosine kinases and serine/threonine kinases have been implicated in signaling Fas-mediated apoptosis (16, 17, 19, 25, 27–30), we subjected all seven phosphoprotein autoantigens to phosphoaminoacid analysis. In each case, phosphorylation was restricted to serine residues (Fig. 4, A–G), implicating one or more serine/threonine protein kinases in the phosphorylation of these autoantigens.
Figure 4.

Autoantigens are phosphorylated exclusively on serine residues during Fas-mediated apoptosis. Jurkat cells were labeled with 32P-orthophosphate, treated with the anti-Fas monoclonal antibody 7C11, and solubilized using NP40 lysis buffer after 2.5 h. Proteins were then precipitated with autoimmune serum, separated on a 12% SDS–polyacrylamide gel, transferred to PVDF, and exposed for autoradiography. Individual phosphoproteins were localized on the membrane, excised, and subjected to acid hydrolysis. Phosphoamino acids were separated by twodimensional electrophoresis in pH 1.9 buffer in the horizontal dimension, followed by pH 3.5 buffer in the vertical dimension before autoradiographic analysis. Individual proteins correspond to those described in Table 1 as follows: (A) patient 1, pp200; (B) patient 1, pp54; (C) patient 7, pp46; (D) patient 11, pp42; (E) patient 3, pp34; (F) patient 8, pp23; and (G) patient 11, pp17. Migration of phosphoaminoacid standards are labeled with circles as follows: phosphoserine (pS), phosphothreonine (pT), phosphotyrosine (pY).
A Protein Kinase Activity Is Precipitated from Apoptotic Lysates Using Selected Patient Sera.
A cascade of stress-activated serine/threonine kinases has been implicated in signaling apoptotic cell death (16, 17, 19, 25, 31). Individual kinases within this cascade are regulated, in part, by phosphorylation. It is therefore possible that stress-activated kinases may be recognized directly by sera derived from patients with autoimmune disease, or may be recruited during apoptosis to preexisting complexes. To test this possibility, lysates from untreated or anti-Fas–treated Jurkat cells were precipitated with individual patient sera, and subjected to an in vitro kinase assay as described (25). Five sera were chosen to encompass all seven phosphoproteins that had been identified in the initial screen using in vivo–labeled apoptotic Jurkat cells (Fig. 1 A and Table 1). In addition, sera from a healthy control patient and patient 6, whose serum is monospecific for the Ro protein, were included for comparison. Fig. 5 A shows that 4/5 ANA + patient sera (i.e., patients 3, 7, 8, and 11) precipitate a kinase whose activity is increased in apoptotic cell extracts compared to untreated cell extracts. The healthy control patient and patient 6 were devoid of kinase activity in this assay. Phosphoproteins migrating at 34 kD (Fig. 5, lanes 4, 6, 8, and 10), 23 kD (lanes 4, 6, 8, and 10), and 46 kD (lane 6) were identified in this assay. The relative migration of these phosphoproteins is similar to that of prominant phosphoproteins identified in the in vivo phosphorylation assay shown in Fig. 1 A. The kinetics with which the kinase (precipitated using serum from patient 7) was activated after Fas ligation was correlated with the induction of DNA fragmentation in the experiment shown in Fig. 5 B. In this experiment, Jurkat cells were cultured in the presence of anti-Fas monoclonal antibodies for the indicated times before processing for DNA fragmentation and in vitro kinase activity. The first appearance of pp46 in the in vitro kinase assay was observed at 90 min (Fig. 5 B), while DNA fragmentation was first observed 120 min after Fas ligation (data not shown). Phosphoamino acid analysis of pp46 showed that the in vitro phosphorylation of pp46 is restricted to serine residues (Fig. 5 C), consistent with the in vivo results shown in Fig. 4 C. A similar kinetic analysis targeting pp34 and pp23 using serum from patient 11 (Fig. 5 A, lanes 9 and 10) gave similar results (data not shown). These results are consistent with the less rigorous time courses presented in Figs. 2 and 3, and suggest that a serine/threonine kinase activated by Fas stimulation is present in the immunoprecipitates from patients 7 and 11 at a time that precedes the onset of DNA fragmentation.
Figure 5.



Autoimmune serum precipitates a serine kinase activity from apoptotic Jurkat lysates. Jurkat cells cultured in the absence (odd numbered lanes) or presence (even numbered lanes) of anti-Fas were solubilized in NP40 lysis buffer after 2.5 h, and precipitated using 3.5 μl of serum derived from the indicated patient. Individual precipitates were subjected to an in vitro kinase reaction at 30°C for 30 min, separated on an SDS–polyacrylamide gel, transferred to nitrocellulose, and subjected to autoradiographic exposure for 2 min. (A) In vitro kinase reaction. Serum derived from the patient number indicated at the top of the figure corresponds to patients described in Table 1. The relative migration of molecular size markers in kilodaltons is indicated on the right side of the panel. (B) Kinetics of kinase activation after Fas ligation as measured using the in vitro kinase reaction performed on immunoprecipitates prepared using serum derived from patient 7. The time in minutes from initial exposure to anti-Fas is indicated at the top of each lane. The position of pp46 is indicated with an arrow on the left side of the panel. (C) Phosphoamino acid analysis of the in vitro phosphorylated 46-kD protein. Migration of phosphoamino acid standards are labeled with circles as follows: phosphoserine (pS), phosphothreonine (pT), phosphotyrosine (pY).
Bcl-2 Overexpression Blocks Apoptosis and Phosphorylation of pp46.
We next asked whether the phosphorylation of pp46 could be blocked by overexpression of the bcl-2 protein, which has been shown to efficiently block apoptosis induced by multiple apoptotic stimuli, including gamma and UV irradiation (32–35). In Fig. 6, Jurkat T cells stably transformed with either bcl-2 (left) or empty vector (right) were labeled with 32P-orthophosphate and subjected to Fas ligation, gamma irradiation, or UV irradiation. Cells were solubilized at the indicated times and lysates were precipitated using serum derived from patient 7. While phosphorylation of pp46 is rapidly induced in Jurkat (neo) control cells in response to gamma irradiation (Fig. 6 B, right), pp46 is absent from Jurkat (bcl-2) transformants treated with this same stimulus (Fig. 6 B, left). Qualitatively similar results are seen with UV irradiation (Fig. 6 C), although a small amount of pp46 is observed in Jurkat (bcl-2) transformants beginning at 4.5 h. Overexpression of bcl-2 effectively inhibited apoptosis in response to these triggers, as judged by the induction of DNA fragmentation (data not shown). In contrast, phosphorylation of pp46 after Fas ligation was relatively unaffected by overexpression of bcl-2 (Fig. 6 A). The induction of DNA fragmentation after Fas ligation was similarly unaffected by overexpression of bcl-2 in these cells (data not shown), supporting the correlation between phosphorylation of pp46 and the induction of apoptosis. A similar inhibitory effect of bcl-2 after gamma and UV irradiation but not anti-Fas treatment, on the phosphorylation of pp54, pp34, and pp17 (Fig. 1 A and Table 1) recognized by serum from patient 11, was also observed (data not shown). Taken together, these results demonstrate that the in vivo phosphorylation of all four autoantigens that were tested correlated with the induction of apoptosis, and is downstream of the inhibitory effects of bcl-2.
Figure 6.
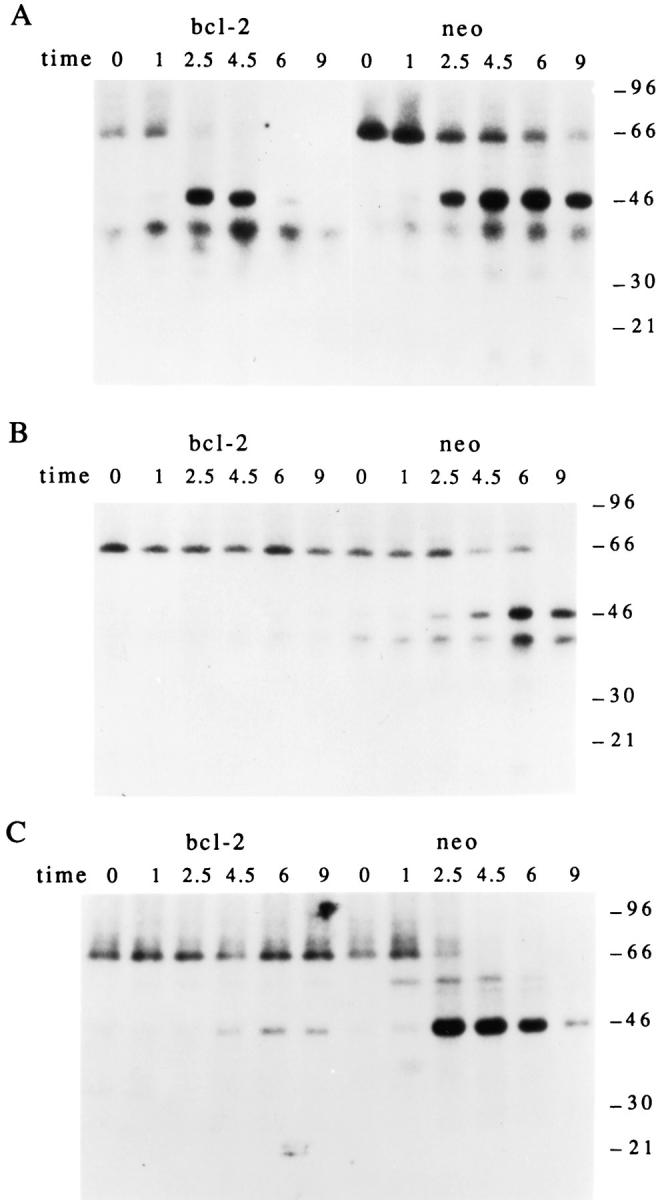
In vivo phosphorylation of pp46 correlates with the induction of apoptosis and is inhibited in Jurkat cells overexpressing bcl-2. Jurkat transformants (bcl-2, left) or Jurkat control transformants (neo, right) were labeled with 32P-orthophosphate, subjected to the indicated apoptotic stimulus, solubilized in NP40 lysis buffer, and precipitated using serum derived from patient 7 before electrophoretic separation. (A) AntiFas treatment; (B) gamma irradiation; (C) UV irradiation. The relative migration of molecular size markers in kilodaltons is indicated on the right side of each panel. The time, in hours, from initial exposure to each stimulus is indicated at the top of each lane.
Discussion
SLE is characterized by the production of autoantibodies that recognize a restricted subset of intracellular proteins and nucleic acids (1). Autoantibodies reactive with single- and double-stranded DNA, as well as nuclear RNP complexes are responsible, in part, for the ability of lupus serum to bind to the nuclei of cultured cells (1, 5). Antinuclear antibodies are almost always found in the serum of patients with SLE, and their presence has both diagnostic and pathogenic implications in this disease. Specific autoantibody profiles are associated with disease subsets (e.g., anti– Jo-1 histidyl tRNA synthetase and myositis) and are predictive of future disease manifestations (e.g., anti–Jo-1 and interstitial lung disease; 36, 37). Other autoantibodies have been shown to be directly pathogenic in animal models and in human disease, including anti-DNA antibodies (immune-complex glomerulonephritis), antiphospholipid antibodies (antiphospholipid antibody syndrome, characterized by arterial and venous thromboses and recurrent fetal loss), and anti-Ro antibodies (congenital heart block in the neonatal lupus syndrome) (36–40).
Evidence continues to mount that defects in apoptosis are at least partially involved in the pathogenesis of SLE. The genetic defects responsible for the lupus-like diseases found in MRL/lpr/lpr and C3H/HeJ-gld/gld mice have been identified as the genes encoding Fas and Fas ligand, respectively, a receptor–ligand pair required for activationinduced death of lymphocytes (41, 42). Additional evidence linking defects in apoptosis to the pathogenesis of SLE comes from studies showing that proteins cleaved by ICE family proteases during apoptosis are common targets for autoantibody production in patients with SLE. This autoantigenic subset includes several DNA repair enzymes, including poly (A) ribose polymerase and the catalytic subunit of DNA-PK (7, 43, 44). Several autoantigens are concentrated at membrane blebs rimming the surface of keratinocytes undergoing apoptosis. Although the role of these membrane blebs in antigen presentation is not known, these results suggest the intriguing possibility that proteins modified during apoptosis may be preferred targets for autoantibody production (6–8, 11, 43, 44). Further support for this hypothesis comes from two recent studies demonstrating that immunization of mice with apoptotic cells leads to the production of autoantibodies, including antibodies directed against DNA-PK (Zhang, C., and S. Schlossman, unpublished data; and 12, 13). This possibility led us to screen serum from patients with autoimmune disease for antibodies reactive with proteins that are phosphorylated during apoptosis. Our results indicate that substrates of serine/threonine kinases activated during stress-induced apoptosis are commonly included in precipitates from apoptotic cells using serum from patients with SLE. Using a more stringent ANA titer of ⩾1:160 as a cutoff value, as opposed to the ⩾1:20 titer used in initial patient selection, demonstrates that at least one new phosphoprotein is observed in 9/10 ANA + sera, and all nine sera from SLE or SLE overlap patients. It should be emphasized, however, that the small number of patients presented in this initial study precludes generalizations about disease associations or prevalence of autoantigen phosphorylation. Future studies using sera from larger numbers of patients with well-defined clinical syndromes and carefully defined serologic characteristics will clarify the importance of autoantigen phosphorylation in autoimmune disease.
Stress-activated serine/threonine kinases (SAP kinases, also referred to as JNK and p38) play an essential role in signaling stress-induced apoptosis (15–17, 30, 31, 45). Several transcription factors (e.g., c-Jun, Elk-1, and ATF-2) are substrates for SAP/JNK/p38, and dominant inhibitory mutations in c-Jun can block stress-induced apoptosis, suggesting that stress-activated kinases influence apoptosis at the transcriptional level. The kinase activity responsible for phosphorylation of pp46 has several similarities to the SAP/JNK kinases, including a requirement for magnesium and manganese but not calcium, resistance to RNase and DNase treatment (data not shown), and identical kinetics of phosphorylation and serine specificity (15–17, 30, 31, 45). Another stress-activated serine/threonine kinase that has been implicated in a signaling cascade leading to apoptosis is FAST kinase (25, 46). FAST kinase phosphorylates TIA-1, but not TIAR, two related RNA-binding proteins that appear to regulate mRNA translation (Kedersha, N., and P. Anderson, unpublished observations) and have been shown to translocate from the nucleus to the cytoplasm during Fasmediated apoptosis (25, 47). We have identified autoantibodies reactive with TIA-1 and TIAR in the sera of ∼2% of patients with a positive ANA (Utz, P.J., and P. Anderson, unpublished observations), consistent with our hypothesis that proteins phosphorylated during apoptosis are preferred targets for autoantibody production. Additional kinases that may be involved in signaling apoptosis include protein kinase C (48), cyclin dependent kinases (49–52), cAMP kinase I (53, 54), casein kinase I (55), Pim-1 (56), Wee-1 (57), and PITSLRE kinases (28). Further characterization of the kinase activities described in this report is ongoing and should allow identification of the responsible kinase(s) and elucidation of their role in autoantibody production and programmed cell death.
The identity of the seven kinase substrates described in this report is not known. Over 100 autoantigens have been identified to date, some of which are known phosphoproteins, including the ribosomal proteins P0, P1, and P2 that are similar in size to the pp17 doublet and pp42 (1). It is possible that these substrates are novel, apoptosis-specific proteins, and perhaps are part of the core apoptotic machinery. A second possibility is that the observed bands may represent proteolytic cleavage products of larger phosphoproteins, as would be predicted for a phosphoprotein such as DNA-PK. The disappearance of the weakly (and variably) phosphorylated 68-kD band precipitated from nonapoptotic cell lysates with serum from patient 7 (Figs. 1 A, 5, and 6) and the subsequent appearance of pp46 in precipitates from apoptotic lysates support this possibility, although the marked increase in phosphorylation of pp46 both in vivo and in vitro argues strongly for a new phosphorylation event. Cleavage and phosphorylation are not necessarily mutually exclusive events, and it remains plausible that serine phosphorylation during apoptosis may target these autoantigens or other proteins in a macromolecular complex for cleavage by an ICE-like protease. Comparison of the sizes of the phosphoproteins, presented in Fig. 1 and Table 1, with the published sizes of ICE-like protease cleavage products demonstrates similarities with U1-70 kD (40-kD product) and pp42 (6); nuclear lamin B (68-kD precursor and 45-kD product) and pp46 (10, 58); UBF/Nor-1 (55-, 35-, 32-, and 24-kD products) and pp54, pp34, and pp23, respectively (9); and an unidentified protein fragment of 35-kD (patient RW) and pp34 (7). A focus of future studies will be to identify each of these seven phosphoproteins and to identify their role in apoptosis and autoantibody production.
In vivo autoantigen phosphorylation was inhibited in cells overexpressing bcl-2 after UV or gamma irradiation, but not after Fas stimulation. Programmed cell death, as assayed by DNA fragmentation and characteristic cell morphologic changes, correlated precisely with antigen phosphorylation for each stimulus. This, together with the observation that none of the autoantigens was phosphorylated in response to CD3 stimulation, suggests that the phosphorylation of autoantigens is specifically correlated with the activation of intrinsic cell death pathway(s) and is not an epiphenomenon associated with stress stimuli. Although the molecular mechanisms by which bcl-2 inhibits apoptosis are not known, it appears to act at a signaling step preceding the activation of the protease apopain/CPP32 (32–35). Recent results showing physical interactions between bcl-2 family members and serine/threonine kinases involved in signaling cell growth (e.g., raf-1) suggest that activation of kinase cascades similar to the cascade (s) described in this report might precede the activation of ICE family proteases under some conditions (59–61).
The self antigens recognized by autoantibodies found in lupus serum comprise a small subset of total cellular proteins. It is striking that many of these antigens are substrates for proteases and kinases involved in signaling or execution of apoptotic cell death. Why are proteins modified during apoptosis preferred targets for autoantibody production in patients with SLE? Perhaps a clue comes from the observation that some autoantigens are concentrated at membrane blebs formed at the surface of apoptotic cells (8, 11). Phosphorylation during apoptosis of the protein constituents of macromolecular complexes such as RNPs may produce neoepitopes or may target individual proteins for proteolysis and/or translocation to membrane blebs, as has been observed for the U1-70–kD protein (6, 8). Although intracellular antigens presented on the surface of apoptotic cells might be recognized by self-reactive B lymphocytes, a productive immune response would not be generated in the absence of antigen-specific helper T cells. It is possible, however, that proteolysis or phosphorylation of selected proteins during apoptosis produces neoepitopes to which T cells are not tolerized. In the special case in which the apoptotic cell is also an APC, priming of naive T cells may ensue. T cell recognition of modified self proteins presented by apoptotic APCs could drive the differentiation and expansion of autoreactive B cells, resulting in autoantibody production. This hypothesis requires that APCs from patients with SLE are unusually sensitive to stress-induced apoptosis. This sensitivity could be conferred by a combination of genetic and environmental factors.
Acknowledgments
The authors thank members of the laboratories of P. Anderson and M. Streuli for insights and helpful comments; V. Shifrin, Q. Medley, S. Porcelli, and S. Schlossman for critical review of the manuscript; the Brigham & Women's Hospital Clinical Immunology Laboratory, P. Fraser, and J. Jackson for providing patient serum; N. Kedersha and M. Robertson for providing anti–bcl-2 and anti-Fas (7C11), respectively; and J. Reed for the gift of the bcl-2- and neo-overexpressing Jurkat cells.
This work was supported in part by National Institutes of Health training grant T32 AI07306 to Brigham & Women's Hospital, Division of Rheumatology and Immunology (P.J. Utz); the Arthritis Foundation (P.J. Utz and P. Anderson); the National Institutes of Health grants AI33600 and CA67929 (P. Anderson); and the Peabody Foundation. P. Anderson is a Scholar of the Leukemia Society of America.
Footnotes
1 Abbreviations used in this paper: ANA, antinuclear antibodies; DNA-PK, DNA-dependent protein kinase; HI-FCS, heat-inactivated FCS; ICE, IL-1β converting enzyme; NP40, nonidet P40; PVDF, polyvinylidene difluoride; RA, rheumatoid arthritis; RAM, rabbit anti–mouse; RNP, ribonucleoprotein; SAP, stress-activated protein; SD, Sjögren's disease; SLE, systemic lupus erythematosus.
References
- 1.von Muhlen CA, Tan EM. Autoantibodies in the diagnosis of systemic rheumatic diseases. Semin Arthritis Rheum. 1995;24:323–358. doi: 10.1016/s0049-0172(95)80004-2. [DOI] [PubMed] [Google Scholar]
- 2.Astaldi-Ricotti G, Bestagno M, Cerino A, Negri C, Caporali R, Cobianchi F, Longhi M, Montecucco C. Antibodies to hnRNP core protein A1 in connective tissue diseases. J Cell Biochem. 1989;40:43–47. doi: 10.1002/jcb.240400105. [DOI] [PubMed] [Google Scholar]
- 3.Gold HA, Topper JN, Clayton D, Craft J. The RNA processing enzyme RNase MRP is identical to the Th RNP and related to RNase P. Science (Wash DC) 1989;245:1377–1380. doi: 10.1126/science.2476849. [DOI] [PubMed] [Google Scholar]
- 4.Montecucco M, Caporali R, Negri C, deGennaro F, Cerino A, Bestagno M, Cobianchi F, Astaldi-Ricotti G. Antibodies from patients with rheumatoid arthritis and systemic lupus erythematosus recognize different epitopes of a single heterogeneous nuclear RNP core protein. Arthritis Rheum. 1990;33:180–186. doi: 10.1002/art.1780330205. [DOI] [PubMed] [Google Scholar]
- 5.Van Veenrooij W, Sillekens P. Small-nuclear RNA-associated proteins: autoantigens in connective tissue diseases. Clin Exp Rheum. 1989;7:635–639. [PubMed] [Google Scholar]
- 6.Casciola-Rosen LA, Miller DK, Anhalt GJ, Rosen A. Specific cleavage of the 70 kDa protein component of the U1 small nuclear riboprotein is a characteristic biochemical feature of apoptotic cell death. J Biol Chem. 1994;269:30757–30760. [PubMed] [Google Scholar]
- 7.Casciola-Rosen LA, Anhalt GJ, Rosen A. DNA-dependent protein kinase is one of a subset of autoantigens specifically cleaved early during apoptosis. J Exp Med. 1995;182:1625–1634. doi: 10.1084/jem.182.6.1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Casciola-Rosen LA, Anhalt G, Rosen A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface blebs on cultured keratinocytes. J Exp Med. 1994;179:1317–1330. doi: 10.1084/jem.179.4.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Casiano CA, Martin SJ, Green DR, Tan EM. Selective cleavage of nuclear autoantigens during CD95 (Fas/Apo-1)–mediated T cell apoptosis. J Exp Med. 1996;184:765–770. doi: 10.1084/jem.184.2.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neamati N, Fernandez A, Wright S, Kiefer J, McConkey DJ. Degradation of lamin B1 precedes oligonucleosomal DNA fragmentation in apoptotic thymocytes and isolated thymocyte nuclei. J Immunol. 1995;154:3788–3795. [PubMed] [Google Scholar]
- 11.Golan TD, Elkon KB, Gharavi AE, Krueger JG. Enhanced membrane binding of autoantibodies to cultured keratinocytes of systemic lupus erythematosus patients after ultraviolet B/ultraviolet A irradiation. J Clin Invest. 1992;90:1067–1076. doi: 10.1172/JCI115922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang C, Ao A, Seth A, Schlossman S. A mitochondrial membrane protein defined by a novel monoclonal antibody is preferentially detected in apoptotic cells. J Immunol. 1996;157:3980–3987. [PubMed] [Google Scholar]
- 13.Mevorach D, Zhou J, Elkon K. Immunization of mice with apoptotic cells induces low levels of autoantibodies. Arthritis Rheum. 1996;39:143s. [Google Scholar]
- 14.Alderson M, Tough T, Davis-Smith T, Braddy S, Falk B, Schooley K, Goodwin R, Smith C, Ramsdell F, Lynch D. Fas ligand mediates activation-induced cell death in human T lymphocytes. J Exp Med. 1995;181:71–77. doi: 10.1084/jem.181.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen Y-R, Meyer CF, Tan T-H. Persistent activation of c-Jun N-terminal kinase 1 (JNK1) in gamma radiation-induced apoptosis. J Biol Chem. 1996;271:631–634. doi: 10.1074/jbc.271.2.631. [DOI] [PubMed] [Google Scholar]
- 16.Kyriakis J, Banerjee P, Nikolakaki E, Dai T, Rubie E, Ahmad M, Avruch J, Woodgett J. The stress-activated protein kinase subfamily of c-jun kinases. Nature (Lond) 1994;369:156–160. doi: 10.1038/369156a0. [DOI] [PubMed] [Google Scholar]
- 17.Kyriakis J, Avruch J. Protein kinase cascades activated by stress and inflammatory cytokines. Bioessays. 1996;18:567–577. doi: 10.1002/bies.950180708. [DOI] [PubMed] [Google Scholar]
- 18.Ju S, Panka D, Cui J, Ettinger R, el-Khatib M, Sherr D, Stanger B, Marshak-Rothstein A. Fas (CD95)/FasL interactions required for programmed cell death after T-cell activation. Nature (Lond) 1995;373:444–448. doi: 10.1038/373444a0. [DOI] [PubMed] [Google Scholar]
- 19.Verheij M, Bose R, Lin X, Yao B, Jarvis W, Grant S, Birrer M, Szabo E, Zon L, Kyriakis J, et al. Requirement for ceramide-initiated SAPK/JNK signalling in stress-induced apoptosis. Nature (Lond) 1996;380:75–79. doi: 10.1038/380075a0. [DOI] [PubMed] [Google Scholar]
- 20.Mathias S, Dressler KA, Kolesnick RN. Characterization of a ceramide-activated protein kinase: stimulation by tumor necrosis factor alpha. Proc Natl Acad Sci USA. 1991;88:10009–10013. doi: 10.1073/pnas.88.22.10009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karwan R, Bennett JL, Clayton DA. Nuclear RNase MRP processes RNA at multiple discrete sites: interaction with an upstream G box is required for subsequent downstream cleavages. Genes Dev. 1991;5:1264–1276. doi: 10.1101/gad.5.7.1264. [DOI] [PubMed] [Google Scholar]
- 22.Laemmli E. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature (Lond) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 23.Harlow, E., and D. Lane. 1988. Immunoblotting. In Antibodies: A Laboratory Manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. 474–510 pp.
- 24.Coligan J, Kruisbeek A, Margulies D, Shevach E, Strober W. Two dimensional phosphopeptide mapping. Curr Prot Immunol. 1994;3:11.2.1–11.2.8. [Google Scholar]
- 25.Tian Q, Taupin J, Elledge S, Robertson M, Anderson P. Fas-activated serine/threonine kinase (FAST) phosphorylates TIA-1 during Fas-mediated apoptosis. J Exp Med. 1995;182:865–874. doi: 10.1084/jem.182.3.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tan E, Cohen A, Fries J, Masi A, McShane D, Rothfield N, Schaller J, Talal N, Winchester R. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25:1272–1277. doi: 10.1002/art.1780251101. [DOI] [PubMed] [Google Scholar]
- 27.Eischen C, Dick C, Leibson P. Tyrosine kinase activation provides an early and requisite signal for Fas-induced apoptosis. J Immunol. 1994;153:1947–1954. [PubMed] [Google Scholar]
- 28.Lahti J, Xiang J, Heath L, Campana D, Kidd V. PITSLRE protein kinase activity is associated with apoptosis. Mol Cell Biol. 1995;15:1–11. doi: 10.1128/mcb.15.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Migita K, Eguchi K, Kawabe Y, Nagataki S. Tyrosine phosphorylation participates in peripheral T-cell activation and programmed cell death in vivo. Immunology. 1995;85:550–555. [PMC free article] [PubMed] [Google Scholar]
- 30.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science (Wash DC) 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 31.Gjertsen B, Doskeland S. Protein phosphorylation in apoptosis. Biochim Biophys Acta. 1995;1269:187–199. doi: 10.1016/0167-4889(95)00117-b. [DOI] [PubMed] [Google Scholar]
- 32.Boise L, Gottschallk A, Quintans J, Thompson C. Bcl-2 and Bcl-2–related proteins in apoptosis regulation. Curr Top Microbiol Immunol. 1995;200:107–121. doi: 10.1007/978-3-642-79437-7_8. [DOI] [PubMed] [Google Scholar]
- 33.Itoh N, Tsujimoto Y, Nagata S. Effect of bcl-2 on Fas antigen-mediated cell death. J Immunol. 1993;151:621–627. [PubMed] [Google Scholar]
- 34.Reed J. Bcl-2 and the regulation of programmed cell death. J Cell Biol. 1994;124:1–6. doi: 10.1083/jcb.124.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sentman C, Shutter J, Hockenberry D, Kanagawa O, Korsmeyer S. Bcl-2 inhibits multiple forms of apoptosis but not negative selection in thymocytes. Cell. 1991;67:879–888. doi: 10.1016/0092-8674(91)90361-2. [DOI] [PubMed] [Google Scholar]
- 36.Goldstein R, Duvic M, Targoff I, Reichlin M, McMenemy A, Reveille J, Warner N, Pollack M, Arnett F. HLA-D region genes associated with autoantibody responses to histidyl-transfer RNA synthetase (Jo-1) and other translation-related factors in myositis. Arthritis Rheum. 1990;33:1240–1248. doi: 10.1002/art.1780330826. [DOI] [PubMed] [Google Scholar]
- 37.Venables P. Polymyositis-associated overlap syndromes. Br J Rheumatol. 1996;35:305–308. doi: 10.1093/rheumatology/35.4.305. [DOI] [PubMed] [Google Scholar]
- 38.Koffler D, Schur P, Kunkel H. Immunologic studies concerning the nephritis of systemic lupus erythematosus. J Exp Med. 1967;126:607–623. doi: 10.1084/jem.126.4.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee L. Maternal autoantibodies and pregnancy. II. The neonatal lupus syndrome. Bailliere's Clin Rheum. 1990;4:69–84. doi: 10.1016/s0950-3579(05)80244-4. [DOI] [PubMed] [Google Scholar]
- 40.Branch W, Dudley D, Mitchell M. IgG fractions from patients with antiphospholipid antibodies cause fetal death in BALB/c mice: a model for autoimmune fetal loss. Am J Obstet Gynecol. 1990;163:210–216. doi: 10.1016/s0002-9378(11)90700-5. [DOI] [PubMed] [Google Scholar]
- 41.Nagata S, Suda T. Fas and Fas ligand: lpr and gld mutations. Immunol Today. 1995;16:39–43. doi: 10.1016/0167-5699(95)80069-7. [DOI] [PubMed] [Google Scholar]
- 42.Nagata S, Golstein P. The Fas death factor. Science (Wash DC) 1995;267:1449–1456. doi: 10.1126/science.7533326. [DOI] [PubMed] [Google Scholar]
- 43.Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle M, Lazebnik YA. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature (Lond) 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- 44.Tewari M, Quan L, O'Rourke K, Desnoyers S, Zeng Z, Beidler D, Poirier G, Salvesen G, Dixit V. Yama/CPP32 beta, a mammalian homologue of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly (ADP ribose) polymerase. Cell. 1995;81:801–809. doi: 10.1016/0092-8674(95)90541-3. [DOI] [PubMed] [Google Scholar]
- 45.Derijard B, Hibi M, Wu I-H, Barrett T, Bing S, Deng T, Karin M, Davis R. JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell. 1994;76:1025–1037. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- 46.Tian Q, Streuli M, Saito H, Schlossman S, Anderson P. A polyadenylate binding protein localized to the granules of cytolytic lymphocytes induces DNA fragmentation in target cells. Cell. 1991;67:629–639. doi: 10.1016/0092-8674(91)90536-8. [DOI] [PubMed] [Google Scholar]
- 47.Taupin J-L, Tian Q, Kedersha N, Robertson M, Anderson P. The RNA-binding protein TIAR is translocated from the nucleus to the cytoplasm during Fas-mediated apoptotic cell death. Proc Natl Acad Sci USA. 1995;92:1629–1633. doi: 10.1073/pnas.92.5.1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Emoto Y, Manome Y, Meinhardt G, Kisaki H, Kharbanda S, Robertson M, Ghayur T, Wong W, Kamen R, Weichselbaum R. Proteolytic activation of protein kinase C delta by an ICE-like protease in apoptotic cells. EMBO (Eur Mol Biol Organ) J. 1995;14:6148–6156. doi: 10.1002/j.1460-2075.1995.tb00305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang J, Walsh K. Resistance to apoptosis conferred by Cdk inhibitors during myocyte differentiation. Science (Wash DC) 1996;273:359–361. doi: 10.1126/science.273.5273.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Meikrantz W, Gisselbrecht S, Tam S, Schlegel R. Activation of cyclin A-dependent protein kinases during apoptosis. Proc Natl Acad Sci USA. 1994;91:3754–3758. doi: 10.1073/pnas.91.9.3754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gao C, Zelenka P. Induction of cyclin B and H1 kinase activity in apoptotic PC12 cells. Exp Cell Res. 1995;219:612–618. doi: 10.1006/excr.1995.1271. [DOI] [PubMed] [Google Scholar]
- 52.Shi L, Nishioka WK, Th'ng J, Bradbury EM, Litchfield DW, Greenberg AH. Premature p34 cdc2 activation required for apoptosis. Science (Wash DC) 1994;263:1143–1145. doi: 10.1126/science.8108732. [DOI] [PubMed] [Google Scholar]
- 53.Duprez E, Gjertsen B, Bernard O, Lanotte M, Soskeland S. Antiapoptotic effect of heterozygously expressed mutant RI (Ala336→ Asp) subunit of cAMP kinase I in a rat leukemia cell line. J Biol Chem. 1993;268:8332–8340. [PubMed] [Google Scholar]
- 54.Vintermyr I, Gjertsen B, Lanotte M, Soskeland S. Microinjected catalytic subunit of cAMP-dependent protein kinase induces apoptosis in myeloid leukemia (IPC-81) cells. Exp Cell Res. 1993;206:157–161. doi: 10.1006/excr.1993.1132. [DOI] [PubMed] [Google Scholar]
- 55.Beyaert R, Vanhaesebroeck B, Declercq W, Van Lint J, Vandenabele P, Agostinis P, Vandenheede J, Fiers W. Casein kinase-1 phosphorylates the p75 tumor necrosis factor receptor and negatively regulates tumor necrosis factor signaling for apoptosis. J Biol Chem. 1995;270:23293–23299. doi: 10.1074/jbc.270.40.23293. [DOI] [PubMed] [Google Scholar]
- 56.Moroy T, Grzeschiczek A, Petzold S, Hartmann K-U. Expression of a Pim-1 transgene accelerates lymphoproliferation and inhibits apoptosis in lpr/lpr mice. Proc Natl Acad Sci USA. 1993;90:10734–10738. doi: 10.1073/pnas.90.22.10734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen G, Shi L, Litchfield D, Greenberg A. Rescue from granzyme B–induced apoptosis by Weel kinase. J Exp Med. 1995;181:2295–2300. doi: 10.1084/jem.181.6.2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Oberhammer FA, Hochegger K, Froschl G, Tiefenbacher R, Pavelka M. Chromatin condensation during apoptosis is accompanied by degradation of lamin A + B without enhanced activation of cdc2 kinase. J Cell Biol. 1994;126:827–837. doi: 10.1083/jcb.126.4.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen C, Faller D. Phosphorylation of bcl-2 protein and association with p21 ras in ras-induced apoptosis. J Biol Chem. 1996;271:2376–2379. doi: 10.1074/jbc.271.5.2376. [DOI] [PubMed] [Google Scholar]
- 60.Wang H, Millan J, Cox A, Der C, Rapp U, Beck T, Zha H, Reed J. R-ras promotes apoptosis caused by growth factor deprivation via a bcl-2 suppressible mechanism. J Cell Biol. 1995;129:1103–1114. doi: 10.1083/jcb.129.4.1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang H, Takayama S, Rapp U, Reed J. Bcl-2 interacting protein, BAG-1, binds to and activates the kinase Raf-1. Proc Natl Acad Sci USA. 1996;93:7063–7068. doi: 10.1073/pnas.93.14.7063. [DOI] [PMC free article] [PubMed] [Google Scholar]