

Abstract
To explore the role of the interleukin (IL)-1β converting enzyme (ICE) in neuronal apoptosis, we designed a mutant ICE gene (C285G) that acts as a dominant negative ICE inhibitor. Microinjection of the mutant ICE gene into embryonal chicken dorsal root ganglial neurons inhibits trophic factor withdrawal–induced apoptosis. Transgenic mice expressing the fused mutant ICE-lacZ gene under the control of the neuron specific enolase promoter appeared neurologically normal. These mice are deficient in processing pro–IL-1β, indicating that mutant ICEC285G blocks ICE function. Dorsal root ganglial neurons isolated from transgenic mice were resistant to trophic factor withdrawal–induced apoptosis. In addition, the neurons isolated from newborn ICE knockout mice are similarly resistant to trophic factor withdrawal–induced apoptosis. After permanent focal ischemia by middle cerebral artery occlusion, the mutant ICEC285G transgenic mice show significantly reduced brain injury as well as less behavioral deficits when compared to the wild-type controls. Since ICE is the only enzyme with IL-1β convertase activity in mice, our data indicates that the mutant ICEC285G inhibits ICE, and hence mature IL-1β production, and through this mechanism, at least in part, inhibits apoptosis. Our data suggest that genetic manipulation using ICE family dominant negative inhibitors can ameliorate the extent of ischemia-induced brain injury and preserve neurological function.
Apoptosis or programmed cell death is a cellular suicide mechanism under internal cellular control (1, 2). The genetic control of programmed cell death is best understood in the nematode Caenorhabditus elegans (3). Mutations in the gene ced-3 eliminate essentially all programmed cell death that occur during the development of C. elegans (4). Genetic mosaic analysis showed that ced-3 acts cell autonomously to induce cell death and thus, ced-3 is an essential component in the cellular suicide mechanism of C. elegans (5). Members of the IL-1β converting enzyme (ICE)1 family are mammalian homologues of the C. elegans ced-3 gene product (6). Microinjection of crmA, a serpin encoded by the cowpox virus that is a specific inhibitor of ICE, inhibited neuronal cell death induced by trophic factor deprivation (7). Peptide inhibitors of the ICE family delay motor neuron death in vitro and in vivo (8). Thus, the ICE protease family plays an important role in mammalian neuronal apoptosis.
Traditionally, ischemia-mediated neuronal cell death has been attributed to necrosis, rather than to apoptosis. This is based on the morphological feature of dying neurons of postischemic brain that include swelling and disintegration of cell membrane, rather than typical cellular shrinkage and nuclear changes seen in apoptosis. Recently, however, the conventional view that necrosis is the major, if not the only, mechanism of ischemia-mediated neuronal degeneration has been challenged. Evidence of activation of apoptotic mechanisms in postischemic cerebral tissue of adult animals has been detected. Internucleosomal cleavage of DNA has been observed both after global (9, 10) and focal (11–13) occlusions. These studies suggest that apoptosis may play an important role in postischemic neuronal cell death. It is not clear, however, which are the genetic and biochemical pathways mediating neuronal apoptotic cell death induced by ischemic insult.
While the critical role of ICE-like proteases in apoptosis has been well established, the role of ICE itself in apoptosis remains controversial. ICE knock out mutant mice generated by gene targeting techniques were found to be only partially defective in apoptosis induced by anti-Fas antibody (14). On the other hand, we and others have found elevated levels of mature IL-1β after apoptotic cell death indicating activation of ICE in apoptosis, since ICE is likely the only enzyme in vivo and in vitro with IL-1β convertase activity (14–18). We have previously demonstrated that binding of endogenously produced mature IL-1β to its type 1 receptor plays an important role in apoptosis (19). We have shown that replacing the cysteine in the active site of ICE with a glycine (C285G) obliterates its ability to mediate cell death (20). The cysteine residue in the active site is required for the IL-1β convertase and the autoprocessing activity of ICE (21). We demonstrate here that ICEC285G mutant is a dominant negative inhibitor of ICE that can inhibit processing of pro–IL-1β by ICE in vivo. Expression of mutant ICEC285G in dorsal root ganglial (DRG) neurons, either by microinjection or in transgenic mice, inhibits trophic factor withdrawal–induced apoptosis. In addition, DRG neurons of ICE knockout mice are also resistant to trophic factor deprivation-induced apoptosis, consistent with the notion that mutant ICEC285G inhibits ICE directly. Finally, we show here that transgenic mice expressing the ICEC285G mutant under the control of neuron specific enolase (NSE) promoter are resistant to neuronal injury induced by cerebral ischemia.
Materials and Methods
Microinjection of β-Actin-M17Z into Chicken Embryonic DRG Neurons.
The experiments were performed essentially as described by Gagliardini et al. (7). Primary cultures of chicken embryonic DRG neurons were isolated under sterile conditions from day 10 embryos (Spafas Inc., Preston, CT). DRGs were dissociated by incubation in trypsin for 15 min at 37°C and trituration. Dissociated neurons were plated on poly-l-lysine– (30 mg/ml for 1 h; Sigma Chemical Co., St. Louis, MO) and laminin– (Sigma Chemical Co.; 20 mg/ml for 2 h) coated chamber slides. DRG neurons were cultured for 2 d in F12 medium (GIBCO BRL, Gaithersburg, MD) containing 10% fetal bovine serum (Hyclone Labs., Logan, UT), penicillin (100 U/ml; GIBCO BRL), streptomycin (100 mg/ml; Sigma Chemical Co.), and 5 mM cytosine β-d-arabinose (Sigma Chemical Co.) supplemented with nerve growth factor (NGF; 10 ng/ml; Sigma Chemical Co.). Neuron injection was performed with a microinjector (model 5242; Eppendorf Inc., Fremont, CA), with glass micropipettes loaded with 1 mg/ml plasmid DNA in Tris/EDTA buffer and 5% rhodamine dye (10 kD rhodamine-isothiocyanite–labeled dextran; Molecular Probes Inc., Eugene, OR), dissolved in 0.2 M KCl. The construction of the fused mutant ICE (C285G)-lacZ plasmid (β-actinM17Z) was described by Miura et al. (20). 3 h after injection, the NGF-containing medium was replaced with NGF- and serumfree medium in the presence of sufficient mouse monoclonal antibody against NGF (Boehringer Mannheim, Indianapolis, IN). The medium was changed daily. Live injected neurons were counted on days 0, 3, and 6.
Construction of NSE-M17Z Plasmid and Generation of Transgenic Mice.
pNSE-M17Z-lacZ construct was made by digesting pNSE-lacZ with SalI and ClaI, which removed a 0.8-kb SalI/ ClaI fragment. The SalI/ClaI digested pNSE-lacZ vector was ligated with a 2-kb SalI/ClaI insert from BSM17Z that contains the mutant ICE (C285G) and the part of lacZ that was removed in the SalI/ClaI digest of the pNSE/lacZ vector. The resulting construct was named pJ655. To generate transgenic mice, pJ655 was linearized by XmnI digestion and gel purified. 14 transgenic mice lines were generated by DNX (Princeton, NJ). Founder mice were SV-129/C57BL/6 hybrid. Initially, five lines were selected (7506, 7512, 7516, 7538, and 7539) based on highest DNA copy number in the genome.
Genotyping and X-galactosidase Assay of M17Z Mice.
Tail DNA was isolated and genotyping was performed using the following PCR primers targeted to the Ice/lacZ fusion (M17Z-F: 5′TGCCCAAGCTTGAAAGACAAGCCC3′, lacZ-R: 5′CTGGCGAAAGGGGGATGTGCTG3′). X-gal staining was performed by removing the vertebral column and sectioning it in a sagital plane. Tissue was fixed for 5 min on ice (0.2% glutaraldehyde in 0.1 M phosphate buffer, 2% formaldehyde, 5 mM EGTA, pH 7.3, 2 mM MgCl2) followed by three 30 min washes at 4°C (0.1 M phosphate buffer, pH 7.3, 2 mM MgCl2, 0.1% sodium deoxycholate, 0.2% NP40). The tissue was then stained overnight with X-gal at 37°C (rinse solution with 1 mg/ml X-gal in DMSO, 5 mM K ferrocyanide, 5 mM K ferricyanide), and then sectioned in a cryostat (40 μm). Photomicrograph was taken in a light microscope (×100) under oil immersion.
Facial Nucleus Neuronal Count.
Facial motorneurons were counted as described by Martinou et al. (22). Briefly, 40 μm brainstem sections of 6-wk-old mice were stained with cresyl violet. Neurons were counted double blindly in both facial nuclei. All sections were counted and no correction was applied.
Pro–IL-1β Processing After Systemic LPS Administration.
LPS from Escherichia coli serotype 0111:B4 was dissolved in 0.1 M PBS (pH 7.4). Each mouse (transgenic 29.5 ± 1.2 g, n = 11; wild type 29.6 ± 2.1 g, n = 7) was injected intraperitoneally either with 10 μg of LPS/g body weight, or with equivalent volume of PBS at 0 and 12 h, and killed by decapitation under halothane anesthesia 2 h after the second injection. Brains were rapidly dissected and frozen in 1.5 ml Eppendorf tubes in liquid nitrogen. Brain tissue concentration of mature IL-1β was determined in duplicate using an ELISA kit (IntertestTM-1βX, Lot B6499F; Genzyme Corp., Cambridge, MA). Brains without olfactory bulbs and cerebellum were homogenized in two volumes of ice-cold 0.1 M PBS containing 2 mM phenylmethylsulfonylfluoride, 1 μg/ml leupeptin, 1 μg/ ml antipain, 1 μg/ml aprotinin, 1 μg/ml pepstatin, 0.05% (wt/ vol) sodium azide and 4 mM ethylenediaminetetraacetic acid using a microtube homogenizer (Sigma Pellet Pestle; Sigma Chemical Co.) for 15 s. The homogenates were centrifuged for 30 min at 50,000 g, and 100 μl of the supernatant was used for each determination. Absorbance was read at 450 nm with an ELISA plate reader (Multiskan MCC/340 MK II; Titertek, Elfab Oy, Finland). Values shown are as mean ± SEM.
Newborn DRG Neuron Trophic Factor Deprivation.
The experiments were performed as described by Friedlander et al. (19). Postnatal day 1 mouse DRG neurons were isolated, dissociated with trypsin for 1 h at 37°C, and plated in an eight-chamber poly- l-lysine/laminin– (Sigma Chemical Co.) coated slide. Wells were seeded at a density of ∼1,000 neurons/well (eight wells/mouse). Neurons were cultured in Ham's F-12 media supplemented with 20% FCS (BioWhittaker, Inc., Walkersville, MD), NGF (200 ng/ml) (Sigma Chemical Co.), brain-derived neurotrophic factor (100 ng/ml; Preprotech, Rocky Hill, NJ), glutamine (2 mM), and penicillin/streptomycin. The medium was replaced with either trophic factor–containing medium (TF[+] = 20% FCS and 200 ng/ml NGF) or trophic factor–deficient medium (TF[−] = serum- and NGF-free medium in the presence of a saturating concentration of mouse NGF monoclonal antibody (100 ng/ml; Boehringer Mannheim).
Permanent Middle Cerebral Artery Occlusion.
Experiments were done as described by Hara et al. (23), except that the occlusion of the middle cerebral artery (MCA) was continuous for 24 h. Neurological grading: 0, no neurological deficits; 1, failure to extend the right forepaw; 2, circling to the contralateral side; 3, loss of walking or righting reflex. All experiments were done in a double-blinded fashion. Values shown as mean ± SEM.
Results
Mutant ICE Protects DRG Neurons from Trophic Factor Withdrawal–induced Apoptosis.
Survival of DRG neurons in culture requires the presence of trophic factors which include nerve growth factor and serum. In the absence of trophic factor support, DRG neurons undergo apoptosis (24). To determine whether the mutant ICE can inhibit DRG neuronal death induced by trophic factor deprivation, primary cultures of chicken embryonic DRG neurons were microinjected with a construct of the fused mutant ICEC285G-lacZ gene under the control of the β-actin promoter (β-actinM17Z). Neurons were co-injected with rhodamine-isothiocyanate dextran as a marker and with Hoechst dye to determine neuronal nuclear morphology. After trophic factor removal, control neurons microinjected with the β-actinlacZ construct survived 22.5 and 6.0% after 3 and 6 d in culture, respectively. No significant difference was detected when compared to cells injected with dye alone. In contrast, neurons injected with β-actin-M17Z survived 85.0 and 81.0% after 3 and 6 d in culture, respectively (Fig. 1). These results showed that the mutant ICE gene inhibits DRG neuronal cell death induced by trophic factor deprivation, suggesting that mutant ICE may be able to suppress the activities of wild-type ICE or ICE-like proteases.
Figure 1.

Microinjection of β-actin-M17Z protects embryonic chicken DRG neurons from trophic factor withdrawal–induced apoptosis. Survival represents the percentage of injected E10 chick DRG neurons remaining alive at 3 and 6 d (100% at day 0) after trophic factor deprivation. A total of 783 neurons were injected with vector alone (solid bar), 421 with dye alone (empty bar), and 850 with β-actin-M17Z (hatched bar). Each condition was done on at least four independent experiments. Results are expressed as means ± SEM.
Generation of Transgenic Mice Expressing the Mutant ICE (C285G) Protein.
To determine whether the mutant ICE gene can also act as an inhibitor of apoptosis in vivo, and to further evaluate its mechanism of action, we established transgenic mouse lines expressing the fused mutant ICEC285GlacZ gene under the control of the NSE promoter (NSEM17Z). Transgenic mice expressing either the lacZ or bcl-2 genes under control of the NSE promoter have been well characterized, and transgene expression has been detected throughout the nervous system (22, 25, 26). PCR was used for genotyping the NSE-M17Z transgenic mice, and protein expression was detected by X-gal staining (Fig. 2). Founder mice from five different lines were crossed with C57BL/6 mice. The expression of NSE-M17Z was detected in the first and second generations of offspring, which were used in the experiments described below.
Figure 2.


(a) X-gal staining of a DRG from transgenic 6-wk-old mouse progeny of the 7539 founder (progeny of the 7512 founder had similar staining pattern). (b) PCR genotyping of transgenic lines. M17Z is the original NSE-M17Z plasmid which was used to make the transgenic mouse, used here as a PCR positive control. 7512 and 7539 are positive transgenic lines.
NSE-M17Z Transgenic Mice Are Developmentally Normal.
NSE-M17Z mice are viable and their embryonic development appears normal. Developmental apoptosis does not appear to be inhibited in these mutant ICEC285G transgenic mice, as evidenced by equal brain size, normal behavior, as well as equal number of neurons in the facial motor nucleus, when compared to their wild-type littermates (wild type [n = 6] 2,323 ± 77, transgenic [n = 6] 2,355 ± 84). The lack of an embryonic phenotype in the mutant ICEC285G mice suggests that mutant ICE does not block neuronal apoptosis during development. Alternatively, since the NSE promoter is active at low levels during embryogenesis, the expression of the mutant ICE gene might not reach a threshold sufficient to block developmental cell death (25).
Mutant ICE Acts In Vivo as a Dominant Negative Inhibitor of ICE.
We next evaluated whether the mutant ICEC285G may act as a dominant negative ICE inhibitor. ICE knockout mice were almost completely defective in processing pro–IL-1β and ICE is the only protease identified so far that can process pro–IL-1β (14, 17). If the mutant ICE transgenic mice have a defect in secreting mature IL-1β, this would provide strong evidence that mutant ICEC285G can act as a dominant negative inhibitor of ICE. Systemic injection of LPS induces release of mature IL-1β, and ICE knockout mice generated by gene-targeting technique were unable to release mature IL-1β upon LPS stimulation (14, 17). To determine if our mutant ICEC285G transgenic mice are also defective in secreting mature IL-1β, we injected LPS intraperitoneally into the mutant ICEC285G transgenic mice and determined the levels of mature IL-1β in whole brain lysates using an ELISA kit which specifically detects mature IL-1β. After the systemic LPS challenge, whole brain lysates of mutant ICEC285G transgenic mice contained 74.7% less mature IL-1β as compared to that of LPS-injected wildtype mice. In control wild-type mice injected intraperitoneally with PBS, there was low but detectable levels of mature IL-1β in the brain (4.0 pg/g brain), whereas this cytokine was undetectable in the brain lysate of PBS-injected mutant ICEC285G mice (Fig. 3). Thus, mutant ICEC285G can act as an effective inhibitor of pro–IL-1β processing, strongly suggesting that mutant ICEC285G is a dominant negative inhibitor of ICE itself.
Figure 3.

Whole brain lysates of NSE-M17Z mice are deficient in processing pro–IL-1β after systemic LPS administration. LPS was injected intraperitoneally (10 μg/g body weight) and 2 h before killing (wild type, n = 4; NSE-M17Z, n = 5). PBS was injected as a control (wild type, n = 3; NSE-M17Z, n = 6). Brains were dissected, and mature IL-1β concentration was determined using an ELISA kit specific for mature IL-1β. Results are expressed as means ± SEM.
Newborn DRG Neurons from the Mutant ICEC285G Transgenic and ICE Knockout Mice Are Resistant to Trophic Factor Withdrawal–induced Apoptosis.
We have previously demonstrated that a CrmA-sensitive pathway, as well as ICE activation followed by endogenously produced mature IL-1β receptor binding, play important roles in trophic factor withdrawal–mediated DRG neuron apoptosis (7, 19). In this same model, we investigated whether DRG neurons isolated from mutant ICEC285G mice were resistant to cell death. DRG neurons were isolated from newborn mutant ICEC285G transgenic and wild-type mice, and cultured in the presence or absence of trophic factor. The survival of wild-type and NSE-M17Z mice DRG neurons in the presence of trophic factor were >95% after 24 h in culture. Removal of trophic factor induces 79.7% of neurons to die within 24 h. DRG neurons from two transgenic lines (7512 and 7539) were significantly protected from apoptosis in culture (48.6% cell death in 24 h) after trophic factor removal as compared to their wild-type littermates (Fig. 4 a). If resistance of DRG neurons isolated from mutant ICEC285G mice to trophic factor deprivation–induced apoptosis can be attributed to the ability of mutant ICE to inhibit endogenous ICE activity, a prediction would be that DRG neurons from ICE knockout mice should also be resistant to neuronal cell death induced by trophic factor deprivation. To test this hypothesis, we examined whether DRG neurons from ICE knockout mice are also protected from trophic factor withdrawal–mediated apoptosis. Newborn DRG neurons were isolated from mutant ICE knockout and control wild-type mice. As shown in Fig. 4 b, DRG neurons isolated from ICE knockout mice are similarly resistant to cell death induced by trophic factor deprivation as our mutant ICEC285G transgenic mice. These results suggest that mutant ICEC285G acts as a dominant negative inhibitor of ICE for the suppression of DRG neuronal cell death–induced apoptosis and that ICE plays an important role in DRG neuronal cell death induced by trophic factor deprivation.
Figure 4.


Survival in vitro of NGF-dependent DRG neurons isolated from (a) mutant ICEC285G transgenic and (b) ICE knockout newborn mice are protected from trophic factor withdrawal–mediated apoptosis. Survival represents the percentage of neurons remaining alive after 24 h of serum deprivation (100% at day 0). (a) The results are the average three double blindly scored independent experiments using newborn mice from lines 7512 and 7539. Neurons from each mouse were plated separately, and at least 400 neurons were counted per well. Results are expressed as means ± SEM. (b) Results are from an experiment double blindly scored performed in quadruplicate, from DRG neurons isolated from four wild-type and four ICE knockout newborn mice. At least 500 neurons were counted per well. Results are expressed as means ± SD.
NSE-M17Z Transgenic Mice Are Resistant to Cerebral Ischemic Injury.
To determine if ICE plays a role in apoptosis induced by ischemic insult and if the mutant ICEC285G may act to reduce ischemic brain injury, we investigated whether the mutant ICEC285G transgenic mice were protected in a mouse focal cerebral ischemia model where apoptotic cell death has been reported (27). We performed permanent MCA occlusion in 14 wild-type and 11 transgenic mice, progeny of five different founder mice (7509, 7512, 7516, 7538, and 7539). Mice were scored neurologically 30 min and 24 h after the occlusion. In the initial 30 min evaluation, there was no significant difference in the neurological score. During the ensuing 24 h, however, wild-type mice remained impaired, while transgenic mice improved neurologically (Fig. 5 A). Mice were killed at 24 h, and infarct volume was quantified with 4% 2,3,5-triphenyltetrazolium chloride. Infarct volume was significantly smaller in NSE-M17Z (66 ± 11 mm3 [n = 11]) when compared to the wild-type littermate mice (125 ± 5 mm3 [n = 14]; Fig. 5 B). Physiologic parameters were recorded in a separate set of transgenic and wild-type mice. Blood pressure, arterial blood gases (PO2, PCO2, and blood pH), regional cerebral blood flow, and body temperature did not significantly differ in the two sets of mice, before and throughout 30 min of ischemia (Fig. 5 C). Thus, expression of mutant ICEC285G, a dominant negative inhibitor of ICE, protects neurons from ischemic insult.
Figure 5.
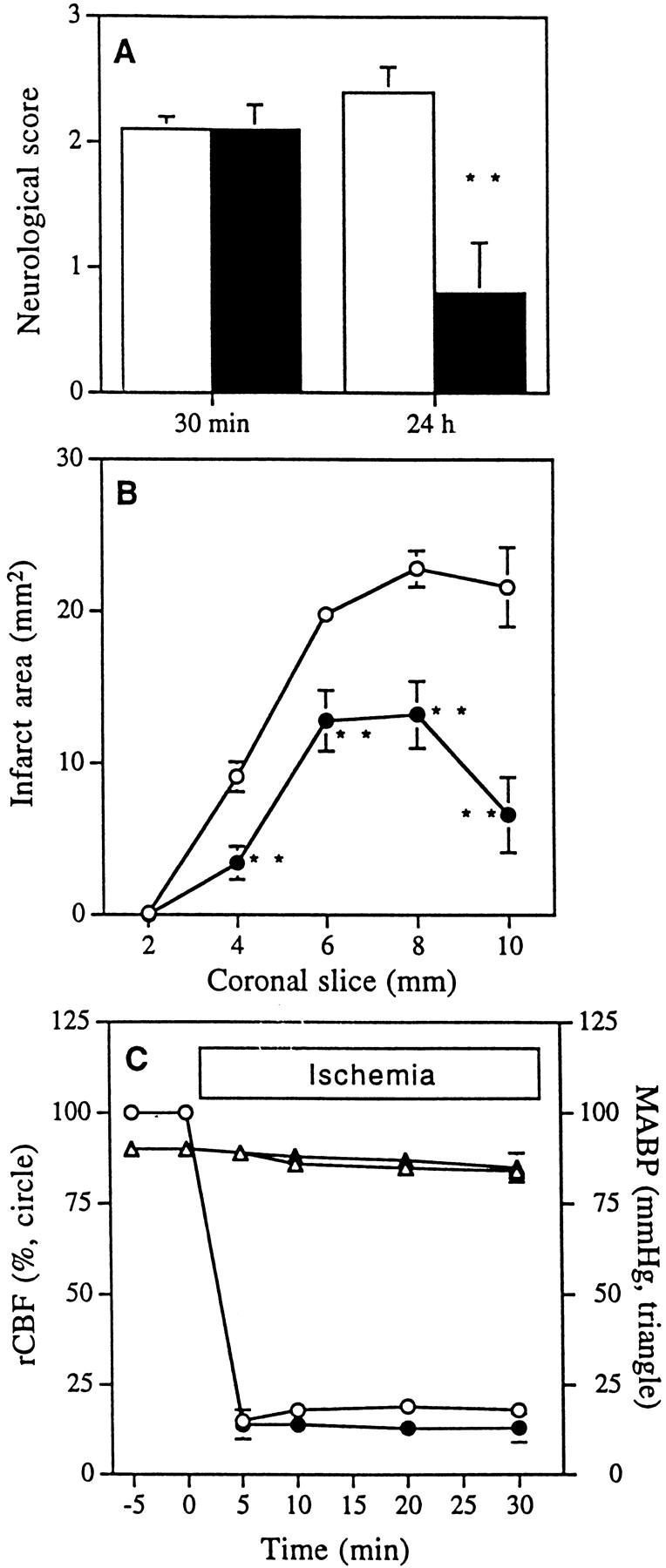
Protection from permanent MCA occlusion–mediated infarct in NSE-M17Z (black) compared to wild-type (white) mice. (A) Neurological grading 30 min and 24 h after occlusion. Neurological grading: 0, no neurological deficits; 1, failure to extend the right forepaw; 2, circling to the contralateral side; 3, loss of walking or righting reflex. (B) Infarct area assessed at 24 h. (C) Regional cerebral blood flow (rCBF), and mean arterial blood pressure (MBP) of wild-type and transgenic mice during 30 min of ischemia (**, P <0.01).
Discussion
We show here that the mutant ICEC285G inhibits apoptosis in two different species (chicken and mouse) and under the control of two different promoters (β-actin and NSE). We presented convincing evidence that mutant ICEC285G acts as a dominant negative inhibitor of ICE by inhibiting processing of pro–IL-1β. X-ray crystallography analysis showed that ICE exists as a dimer of two p20 and two p10 subunits processed from two p45 precursor molecules (28). Expression of a catalytically inactive mutant of ICE may result in formation of inactive dimers which will inhibit endogenous wild-type ICE function. Since DRG neurons from ICE knockout mice were protected from trophic factor deprivation–induced apoptosis as were DRG neurons from NSEM17Z transgenic mice, we believe that, at least in DRG neurons, the mutant ICEC285G prevented neuronal cell death by inhibiting ICE activity. This is consistent with our previous data that demonstrate that the addition of the IL-1 receptor antagonist, a naturally existing IL-1β antagonist, inhibited mouse DRG neuronal cell death induced by trophic factor deprivation, suggesting that release of mature IL-1β processed by ICE plays an active role in apoptosis induced by trophic factor deprivation (19).
We determined the role of ICE-like proteases in apoptosis induced by cerebral ischemia in a mouse permanent focal stroke model. We found that NSE-M17Z transgenic mice suffered less tissue injury and less behavioral changes as a result of ischemic injury, compared to the wild-type mice. Although we cannot rule out the possibility that the reduction of neuronal damage by mutant ICE is due to its ability to inhibit other members of the ICE family, evidence suggests that ICE itself plays an important role in cerebral ischemia-induced cell death. Elevated levels of IL-1β are detected after cerebral ischemia (29). In addition, intraventricular administration of the IL-1 receptor antagonist decreases infarct size after permanent MCA occlusion (30). We have also demonstrated that endogenously produced mature IL-1β plays an important role in hypoxia-mediated apoptosis in vitro (19). These results suggest the involvement of ICE and of mature IL-1β receptor binding in the mechanism of ischemia-induced cell death. Our results further corroborate the notion that ICE plays an important role in apoptosis induced by ischemic injury. After exposure to certain death stimuli, the ICE cell death cascade is activated. As demonstrated here, and in previous studies, apoptosis may be inhibited by blocking the ICE cell death cascade, either the activation of pro-ICE, the function of active ICE, or the product of ICE activity which is mature IL-1β (7, 19, 31). We cannot rule out, however, that mutant ICEC285G may also cross-inhibit other cell death gene products, since subunits of different ICE family members sharing significant sequence homology may bind to each other forming heterooligomeres (21). Due to the limited availability of the ICE knockout mice to us, we were unable to examine directly if the ICE knockout mice are resistant to ischemic injury. Since embryonic development of the mutant ICEC285G and of ICE knockout mice are normal and no significant defect in embryonic apoptosis was uncovered (14, 17), a nonredundant function of ICE in developmental apoptosis has been ruled out. We demonstrated here that transgenic mice expressing a dominant negative mutant ICE are significantly protected from neuronal cell death induced by ischemic insult, suggesting that ICE may play an important role in pathological cell death. Our results suggest that ischemic-induced injury, and possibly other disorders featuring apoptosis, can be treated with inhibitors aimed at modulating the activity of the ICE protease family to reduce tissue injury and preserve brain function.
Acknowledgments
We thank Dr. Winnie Wong of BASF for permission to use ICE knockout mice for DRG experiments. We thank Dr. O. Isacson for help with the histological sections.
Footnotes
J. Yuan was supported in part by grants from the National Institute of Aging, National Institute of Neurological Disorders and Stroke, and Bristol Myer-Squibb. R.M. Friedlander was supported by a postdoctoral training fellowship from National Institutes of Health and by an Upjohn sponsored award from the Joint Section on Cerebrovascular Surgery (Congress of Neurological Surgeons and the American Association of Neurological Surgeons) and from American Brain Tumor Association. M.A. Moskowitz, H. Hara, and K.B. Fink were supported by a grant from the National Institute of Neurological Disorders Interdepartmental Stroke Program Project (NS10828). K.B. Fink was supported by the Deutsche Forschungs Gemelnschaft (F:600/2-1). M.C. Fishman and W. Li were supported by a grant from Bristol Myer-Squibb. A.H. Greenberg and G. MacDonald were supported by a grant from the National Cancer Institute of Canada and the Medical Research Council of Canada.
1 Abbreviations used in this paper: DRG, dorsal root ganglion; ICE, IL-1β converting enzyme; MCA, middle cerebral artery; NGF, nerve growth factor; NSE, neuron specific enolase.
R. Friedlander and V. Gagliardini contributed equally to this work.
References
- 1.Wyllie AH, Kerr JFR, Currie A. Cell death: the significance of apoptosis. Int Rev Cytol. 1980;68:251–306. doi: 10.1016/s0074-7696(08)62312-8. [DOI] [PubMed] [Google Scholar]
- 2.Yuan J. Molecular control of life and death. Curr Opin Cell Biol. 1995;7:211–4. doi: 10.1016/0955-0674(95)80030-1. [DOI] [PubMed] [Google Scholar]
- 3.Ellis HM, Horvitz HR. Genetic control of programmed cell death in the nematode C. elegans. . Cell. 1986;44:817–829. doi: 10.1016/0092-8674(86)90004-8. [DOI] [PubMed] [Google Scholar]
- 4.Ellis RE, Yuan JY, Horvitz HR. Mechanisms and functions of cell death. Annu Rev Cell Biol. 1991;7:663–698. doi: 10.1146/annurev.cb.07.110191.003311. [DOI] [PubMed] [Google Scholar]
- 5.Yuan JY, Horvitz HR. The Caenorhabditis elegans genes ced-3 and ced-4act cell autonomously to cause programmed cell death. Dev Biol. 1990;138:33–41. doi: 10.1016/0012-1606(90)90174-h. [DOI] [PubMed] [Google Scholar]
- 6.Yuan Y, Shaham S, Ledoux S, Ellis HM, Horvitz HR. The C. elegans cell death gene ced-3encodes a protein similar to mammalian interleukin-1β-converting enzyme. Cell. 1993;75:641–652. doi: 10.1016/0092-8674(93)90485-9. [DOI] [PubMed] [Google Scholar]
- 7.Gagliardini V, Fernandez PA, Lee RKK, Drexler HCA, Rotello RJ, Fishman MC, Yuan J. Prevention of vertebrate neuronal cell death by the crmA gene. Science (Wash DC) 1994;263:826–828. doi: 10.1126/science.8303301. [DOI] [PubMed] [Google Scholar]
- 8.Milligan CE, Prevette D, Yaginuma H, Homma S, Cardwell C, Fritz LC, Tomaselli KJ, Oppenheim RW, Schwartz LM. Peptide inhibitors of the ICE protease family arrest programmed cell death of motorneurons in vivo and in vitro. Neuron. 1995;15:385–393. doi: 10.1016/0896-6273(95)90042-x. [DOI] [PubMed] [Google Scholar]
- 9.Heron A, Pollard H, Dessi F, Moreau J, Lasbennes F, Ben-Ari Y, Charriaut-Marlangue C. Regional variability in DNA fragmentation after global ischemia evidenced by combined histological and gel electrophoresis observations in the rat brain. J Neurochem. 1993;61:1973–1976. doi: 10.1111/j.1471-4159.1993.tb09843.x. [DOI] [PubMed] [Google Scholar]
- 10.MacManus JP, Buchan AM, Hill IE, Rasquinha I, Preston E. Global ischemia can cause DNA fragmentation indicative of apoptosis in rat brain. Neurosci Lett. 1993;164:89–92. doi: 10.1016/0304-3940(93)90864-h. [DOI] [PubMed] [Google Scholar]
- 11.Linnik MD, Zobrist RH, Hatfield MD. Evidence supporting a role for programmed cell death in focal cerebral ischemia in rats. Stroke. 1993;24:2002–2008. doi: 10.1161/01.str.24.12.2002. [DOI] [PubMed] [Google Scholar]
- 12.Tominaga T, Kure S, Narisawa K, Yoshimoto T. Endonuclease activation following focal ischemic injury in the rat brain. Brain Res. 1993;608:21–26. doi: 10.1016/0006-8993(93)90768-i. [DOI] [PubMed] [Google Scholar]
- 13.MacManus JP, Hill IE, Huang ZG, Rasquinha I, Xue D, Buchan AM. DNA damage consistent with apoptosis in transient focal ischaemic neocortex. Neuroreport. 1994;5:493–496. doi: 10.1097/00001756-199401120-00031. [DOI] [PubMed] [Google Scholar]
- 14.Kuida K, Lippke JA, Ku G, Harding MW, Livingston DJ, Su MSS, Flavell RA. Altered cytokine export and apoptosis in mice deficient in interleukin-1β converting enzyme. Science (Wash DC) 1995;267:2000–2002. doi: 10.1126/science.7535475. [DOI] [PubMed] [Google Scholar]
- 15.Hogquist AH, Nett MA, Unaune ER, Chaplin DD. Interleukin 1 is processed and released during apoptosis. Proc Natl Acad Sci USA. 1991;88:8485–8489. doi: 10.1073/pnas.88.19.8485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zychlinsky A, Fitting C, Cavaillon JM, Sansonetti PJ. Interleukin 1 is released by murine macrophages during apoptosis induced by Shigella flexneri. . J Clin Invest. 1994;94:1328–1332. doi: 10.1172/JCI117452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li P, Allen H, Banerjee S, Franklin S, Herzog L, Johnston C, McDowell J, Paskind M, Rodman L, Salfeld J, et al. Mice deficient in IL-1β–converting enzyme are deficient in production of mature IL-1β and resistant to endotoxic shock. Cell. 1995;80:401–411. doi: 10.1016/0092-8674(95)90490-5. [DOI] [PubMed] [Google Scholar]
- 18.Miura M, Friedlander RM, Yuan J. Tumor necrosis factor–induced apoptosis is mediated by a CrmA-sensitive cell death pathway. Proc Natl Acad Sci USA. 1995;92:8318–8322. doi: 10.1073/pnas.92.18.8318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Friedlander RM, Gagliardini V, Rotello RJ, Yuan J. Functional role of interleukin-1β (IL-1β) in IL-1β– converting enzyme–mediated apoptosis. J Exp Med. 1996;184:717–724. doi: 10.1084/jem.184.2.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miura M, Zhu H, Rotello R, Hartweig EA, Yuan J. Induction of apoptosis in fibroblasts by IL-1β–converting enzyme, a mammalian homologue of the C. eleganscell death gene. Cell. 1993;75:653–660. doi: 10.1016/0092-8674(93)90486-a. [DOI] [PubMed] [Google Scholar]
- 21.Gu Y, Wu J, Faucheu C, Livingston DJ, Su MSS. Interleukin-1β converting enzyme requires oligomerization for activity of processed forms in vivo. EMBO (Eur Mol Biol Organ) J. 1995;14:1923–1931. doi: 10.1002/j.1460-2075.1995.tb07184.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martinou JC, Dubois-Dauphin M, Staple JK, Rodriguez I, Frankowski H, Missotten M, Albertini P, Talabot D, Catsicas S, Pietra C, Huarte J. Overexpression of BCL-2 in transgenic mice protects neurons from naturally occurring cell death and experimental ischemia. Neuron. 1994;13:1017–1030. doi: 10.1016/0896-6273(94)90266-6. [DOI] [PubMed] [Google Scholar]
- 23.Hara H, Huang P, Panahian N, Fishman MC, Moskowitz MA. Reduced brain edema and infarction volume in mice lacking the neuronal isoform of nitric oxide synthetase after transient MCA occlusion. J Cereb Blood Flow Metab. 1996;16:605–611. doi: 10.1097/00004647-199607000-00010. [DOI] [PubMed] [Google Scholar]
- 24.Davies AM. Molecular and cellular aspects of patterning sensory connections in the vertebrate nervous system. Development (Camb) 1987;101:185–208. [PubMed] [Google Scholar]
- 25.Forss-Petter S, Danielson PE, Catsicas S, Battenberg E, Price J, Nerenberg M, Sutcliffe JG. Transgenic mice expressing β-galactosidase in mature neurons under neuron-specific enolase promoter control. Neuron. 1990;5:187–197. doi: 10.1016/0896-6273(90)90308-3. [DOI] [PubMed] [Google Scholar]
- 26.Farlie PG, Dringen R, Rees SM, Kannourakis G, Bernard P. bcl-2 transgene expression can protect neurons against developmental and induced cell death. Proc Natl Acad Sci USA. 1995;92:4397–4401. doi: 10.1073/pnas.92.10.4397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Y, Chopp M, Jiang N, Zaloga C. In situ detection of DNA fragmentation after focal ischemia in mice. Mol Brain Res. 1995;28:164–168. doi: 10.1016/0169-328x(94)00220-9. [DOI] [PubMed] [Google Scholar]
- 28.Wilson KP, Black J-AF, Thomson JA, Kim EE, Griffith JP, Navia MA, Murcko MA, Chambers SP, Aldape RA, Raybuck SA, Livingston DJ. Structure and mechanism of interleukin-1β converting enzyme. Nature (Lond) 1994;370:270–275. doi: 10.1038/370270a0. [DOI] [PubMed] [Google Scholar]
- 29.Lui T, Mc PC, Donnell, Young PR, White RF, Siren AL, Hallenbeck JM, Barone FC, Feuerstein GZ. Interleukin-1β mRNA expression in ischemic rat cortex. Stroke. 1993;24:1746–1751. doi: 10.1161/01.str.24.11.1746. [DOI] [PubMed] [Google Scholar]
- 30.Relton JK, Rothwell NJ. Interleukin-1 receptor antagonist inhibits ischaemic and excitotoxic neuronal damage in rats. Brain Res Bull. 1992;29:243–246. doi: 10.1016/0361-9230(92)90033-t. [DOI] [PubMed] [Google Scholar]
- 31.Boudreau N, Sympson CJ, Werb Z, Bissell MJ. Suppression of ICE and apoptosis mammary epithelial cells by extracellular matrix. Science (Wash DC) 1995;267:891–893. doi: 10.1126/science.7531366. [DOI] [PMC free article] [PubMed] [Google Scholar]