

Abstract
Immunoglobulins (Ig), particularly IgE, are believed to be crucially involved in the pathogenesis of asthma and, equally, in allergic models of the disease. To validate this paradigm we examined homozygous mutant C57BL/6 mice, which are B cell deficient, lacking all Ig. Mice were immunized intraperitoneally with 10 μg ovalbumin (OVA) plus alum, followed by daily (day 14–20) 30 min exposures to OVA aerosol (OVA/OVA group). Three control groups were run: OVA intraperitoneally plus saline (SAL) aerosol (OVA/SAL group); saline intraperitoneally plus saline aerosol; saline intraperitoneally plus OVA aerosol (n = 6–7). Lung and large airway tissues obtained 24 h after the last OVA or SAL exposure were examined by light microscopy and transmission electron microscopy (TEM). The Ig-deficient mice receiving OVA/ OVA treatment had swollen and discolored lungs and exhibited marked eosinophilia both in large airway subepithelial tissue (49.2 ± 12.0 cells/mm basement membrane [BM] versus OVA/ SAL control 1.2 ± 0.3 cells/mm BM; P <0.001), and perivascularly and peribronchially in the lung (49.3 ± 9.0 cells/unit area versus OVA/SAL control 2.6 ± 0.6 cells/unit area; P <0.001). The eosinophilia extended to the regional lymph nodes. TEM confirmed the subepithelial and perivascular localization of eosinophils. Mucus cells in large airway epithelium increased from 1.5 ± 0.8 (OVA/SAL mice) to 39.5 ± 5.7 cells/mm BM in OVA/OVA treated mice (P <0.001). OVA/SAL mice never differed from the other control groups. Corresponding experiments in wild-type mice (n = 6–7 in each group) showed qualitatively similar but less pronounced eosinophil and mucus cell changes. Macrophages and CD4+ T cells increased in lungs of all OVA/OVA-treated mice. Mast cell number did not differ but degranulation was detected only in OVA/OVA-treated wild-type mice. Immunization to OVA followed by OVA challenges thus cause eosinophil-rich inflammation in airways and lungs of mice without involvement of B cells and Ig.
Airway mucosal inflammation in allergic asthma is characterized by increased numbers of eosinophils, but macrophages and T and B lymphocytes also may be increased (1, 2). The eosinophils are believed to play a central role in the pathogenesis of this disease by releasing proinflammatory mediators/cytokines and proteins that are epithelium toxic (3, 4). Eosinophils express various Ig Fc receptors possibly involved in activation of these cells (5, 6). IgE may mediate eosinophil recruitment and activation also through indirect pathways, i.e., through the release of mast cell mediators/ cytokines (7, 8) and T cell cytokines (9). Coyle and colleagues (9) recently reported that administration of nonanaphylactogenic anti-IgE mAbs (neutralizing serum IgE) before antigen challenge significantly reduced the recruitment of eosinophils into the lungs of actively immunized mice. Through further experiments involving anti-CD23 mAbs and CD23-deficient mice, the authors suggested that this effect was due to inhibition of the IgE–CD23-facilitated antigen presentation to T cells, leading to inhibited secretion of IL-5 (9). Such data agree well with a widely accepted paradigm that allergic eosinophilic asthma is an IgE-dependent disease. This paradigm, which also rests on epidemiological data showing association between elevated IgE levels and bronchial asthma (10, 11), forms the basis of major research lines including development of treatment principles such as anti-IgE and anti–IL-4 (12, 13). However, there are also reports that question a major role of IgE in asthma and in allergic models of asthma. For example, specific IgE titres may not correlate with airway hyperreactivity or pulmonary eosinophilia (14, 15), and anaphylactic death can occur in IgE-deficient mice (16). In the latter study, the authors suggested that other Ig than IgE were involved in this anaphylaxis (16).
This study examines whether or not eosinophilic airway and pulmonary responses may develop in immunized and allergen-exposed mice in the absence of all Ig. Thus, we have used mice that are B cell deficient (lacking all Ig) due to a homozygous targeted disruption of the membrane exon of the Ig μ chain gene (17). We used a protocol that in corresponding wild-type mice produces an established allergic model of eosinophilic asthma (14, 18–20). Hence, this study asks whether B cells and Ig are crucially involved in the development of immunization and allergen exposureinduced eosinophilic pulmonary and airway inflammation.
Materials and Methods
Animals and Study Design.
Homozygous mutant C57BL/6 mice with a targeted disruption of the membrane exon of the Ig μ chain gene (17) (Jackson Laboratory, Bar Harbor, ME), 6–8 mo of age, and wild-type C57BL/6 mice (Bomholtgaard, Denmark), 3 mo of age, were used in the experiments. The present batch of Ig-deficient mice was routinely checked for lack of Ig by immunohistochemistry, using FITC-labeled anti-IgG and anti-IgM antibodies. All mice were kept in well-controlled animal housing facilities and had free access to tap water and pelleted food throughout the experimental period.
Each experimental group consisted of 6–10 animals. Two groups of Ig-deficient mice and two groups of wild-type mice were actively immunized to OVA (day 0). Also, two groups each of the different mice were sham immunized with saline (SAL)1 (day 0). From day 14 to day 20, the groups were challenged either with aerosolized OVA or SAL (Table 1). Animals were sacrificed by i.p. injection of pentobarbital 24 h after last aerosol challenge, and the lungs and extrahilar tracheobronchial airways were rapidly dissected out.
Table 1.
Effects on Eosinophils and Mucus Cells of Immunization and Allergen Challenge in Ig-Deficient and Wild-type Mice
| Groups | Immunization | Challenge | Eosinophils in lung tissue (eos./unit area) | Eosinophils in large airway subepithelial tissue (eos./mm BM) | Mucus cells in large airway epithelium (cells/mm BM) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ig-deficient OVA/OVA | OVA | OVA | 49.3 ± 9.0* | 49.2 ± 12.0* § | 39.5 ± 5.7* § | |||||
| Ig-deficient OVA/SAL | OVA | SAL | 2.6 ± 0.6 | 1.2 ± 0.3 | 1.5 ± 0.8 | |||||
| Ig-deficient SAL/OVA | SAL | OVA | 3.4 ± 0.6 | 1.0 ± 0.3 | 0.3 ± 0.2 | |||||
| Ig-deficient SAL/SAL | SAL | SAL | 3.7 ± 0.8 | 1.0 ± 0.4 | 0.6 ± 0.3 | |||||
| Wild-type OVA/OVA | OVA | OVA | 30.2 ± 10.2* | 12.2 ± 5.8‡ | 29.3 ± 6.2§ | |||||
| Wild-type OVA/SAL | OVA | SAL | 1.5 ± 0.9 | 0.9 ± 0.4 | 4.7 ± 2.7 | |||||
| Wild-type SAL/OVA | SAL | OVA | 1.0 ± 0.2 | 0.9 ± 0.4 | 2.4 ± 1.3 | |||||
| Wild-type SAL/SAL | SAL | SAL | 1.4 ± 0.2 | 0.9 ± 0.4 | 0.4 ± 0.2 |
BM, basement membrane; values are means ± SEM.
P <0.001 compared with the corresponding OVA/SAL control group.
P <0.01.
P <0.05.
Immunization and Challenge.
We have used a protocol slightly modified from that developed by Brusselle and colleagues (20). On the first day of the experiment (day 0), mice were actively immunized to chicken OVA (Grade III; Sigma, St. Louis, MO) via i.p. injection with 10 μg OVA, adsorbed to 1 mg of alum. Control animals were injected i.p. on day 0 with 0.4 ml SAL. From day 14 to day 20, the mice were exposed daily to aerosolized SAL or OVA over a 30-min period by placing groups of 6–12 awake mice in an exposition chamber. The aerosols were generated into the chamber using a nebulizer (Bird 500 ml Inline Micronebulizer driven at 4 bar, Bird Co., Palm Springs, CA). The concentration of OVA in the nebulizer was 1% wt/vol. Immunized wild-type mice were required to develop inflammatory infiltrates after allergen challenge to be included in the present analysis.
Histochemistry.
Lung tissue specimens and extrahilar large airway segments were immersed overnight in Stefanini's fixative (2% paraformaldehyde and 0.2% picric acid in 0.1 M phosphate buffer, pH 7.2), rinsed repeatedly in buffer (Tyrode buffer supplemented with 10% sucrose), frozen in mounting medium (TissueTek, Miles, Inc., Elkhart, IN), and stored at −80°C until sectioning. Eosinophils were detected by histochemical visualization of cyanide-resistant eosinophil peroxidase (EPO) activity (21). In brief, 10-μm cryosections were incubated for 8 min at room temperature in PBS buffer (pH 7.4) supplemented with 3.3-diaminobenzidine tetrahydrochloride (60 mg/100 ml; Sigma), 30% H2O2 (0.3 ml/100 ml), and NaCN (120 mg/100 ml). Slides were then rinsed in water and mounted in Kaisers medium (Merck, Darmstadt, Germany). Eosinophils were identified by their dark brown reaction product. For histochemical detection of mucus-containing cells in lungs and airways, 10-μm cryosections were stained with periodic acid–Schiff reagent (PAS). Distinct purple–red granules could be observed in the mucus-containing cells. For assessment of general airway morphology, sections were stained with hematoxylin and erythrosin. Also, toluidine blue staining was performed to visualize mast cells.
Immunohistochemistry.
Lung tissue specimens were rapidly frozen on dry ice in mounting medium and stored at −80°C until sectioning. 10-μm specimens were cut in a cryostat and the sections placed on chromealum-coated slides and air-dried. To visualize macrophages, sections were fixed in cold acetone diluted 1:2 in distilled water for 30 s, followed by final fixation in cold acetone (100%) for 5 min. After washing in PBS supplemented with 0.25% Triton X-100, the sections were exposed to a monoclonal rat anti–mouse F4/80 antigen antibody (code no. MCA 497; Serotec, Kidlington, UK) overnight at +4°C. Next, after washing, the sections were incubated with a FITC-labeled affinity-purified anti-rat Ig secondary antibody raised in rabbit (Vector Laboratories, Burlingame, CA) 1 h at room temperature. After another washing, the sections were mounted in PBS/glycerine (1:1) and examined in a fluorescence microscope. To visualize CD4+ T cells, unfixed tissue sections were exposed to a rat monoclonal anti–mouse L3T4 (CD4) antibody (code no. 01440; Becton Dickinson, Bedford, MA) for 1 h at room temperature, washed, and then incubated for 1 min with the same secondary antibody as above. After another washing, the sections were mounted as above and examined. In control sections, omitting the primary antibody, no or only slight background staining was found.
Transmission Electron Microscopy.
Lung tissue and proximal tracheal segments were immersed in a fixative consisting of a mixture of 3% formaldehyde and 1% glutaraldehyde in 0.1 M phosphate buffer, pH 7.2. The specimens were fixed overnight and then post fixed in 1% osmium tetroxide for 1 h. Then, they were dehydrated in graded acetone solutions, and embedded in Polybed 812 (Polysciences, Inc., Warrington, NY). For high resolution light microscopy, semithin (1 μm) sections of the embedded tissue were cut and stained with Azur–methylene blue. These sections were used to select areas of interest for electron microscopical examination. Ultrathin sections (60–80 nm) were cut on an LKB MK III ultratome and routinely contrasted with uranyl acetate and lead citrate. The sections were examined using a Philips CM-10 transmission electron microscope.
Quantification and Statistics.
Quantification of eosinophils, mast cells, or PAS-positive cells in the extrahilar airways was performed in one section from each animal. The total number of eosinophils or mast cells within 0–120 μm from the airway epithelial basement membrane and the total number of PAS-positive cells in the large airway epithelium in each section were counted. The total length of the airway epithelial basement membrane in each section was calculated using a computerized image analysis system and the density of eosinophils, mast cells, or PAS-positive cells was expressed as cells/mm basement membrane. For evaluation of the number of eosinophils in pulmonary tissue, ten randomly selected areas (0.04 mm2 each) in one lung section from each animal were examined. The number of eosinophils in the 10 areas was counted at a magnification of ×400 and the mean was expressed as eosinophils/unit area. All quantifications were performed in a blind fashion.
Data are expressed as mean ± SEM. To calculate significance levels between treatment groups Student's t test was used throughout the study. To achieve comparable standard deviations, values were transformed to logarithms before the statistical analysis. P <0.05 were used as the generally accepted level of statistical significance for differences between mean values. In no case were significances between corresponding control groups, i.e., SAL/SAL, SAL/ OVA, and OVA/SAL, obtained.
Results
Eosinophils.
On gross examination during the tissue dissection, the lungs of OVA/OVA-treated animals appeared swollen and discoloured. These animals, of both the Ig deficient and wild type, showed a multifocal perivascular and peribronchial eosinophilic distribution in the lung tissue (Fig. 1, a–b) and marked eosinophilic infiltrates occurred in the large airway tissue and in the tracheobronchial lymph nodes (Table 1). There was a particularly dense infiltration of eosinophils in the tracheobronchial lymph nodes of Igdeficient mice (Fig. 2). Eosinophilic infiltrates were not observed in lung or large airway tissue from control mice of either the wild-type or the Ig-deficient type (Fig. 1, c–d; Table 1).
Figure 1.
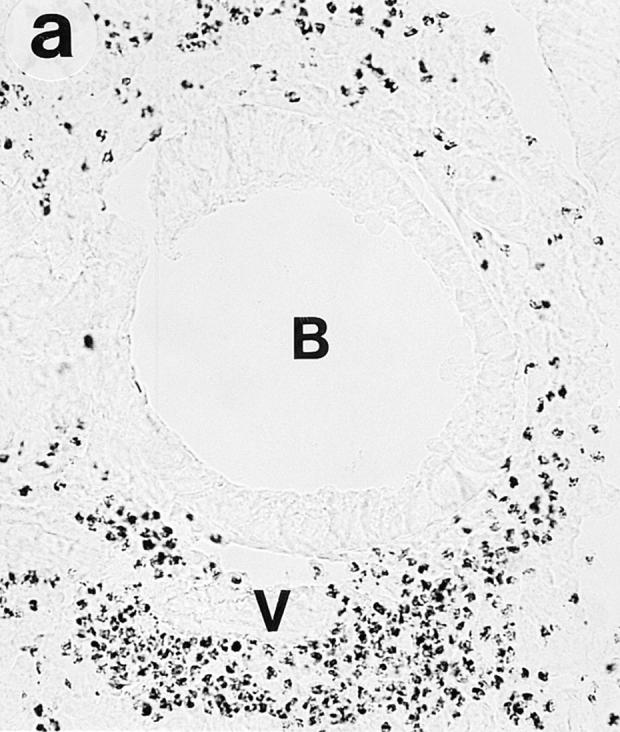
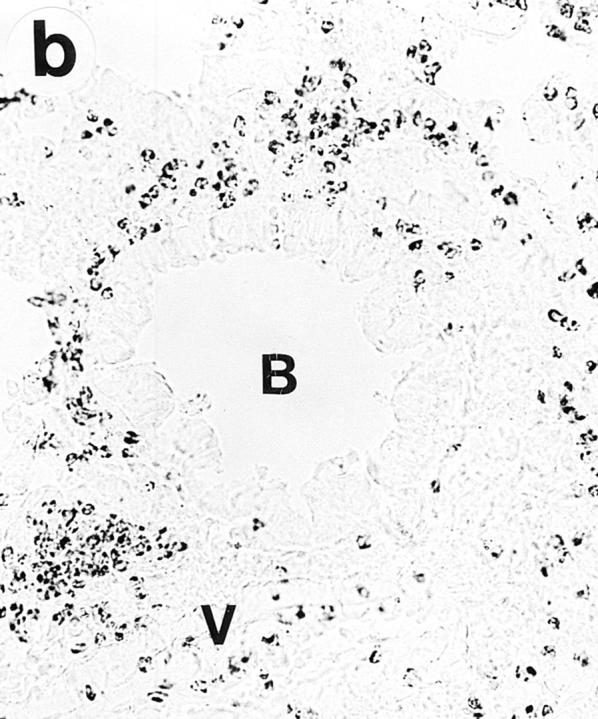
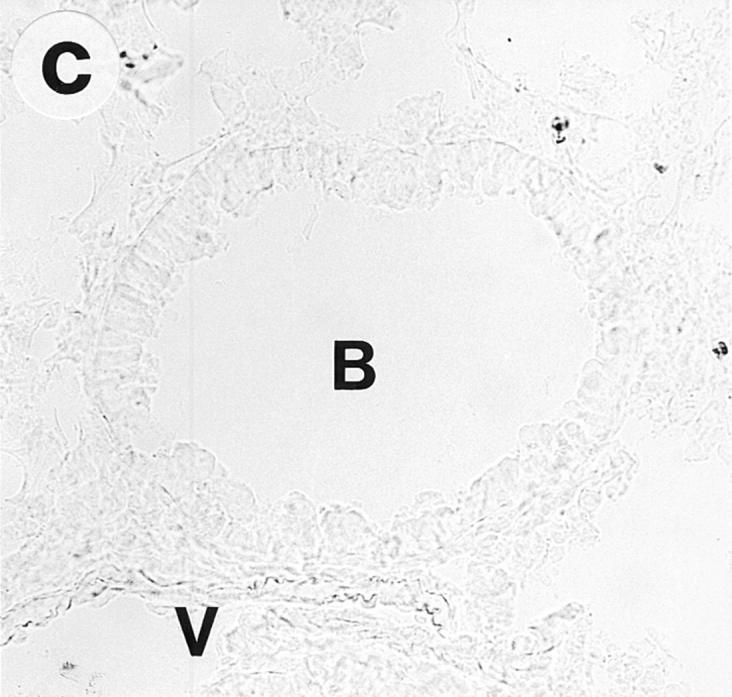
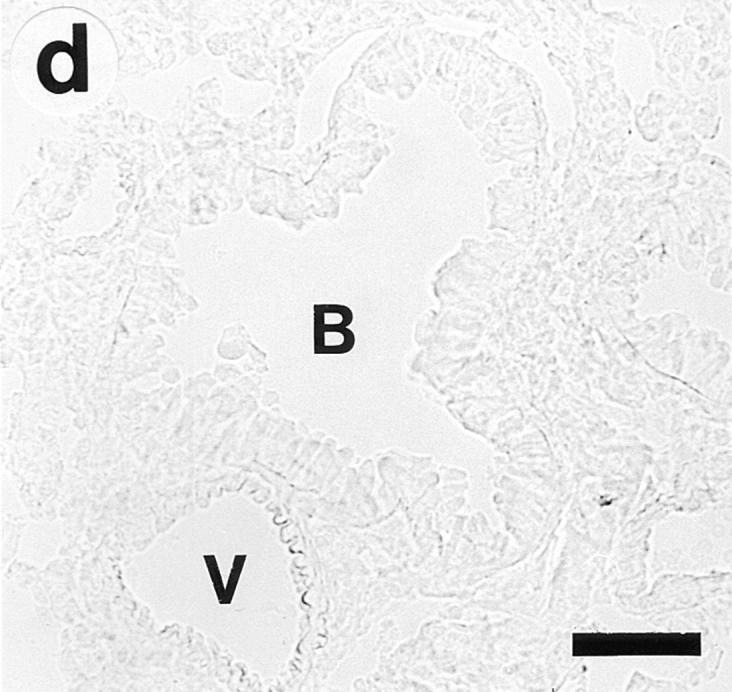
Lung histology. Eosinophils are visualized by histochemical demonstration of cyanide-resistant eosinophil peroxidase activity. Multifocal perivascular and peribronchial eosinophilic distribution in the lung tissue is seen in immunized and OVAchallenged Ig-deficient (a) and wild-type (b) mice. No eosinophilic infiltrates were observed in lung tissue from control mice of either the Ig-deficient (c) or the wild-type (d) mice. B, bronchus; V, blood vessel. Bar, 60 μm.
Figure 2.

Light micrograph, demonstrating the dense infiltration of eosinophils in tracheobronchial lymph nodes of OVA/OVA-treated B cell– deficient mice. Eosinophils are detected by histochemical visualization of cyanide-resistant eosinophil peroxidase activity. TL, tracheal lumen; LN, lymph node. Bar, 150 μm.
Ultrastructural examination confirmed the infiltration of eosinophils in the subepithelial tissue of the trachea, bronchi, and bronchioles and around smaller vessels in immunized and OVA-challenged mice. Also, to a much lesser extent, eosinophils were observed in the alveolar walls. Very few eosinophils were detected in the airway epithelium. Ultrastructural signs of activation/degranulation of eosinophils were not found in the lung or large airway tissue of OVA/OVA-treated mice, i.e., the secondary granules had a central electron-dense core surrounded by a lighter area and clusters of free extracellular eosinophil granules (as would occur secondary to eosinophil lysis) were not detected. Numerous lymphocytes, macrophages, and a few neutrophils were seen both in large airways and lungs of OVA/OVA-treated mice.
Epithelial Changes.
In OVA/OVA-treated animals (both B cell–deficient and wild-type mice) there was a marked increase in airway epithelial mucus cells (Table 1) and the height of the epithelium appeared to be increased (Fig. 3). The OVA/OVA-treated animals further differed from controls by partial or seemingly complete occlusion of many smaller airways with viscid PAS-positive mucus plugs (Fig. 4). TEM examination of SAL/SAL-treated animals revealed only few cells containing mucus granules (see Fig. 3 a), whereas OVA/OVA-treated mice (both B cell deficient and wild type) exhibited an epithelium with numerous mucus granule–containing cells protruding into the airway lumen (see Fig. 3 b).
Figure 3.
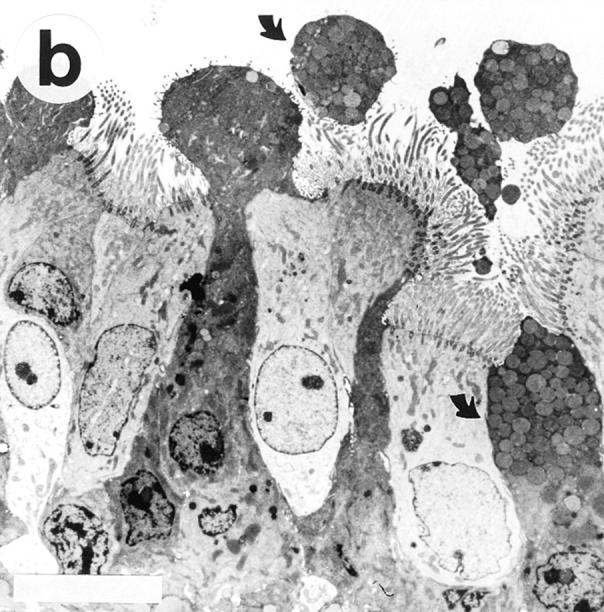
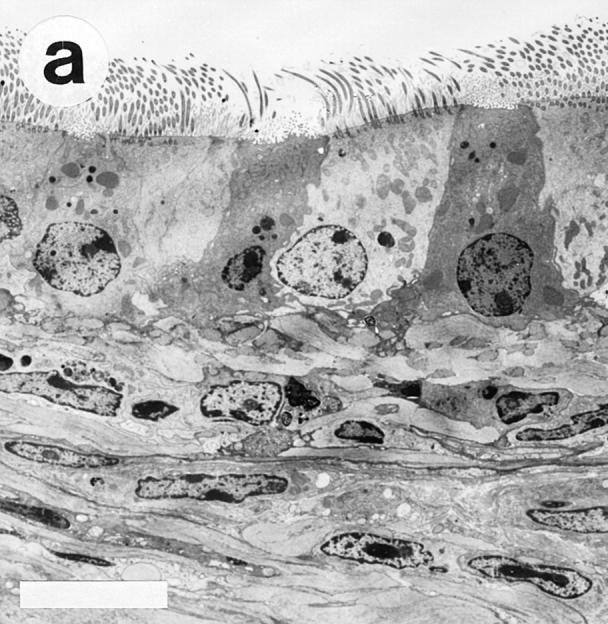
Transmission electron micrographs showing the tracheal epithelium in Ig-deficient mice. The tracheal epithelium in SAL/SAL-treated animals (a) harbored only few scattered cells containing mucus granules. In contrast, OVA/OVA-treated mice (b) exhibited an epithelium with numerous mucus granule– containing cells (arrows), often protruding into the airway lumen. Bars, 10 μm.
Figure 4.


Light micrographs of PAS-stained lung tissue cryosections obtained from immunized Ig-deficient mice 24 h after last challenge with aerosolized SAL (a) or OVA (b). After saline challenge, there are no PAS-positive cells in the airway epithelium or PAS-positive material in the bronchial lumen. In contrast, after ovalbumin challenge there are numerous PAS-positive cells in the airway epithelium and PAS-positive material is present in the bronchial lumen. B, bronchus. Bar, 100 μm.
Immunocytochemistry.
Macrophages partly had a peribronchial and perivascular distribution, partly a homogenous distribution throughout the lung parenchyma in both the wild-type and Ig-deficient OVA/OVA-treated mice. In the control mice (OVA/SAL groups), macrophages appeared less prominent and only the homogenous distribution pattern was observed. CD4+ T cells were found mainly accumulated in the peribronchial and perivascular inflammatory infiltrates of OVA/OVA-treated animals. In control mice (OVA/SAL groups), CD4+ T cells were scarce. No obvious difference in CD4+ T cell distribution pattern was detected between wild type and Ig-deficient mice.
Mast Cells.
There was no significant difference in the number of large airway subepithelial mast cells between OVA/OVA-treated wild-type and Ig-deficient mice (data not shown). Furthermore, there was no difference in mast cell number between OVA/OVA-treated animals and controls (data not shown). In the OVA/OVA-treated wildtype mice a proportion of the mast cells were degranulated, i.e., the cells contained only a few metachromatic granules. This sign of mast cell activation was not detected in the OVA/OVA-treated Ig-deficient mice or in controls.
Discussion
This study demonstrates that B cell–deficient mice, when subjected to an immunization and allergen challenge regime, previously established in wild-type mice (14, 18–20), develop airway and pulmonary eosinophilic inflammation and epithelial phenotypic changes at least as pronounced as in wild-type mice. These data support the notion that B cells are not essential for antigen presentation in the airways (22). Furthermore, they indicate that major characteristic features of allergic mouse models of asthma can develop independent of all Ig. Thus, the present findings challenge the general notion that IgE, or other Ig, is crucially involved in the eosinophilic inflammation of allergic airway disease.
The present mice with a homozygous targeted disruption of the membrane exon of the Ig μ chain gene have already been used in several studies (22–27). They are unable to produce antibodies and the absence of Ig-positive cells and Ig deposition has been repeatedly demonstrated (17, this study). By the use of this knockout mouse, Epstein et al. (22) have shown that B cells are not critical for the priming of T cells in response to protein antigen. The present allergic inflammation thus may involve T cell responses (both priming and secondary responses of T cells) potentially evoked by involvement of airway dendritic cells (28), monocytes/macrophages (29), and eosinophils (30, 31) (but not B cells) as antigen-presenting cells. Using allergic C57BL/6 mice, Kips et al. (18) and Brusselle et al. (19, 20) have demonstrated that eosinophilic pulmonary inflammation and increased IgE levels do not develop in IL-4–deficient mice or in mice lacking CD4+ T lymphocytes. Other findings, including the demonstration that allergen-induced pulmonary eosinophilia can be transferred to naive rats by allergen-specific CD4+ T cells from sensitized animals (32), suggest a crucial role of CD4+ T cells, and their production of IL-4 and IL-5 (33), in causing airway and pulmonary eosinophilia. This study also showed that CD4+ T cells and macrophages were increased by immunization and allergen exposure both in B cell–deficient and wild-type mice.
In allergic guinea pigs, eosinophils may abound in tracheobronchial lymph nodes (34). An intriguingly dense infiltration of eosinophils into the tracheobronchial lymph nodes was now observed in OVA/OVA-treated mice, being especially pronounced in the B cell–deficient mice. In a previous study, Krinzman et al. (35) observed an expansion of the CD4+ T cell population in the tracheobronchial lymph nodes after antigen challenge in immunized mice. As eosinophils may have a capability to function as antigen-presenting cells (30, 31), and because they occur in the regional lymph nodes in allergic airway inflammation, it cannot be excluded that these cells are important for T cell activation in response to protein antigen in the present mice. CD4+ T cells also produce IL-4, which may cause excessive IgE synthesis by B cells (36). IgE antibodies bound to mast cells typically react with antigen to evoke mast cell degranulation, which, as supported by the present observations, probably occurred in the wild-type C57BL/6 mice. Release of proinflammatory mast cell mediators, including eosinophilic chemotactic factors (7, 8), thus may elicit eosinophilic inflammation. However, unreduced levels of eosinophilia in mast cell–deficient mice (9, 20) and in Ig-deficient mice (this study) compared with wild-type animals indicate that the T cells may not rely on this pathway to exert their effects on airway eosinophilia. The present data are compatible with a significant role of CD4+ T cells but indicate that neither IgE nor other Ig, or B cells, are required for development of eosinophilic inflammation in lungs and airways of allergic mice.
The present protocol for immunization and allergen exposure was adopted from Kips et al., and Brusselle et al., (14, 18–20). In wild-type C57BL/6 mice these authors have quantitated several features, including increased total and antigen-specific IgE levels, pulmonary hyperresponsiveness, and increased numbers of lymphocytes and eosinophils in bronchoalveolar lavage fluids (BAL). Interestingly, Kips et al. (14) observed that administration of IL-12, selectively during the challenge period, inhibited both the eosinophilia and the hyperresponsiveness without reducing IgE. This latter finding may be explained by the possibility, demonstrated here, that airway and pulmonary inflammation is Ig-independent in these mice. The present study, which quantitates eosinophils in airway-pulmonary tissues rather than in BAL (see references 14, 18–20), demonstrates that eosinophilic inflammation may be even more pronounced in mice lacking all Ig than in the wild-type mice. Further comparisons involving age-matched groups of mice are warranted to validate this particular point of exaggerated eosinophilia. The present focus on eosinophils in the tissues (airway, pulmonary, lymph node) further revealed a paucity of epithelial eosinophils, equally in Ig-deficient and wild-type mice groups. This is remarkably different from human allergic airway diseases in which eosinophils characteristically are present in quite large numbers in the epithelium (4). Another feature of the human diseases (asthma and allergic rhinitis) is the occurrence of eosinophil lysis and the distribution of clusters of free eosinophil granules (Cfegs) in the airway mucosa (37). This sign of ultimate activation of eosinophils was not detected in the present study, in any group of animals, employing two methods (cyanide-resistant eosinophil peroxidase-staining and transmission electron microscopy [TEM]) that otherwise well identify Cfegs (38). Also, the intact eosinophils exhibited ultrastructural characteristics of nonactivated eosinophils including dense granules and smooth cell membranes in both Ig-deficient and wild-type mice (this study). Rarity of epithelial eosinophils and lack of clearly activated eosinophils may characterize also other mice models of allergic asthma (39), but so far have received little attention.
Abundance of PAS-positive epithelial cells in allergenchallenged mice has been reported previously (40, 41) and this feature was selected as one of the primary quantitative indices of airway inflammation in the present study. We could demonstrate dramatic increases in the number of airway epithelial mucus cells in the OVA/OVA-treated animals, not least in the Ig-deficient mice. As evidenced by PASpositive material in the airway lumen, and as supported by TEM observations, these cells were actively secreting in both wild-type and Ig-deficient OVA/OVA-treated animals. Indeed, several small airways of the allergic animals were occluded by the PAS-positive secretions. Such epithelial changes may be the result of several nonspecific inflammatory mechanisms. However, the similarity in appearance of the present epithelial changes agree with the possibility that allergic inflammatory processes may not differ greatly between wild-type animals and animals lacking B cells and Ig. One potential cause of mucus cell generation is epithelial damage–repair processes. However, our histological analysis revealed neither damage nor the occurrence of poorly differentiated repair cells in the epithelium (see reference 42). In normal mice mucus-containing cells are rare, although the common Clara cells occasionally may contain PAS-positive granules (43). The possibility that Clara cells may undergo transformation to the present mucus cells, which are packed with PAS-positive granules, now remains an attractive conjecture.
In conclusion, this study has demonstrated that immunization and allergen exposure-induced airway and pulmonary eosinophilic inflammation develops in mice that lack B cells and all Ig. These data challenge basic concepts of allergic airway diseases and question the curative potential of treatments aimed at inhibition of Ig and/or B cells.
Acknowledgments
This work was supported by the Swedish Medical Research Council (06P-11813, 8308, 4499), The Medical faculty, Lund University, and Astra-Draco, Lund, Sweden.
Footnotes
1 Abbreviations used in this paper: BAL, bronchoalveolar lavage fluids; Cfegs, clusters of free eosinophil granules; EPO, eosinophil peroxidase; PAS, periodic acid–Schiff reagent; SAL, saline; TEM, transmission electron microscopy.
References
- 1.Bradley BL, Azzawi M, Jacobson M, Assoufi B, Collins JV, Irani A-MA, Schwartz LB, Durham SR, Jeffery PK, Kay AB. Eosinophils, T-lymphocytes, mast cells, neutrophils, and macrophages in bronchial biopsy specimens from atopic subjects with asthma: comparison with biopsy specimens from atopic subjects without asthma and normal control subjects and relationship to bronchial hyperresponsiveness. J Allergy Clin Immunol. 1991;88:661–674. doi: 10.1016/0091-6749(91)90160-p. [DOI] [PubMed] [Google Scholar]
- 2.Kidney JC, Wong AG, Efthimiadis A, Morris MM, Sears MR, Dolowich J, Hargreave FE. Elevated B cells in sputum of asthmatics: close correlation with eosinophils. Am J Respir Crit Care Med. 1996;153:540–544. doi: 10.1164/ajrccm.153.2.8564094. [DOI] [PubMed] [Google Scholar]
- 3.Seminario M-C, Gleich GJ. The role of eosinophils in the pathogenesis of asthma. Curr Opin Immunol. 1994;6:860–864. doi: 10.1016/0952-7915(94)90005-1. [DOI] [PubMed] [Google Scholar]
- 4.Frigas, E., S. Motojima, and G.J. Gleich. 1991. The eosinophilic injury to the mucosa of the airways in the pathogenesis of bronchial asthma. Eur. Respir. J. 4 (Suppl.):123–135. [PubMed]
- 5.Gounni AS, Lamkhioued B, Ochiai K, Tanaka Y, Delaporte E, Capron A, Kinet J-P, Capron M. Highaffinity IgE receptor on eosinophils is involved in defence against parasites. Nature (Lond) 1994;367:183–186. doi: 10.1038/367183a0. [DOI] [PubMed] [Google Scholar]
- 6.Tomassini M, Tsicopoulos A, Tai PC, Gruart V, Tonnel A-B, Prin L, Capron A, Capron M. Release of granule proteins by eosinophils from allergic and nonallergic patients with eosinophilia on immunoglobulin-dependent activation. J Allergy Clin Immunol. 1991;88:365–375. doi: 10.1016/0091-6749(91)90099-a. [DOI] [PubMed] [Google Scholar]
- 7.Plaut M, Pierce JH, Watson CJ, Hanley-Hyde J, Nordan RP, Paul WE. Mast cell lines produce lymphokines in response to cross-linkage of Fcε RI or to calcium ionophores. Nature (Lond) 1989;339:64–67. doi: 10.1038/339064a0. [DOI] [PubMed] [Google Scholar]
- 8.Galli SJ. New concepts about the mast cell. N Engl J Med. 1993;328:257–265. doi: 10.1056/NEJM199301283280408. [DOI] [PubMed] [Google Scholar]
- 9.Coyle AJ, Wagner K, Bertrand C, Tsuyuki S, Bews J, Heusser C. Central role of immunoglobulin (Ig) E in the induction of lung eosinophil infiltration and T helper 2 cell cytokine production: inhibition by a non anaphylactogenic anti-IgE antibody. J Exp Med. 1996;183:1303–1310. doi: 10.1084/jem.183.4.1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sears MR, Burrows B, Flannery EM, Herbison GP, Hewitt CJ, Holdaway MD. Relation between airway responsiveness and serum IgE in children with asthma and in apparently normal children. N Engl J Med. 1991;325:1067–1071. doi: 10.1056/NEJM199110103251504. [DOI] [PubMed] [Google Scholar]
- 11.Burrows B, Martinez FD, Halonen M, Barbee RA, Cline MG. Association of asthma with serum IgE levels and skin-test reactivity to allergens. N Engl J Med. 1989;320:271–277. doi: 10.1056/NEJM198902023200502. [DOI] [PubMed] [Google Scholar]
- 12.Fick, R.B., M. Laviolette, L.-P. Boulet, J. Ruppel, M. Schoenhoff, J. Cote, F. Deschesnes, J. Reimann, and P. Boche. 1996. Bronchoalveolar lavage fluid (BALF) levels of IgE and an anti-IgE monoclonal antibody (Mab-E) following systemic administration and allergen-induced early asthmatic response (EAR). Am. J. Respir. Crit. Care Med. 153:A533. (Abstr.).
- 13.Zhou, C.Y., G.A. Koenig, I.C. Crocker, F.A. Romero, A.K. Bewtra, and R.G. Townley. 1996. Regulation of IgE production by anti-IL-4 in mouse asthma model. Am. J. Respir. Crit. Care Med. 153:A221. (Abstr.)
- 14.Kips JC, Brusselle GJ, Joos GF, Peleman RA, Tavernier JH, Devos RR, Pauwels RA. Interleukin-12 inhibits antigen-induced airway hyperresponsiveness in mice. Am J Respir Crit Care Med. 1996;153:535–539. doi: 10.1164/ajrccm.153.2.8564093. [DOI] [PubMed] [Google Scholar]
- 15.Renz H, Smith HR, Henson JE, Ray BS, Irvin CG, Gelfand EW. Aerosolized antigen exposure without adjuvant causes increased IgE production and increased airway responsiveness in the mouse. J Allergy Clin Immunol. 1992;89:1127–1138. doi: 10.1016/0091-6749(92)90296-e. [DOI] [PubMed] [Google Scholar]
- 16.Oettgen HC, Martin TR, Wynshaw-Boris A, Deng C, Drazen JM, Leder P. Active anaphylaxis in IgEdeficient mice. Nature (Lond) 1994;370:367–370. doi: 10.1038/370367a0. [DOI] [PubMed] [Google Scholar]
- 17.Kitamura D, Roes J, Kuhn R, Rajewsky K. A B cell–deficient mouse by targeted disruption of the membrane exon of the immunoglobulin μ chain gene. Nature (Lond) 1991;350:423–426. doi: 10.1038/350423a0. [DOI] [PubMed] [Google Scholar]
- 18.Kips JC, Brusselle GJ, Joos GF, Peleman RA, Devos RR, Tavernier JH, Pauwels RA. Importance of interleukin-4 and interleukin-12 in allergen-induced airway changes in mice. Int Arch Allergy Immunol. 1995;107:115–118. doi: 10.1159/000236947. [DOI] [PubMed] [Google Scholar]
- 19.Brusselle GJ, Kips JC, Joos GF, Bluethmann H, Pauwels RA. Allergen-induced airway inflammation and bronchial responsiveness in wild-type and interleukin-4deficient mice. Am J Respir Cell Mol Biol. 1995;12:254–259. doi: 10.1165/ajrcmb.12.3.7873190. [DOI] [PubMed] [Google Scholar]
- 20.Brusselle GJ, Kips JC, Tavernier JH, Van der Heyden JG, Cuvelier CA, Pauwels RA, Bluethmann H. Attenuation of allergic airway inflammation in IL-4 deficient mice. Clin Exp Allergy. 1994;24:73–80. doi: 10.1111/j.1365-2222.1994.tb00920.x. [DOI] [PubMed] [Google Scholar]
- 21.Ten RM, Pease LR, McKean DJ, Bell MP, Gleich GJ. Molecular cloning of the eosinophil peroxidase: evidence for the existence of a peroxidase multigene family. J Exp Med. 1989;169:1757–1769. doi: 10.1084/jem.169.5.1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Epstein MM, Di Rosa F, Jankovic D, Sher A, Matzinger P. Successful T cell priming in B cell–deficient mice. J Exp Med. 1995;182:915–922. doi: 10.1084/jem.182.4.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Di Rosa F, Matzinger P. Long-lasting CD8 T cell memory in the absence of CD4 T cells or B cells. J Exp Med. 1996;183:2153–2163. doi: 10.1084/jem.183.5.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Asano MS, Ahmed R. CD8 T cell memory in B cell–deficient mice. J Exp Med. 1996;183:2165–2174. doi: 10.1084/jem.183.5.2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Phillips JA, Romball CG, Hobbs MV, Ernst DN, Shultz L, Weigle WO. CD4+T cell activation and tolerance induction in B cell knockout mice. J Exp Med. 1996;183:1339–1344. doi: 10.1084/jem.183.4.1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Constant S, Schweitzer N, West J, Ranney P, Bottomly K. B lymphocytes can be competent antigen-presenting cells for priming CD4+T cells to protein antigens in vivo. J Immunol. 1995;155:3734–3741. [PubMed] [Google Scholar]
- 27.Vordermeier HM, Venkataprasad N, Harris DP, Ivanyi J. Increase of tuberculous infection in the organs of B cell–deficient mice. Clin Exp Immunol. 1996;106:312–316. doi: 10.1046/j.1365-2249.1996.d01-845.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Holt PG. Regulation of antigen-presenting cell function(s) in lung and airway tissues. Eur Respir J. 1993;6:120–129. [PubMed] [Google Scholar]
- 29.Gant V, Cluzel M, Shakoor Z, Rees PJ, Lee TH, Hamblin AS. Alveolar macrophage accessory cell function in bronchial asthma. Am Rev Respir Dis. 1992;146:900–904. doi: 10.1164/ajrccm/146.4.900. [DOI] [PubMed] [Google Scholar]
- 30.Del Pozo V, De Andrés B, Martin E, Cárdaba B, Fernández JC, Gallardo S, Tramón P, Leyva-Cobian F, Palomino P, Lahoz C. Eosinophil as antigen-presenting cell: activation of T cell clones and T cell hybridoma by eosinophils after antigen processing. Eur J Immunol. 1992;22:1919–1925. doi: 10.1002/eji.1830220736. [DOI] [PubMed] [Google Scholar]
- 31.Weller PF, Rand TH, Barrett T, Elovic A, Wong DTW, Finberg RW. Accessory cell function of human eosinophils. HLA-DR-dependent, MHC-restricted antigen-presentation and IL-1α expression. J Immunol. 1993;150:2554–2562. [PubMed] [Google Scholar]
- 32.Watanabe A, Mishima H, Renzi PM, Xu L-J, Hamid Q, Martin JG. Transfer of allergic airway responses with antigen-primed CD4+ but not CD8+T cells in Brown norway rats. J Clin Invest. 1995;96:1303–1310. doi: 10.1172/JCI118165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Foster PS, Hogan SP, Ramsay AJ, Matthaei KI, Young IG. Interleukin 5 deficiency abolishes eosinophilia, airways hyperreactivity, and lung damage in a mouse asthma model. J Exp Med. 1996;183:195–201. doi: 10.1084/jem.183.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Erjefält, J.S., M. Korsgren, M.C. Nilsson, F. Sundler, and C.G.A. Persson. 1997. Accumulation and activation of fibrinogen, neutrophils, and eosinophils at allergen challenge induced epithelial damage-restitution sites. Clin. Exp. Allergy. In press.
- 35.Krinzman SJ, De Sanctis GT, Cernadas M, Kobzik L, Listman JA, Christiani DC, Perkins DL, Finn PW. T cell activation in a murine model of asthma. Am J Physiol. 1996;271:L476–L483. doi: 10.1152/ajplung.1996.271.3.L476. [DOI] [PubMed] [Google Scholar]
- 36.Finkelman FD, Katona IM, Urban JF, Holmes J, Ohara J, Tung AS, Sample JG, Paul WE. IL-4 is required to generate and sustain in vivo IgE responses. J Immunol. 1988;141:2335–2341. [PubMed] [Google Scholar]
- 37.Persson, C.G.A., J.S. Erjefält, M. Andersson, I. Erjefält, L. Greiff, M. Korsgren, M. Linden, F. Sundler, and C. Svensson. 1996. Epithelium, microcirculation, and eosinophils: new aspects of the allergic airway in vivo. Allergy. In press. [DOI] [PubMed]
- 38.Erjefält JS, Sundler F, Persson CGA. Eosinophils, neutrophils, and venular gaps in the airway mucosa at epithelial removal–restitution. Am J Respir Crit Care Med. 1996;153:1666–1674. doi: 10.1164/ajrccm.153.5.8630618. [DOI] [PubMed] [Google Scholar]
- 39.Eum S-Y, Haile S, Lefort J, Huerre M, Vargraftig BB. Eosinophil recruitment into the respiratory epithelium following antigenic challenge in hyper-IgE mice is accompanied by interleukin 5-dependent bronchial hyperresponsiveness. Proc Natl Acad Sci USA. 1995;92:12290–12294. doi: 10.1073/pnas.92.26.12290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blyth DI, Pedrick MS, Savage TJ, Hessel EM, Fattah D. Lung inflammation and epithelial changes in a murine model of atopic asthma. Am J Respir Cell Mol Biol. 1996;14:425–438. doi: 10.1165/ajrcmb.14.5.8624247. [DOI] [PubMed] [Google Scholar]
- 41.Henderson, W.R., D.B. Lewis, R.K. Albert, Y. Zhang, W.J.E. Lamm, G.K.S. Chiang, F. Jones, P. Eriksen, Y.-T. Tien, M. Jonas et al. The importance of leukotrienes in airway inflammation in a mouse model of asthma. 1996. J. Exp. Med. 184:1483–1494. [DOI] [PMC free article] [PubMed]
- 42.Persson, C.G.A., and J.S. Erjefält. 1997. Airway epithelial restitution after shedding and denudation. In The Lung: Scientific Foundations. R.G. Crystal, J.B. West, E.R. Weibel, and P.J. Barnes, editors. Lippincott-Raven, Philadelphia, PA. 2611–2627.
- 43.Pack RJ, Al-Ugaily LH, Morris G. The cells of the tracheobronchial epithelium of the mouse: a quantitative light and electron microscope study. J Anat. 1981;132:71–84. [PMC free article] [PubMed] [Google Scholar]