

Abstract
In skeletal muscle excitation–contraction (E–C) coupling, the depolarization signal is converted from the intracellular Ca2+ store into Ca2+ release by functional coupling between the cell surface voltage sensor and the Ca2+ release channel on the sarcoplasmic reticulum (SR). The signal conversion occurs in the junctional membrane complex known as the triad junction, where the invaginated plasma membrane called the transverse-tubule (T-tubule) is pinched from both sides by SR membranes. Previous studies have suggested that junctophilins (JPs) contribute to the formation of the junctional membrane complexes by spanning the intracellular store membrane and interacting with the plasma membrane (PM) in excitable cells. Of the three JP subtypes, both type 1 (JP-1) and type 2 (JP-2) are abundantly expressed in skeletal muscle. To examine the physiological role of JP-1 in skeletal muscle, we generated mutant mice lacking JP-1. The JP-1 knockout mice showed no milk suckling and died shortly after birth. Ultrastructural analysis demonstrated that triad junctions were reduced in number, and that the SR was often structurally abnormal in the skeletal muscles of the mutant mice. The mutant muscle developed less contractile force (evoked by low-frequency electrical stimuli) and showed abnormal sensitivities to extracellular Ca2+. Our results indicate that JP-1 contributes to the construction of triad junctions and that it is essential for the efficiency of signal conversion during E–C coupling in skeletal muscle.
Keywords: dihydropyridine receptor; excitation–contraction coupling; junctophilin; ryanodine receptor; triad junction
Introduction
The junctional membrane complex between the plasma membrane (PM)* and the endoplasmic–sarcoplasmic reticulum (ER–SR) is an important structural foundation for crosstalk between the cell surface and intracellular ionic channels (Pozzan et al., 1994; Berridge, 1998). In skeletal muscle cells, the transverse-tubule (T-tubule) as the invaginated PM and the SR membrane together form the junctional complex, called the triad junction (Flucher, 1992). The dihydropyridine receptor (DHPR) and the ryanodine receptor (RyR) function as the cell surface voltage sensor and the SR Ca2+ release channel, respectively, and the proposed direct coupling between them in the triad junction converts cell surface depolarization to the intracellular Ca2+ signal for contraction without the requirement of Ca2+ influx (Tanabe et al., 1988; Schneider, 1994; Takeshima et al., 1994). The junctional membrane structure of the triad junction is probably required for functional coupling between DHPR and RyR, but neither DHPR nor RyR contributes to the constitution (Takekura et al., 1995; Ikemoto et al., 1997). In addition to the triad, junctional membrane complexes between the PM and ER–SR are shared by excitable cell types, including the diad in cardiac myocytes, peripheral coupling in smooth muscle and immature striated muscle cells, and subsurface cisternae in neurons. It is possible that these junctional membrane structures are composed by a similar molecular mechanism.
Of several transmembrane proteins found in the skeletal muscle triad junction (Fan et al., 1995; Jones et al., 1995; Nishi et al., 1998; Takeshima et al., 1998), junctophilin (JP) seems to be responsible for the formation of the junctional membrane structure. JP is composed of two major domains, a COOH-terminal hydrophobic segment spanning the junctional SR membrane, and the remaining cytoplasmic region interacting specifically with the PM. Moreover, the junctional membrane complex between the ER and the PM was produced by injection of JP cRNA into amphibian embryonic cells (Takeshima et al., 2000). We have identified three JP subtypes derived from different genes, namely JP-1, -2 and -3 in mammalian excitable tissues. JP-1 is found predominantly in skeletal muscle, JP-2 is expressed throughout skeletal, cardiac, and smooth muscle cells, and JP-3 is expressed specifically in the brain (Nishi et al., 2000; Takeshima et al., 2000). Mutant mice lacking JP-2 died in utero due to cardiac failure. Cardiac myocytes from the mutant embryos showed deficiency of peripheral couplings and abnormal Ca2+ transients, indicating that the JP-2–mediated formation of the junctional membrane complex is essential for physiological coupling between DHPR and RyR (Takeshima et al., 2000). In skeletal muscle, both JP-1 and JP-2 mRNAs are abundantly expressed, but functional differences between the JP subtypes have not yet been examined. In this report, we describe the essential role of JP-1 in skeletal muscle based on the morphological and functional experimental results in JP-1 knockout mice.
Results and discussion
Coexpression of JP-1 and JP-2 in skeletal muscle
Our results in RNA blot hybridization have suggested that both JP-1 and JP-2 are expressed in skeletal muscle cells (Takeshima et al., 2000). To further examine the expression, we prepared bacterial fusion proteins carrying the divergent regions of JPs, in which no conserved amino acid sequences are observed among the subtypes, and subtype-specific antibodies were developed using the fusion proteins as antigens. In immunoblot analysis of microsomal preparations from adult mouse tissues, the resulting antibodies were subtype specific, and JP-1 and JP-2 were detected as protein bands showing distinguishable migrations on gel electrophoresis (Fig. 1 A). JP-1 was specifically detected in skeletal muscle, and JP-2 was found in both skeletal and cardiac muscles. The data obtained in the Northern and Western analyses were consistent. The analysis in mouse hindlimb muscle showed that the expression levels of both JP-1 and JP-2 significantly increase during muscle maturation (Fig. 1 B). The induction of JP-1 during muscle development is especially remarkable in that the expression levels were very low in embryos and neonates, but muscles from young adult mice contained abundant JP-1. Histochemical analysis demonstrated that both antibodies to JP-1 and JP-2 specifically reacted with the rows localized at A-I junctions in the longitudinal sections of muscle preparations (Fig. 1 C). Because all muscle cells examined in hindlimb were immunolabeled with both antibodies, the JP subtypes are coexpressed in both fast and slow fibers. Our previous results demonstrated specific localization of JP-1 in the skeletal muscle triad junction (Takeshima et al., 2000). The present results showed that both JP-1 and JP-2 are colocalized in the triad junction.
Figure 1.

Expression of JP subtypes in skeletal muscle. (A) Western blot analysis of JP subtypes in mouse tissues. Total microsomes from adult mouse tissues (15 μg protein each) were analyzed with antibodies specific to JP-1 and JP-2. B, brain; H, heart; K, kidney; L, liver; SM, skeletal muscle. Size markers are shown in kilodaltons. JP-2 was detected as a broad band in skeletal muscle due to comigration with Ca2+-ATPase, the major protein component in the SR. (B) Western blot analysis of JP subtypes during muscle maturation. Total hindlimb microsomes (40 μg protein each) prepared from embryonic day 14 (E14) to postnatal day 28 (P28) mice were analyzed using the subtype-specific antibodies. Although expression of JP-1 in embryos and neonates could be detected in longer exposure (see Fig. 2), the signal densities were markedly lower than those of young adult mice. In contrast with JP-1, induction of JP-2 expression was relatively loose during muscle maturation. (C) Immunohistochemical analysis of JP subtypes in skeletal muscle. A cryosection of hindlimb muscle from adult mouse was labeled by immunofluorescence using antibodies specific to JP-1 (on 543 nm excitation) and JP-2 (on 488 nm excitation). Cytoplasmic rows immunolabeled with both antibodies are localized in identical positions (Merged). Essentially the same staining patterns were observed in all muscle fibers examined in hindlimb muscle. Bar, 10 μm.
Generation of JP-1 knockout mice
DNA segments containing part of the JP-1 gene were isolated from a mouse genomic library using the 5′-terminal region of the cDNA as a probe. The determined first and second exons encode 126– and 253–amino acid residues of JP-1, respectively. In both mouse and human genes, the first introns disrupt an identical position in the primary structure of JP-1 (Nishi et al., 2000). Our in situ hybridization analysis demonstrated that the JP-1 gene was mapped to mouse chromosome 1A2-5 (unpublished data). Several same genes have been mapped to the mouse chromosome 1A2-5 and human chromosome 8q21 containing the JP-1 gene, and the regions are homologous between mouse and human genomes.
The genomic DNA segments were used for constructing a targeting vector (Fig. 2 A). In the vector, the region containing the first protein-coding sequence was replaced by the neomycin resistance gene for positive selection, and the diphtheria toxin gene was attached at the 3′ terminus for negative selection. ES cells were transfected with the targeting vector, and the resultant G418-resistant clones were screened by Southern blot hybridization. Several ES clones were shown to have the expected pattern of arrangement in genomic DNA for homologous recombination in Southern blot screening (unpublished data). Chimeric male mice were produced by injecting the ES cells into blastocysts, and bred to yield mutant mice detected by PCR (Fig. 2 B). The chimeric mice and the heterozygous mutants could be maintained normally under our conventional housing conditions. Neonatal mice homozygous for the mutation were obtained by crossing between heterozygous mice. Western blot analyses showed an absence of JP-1 (Fig. 2 C) in microsomal preparations of hindlimb muscle from the mutant neonates, indicating that the introduced mutation is a null mutation of the gene. On the other hand, normal levels of JP-2 expression were observed in mutant muscle (Fig. 2 D), and fluorescence microscopy showed no detectable difference in subcellular localization of JP-2 between the genotypes (Fig. 2 E).
Figure 2.

Generation of JP-1 knockout mice. (A) Homologous recombination at the mouse JP-1 locus. Restriction enzyme maps of the wild-type allele, targeting vector, and mutant allele are illustrated. The exons in the gene (E1 and E2) are indicated by filled boxes, and the neomycin resistance gene (neo) and diphtheria toxin gene (DTA) are indicated by open boxes; the directions of transcription are shown by arrows. The hybridization probe and PCR primers for detection of the mutant gene are indicated by a hatched box and opened arrows, respectively; the predicted sizes of the DNA fragments in Southern blot analysis and PCR are also shown. (B) Detection of mutant JP-1 allele by PCR. Genomic DNAs from newborn mice were analyzed by PCR using primers indicated in A. Amplified DNA fragments were run on a 1.5% agarose gel, and size markers are indicated in base pairs. Positions of PCR products from wild-type and mutant genes are given. Immunoblot analysis of JP-1 (C) and JP-2 (D) in newborn mice. Microsomal proteins from hindlimb preparations were analyzed using specific antibody against JP-1 in C or JP-2 in D. Positions for target proteins are given, and size markers are indicated in kilodaltons. (E) Immunofluorescence detection of JP-2 in JP-1 knockout muscle. Cryosections of hindlimb muscles from wild-type and JP-1 knockout neonates were examined using an antibody specific to JP-2. No difference in staining pattern was detected between the genotypes. The partial sequence data for the JP-1 gene has been deposited in the database (GenBank/EMBL/DDBJ accession no. AB024446). Bar, 3 μm.
Perinatal lethality in JP-1 knockout mice
The JP-1 knockout neonates could be obtained by crossing between the heterozygous mutants, and therefore they survived fetal development. Mother mice delivered and nursed the knockout neonates normally, and all mutants were pink in color and showed kicking movements, demonstrating apparently normal function of the respiratory and limb motor systems. However, the knockout neonates died within one day after birth, although they could survive for 3–20 h. The knockout neonates exhibited a suckling defect, as no milk was detected in the stomachs. Milk suckling depends mainly on neural reflexes. Touch stimulation around the lip induces suckling-like responses, including rhythmic mouth opening and pursing movements. In the JP-1 knockout neonates, wide opening of the mouth was not observed and the jaw movements seemed to be weak compared with control neonates. Suckling-like responses can be monitored by electromyogram (EMG) recording of jaw muscles (Westneat and Hall, 1992). As shown in Fig. 3 , the JP-1 knockout neonates showed EMG responses similar to controls; the frequencies recorded in wild-type and mutant neonates were 1.10 ± 0.02 and 1.08 ± 0.02 Hz (mean ± SEM), respectively. Hence, the mutant mice retain the neural circuit responsible for the suckling reflex including the central pattern generator localized in the brain stem (Nakamura and Katakura, 1995). On the other hand, newborn mice open their mouths and make squeaky voices and kick and struggle in response to pinches of the tail or limb by forceps. Although the JP-1 knockout neonates showed normal responses of struggling, they developed infirm voices and did not open their mouths widely. These observations likely suggest that the contractile activity of jaw muscles is impaired in mutant mice.
Figure 3.

EMG response of jaw muscle induced by tactile stimulation. (A) The stimulation on the tongue evoked rhythmic EMG responses likely corresponding to suckling in wild-type neonates. Similar responses were detected in the JP-1 knockout neonates (B), although the baseline activity was higher than control and therefore the recording trace was rather noisy. It may be presumed that reduced contractions of jaw muscles did not produce modulative feedback regulation to the reflex system through muscle spindle cells in the mutant neonates. The horizontal bar, upper trace and lower trace show duration of stimulation, rectified and moving average EMG activity, and EMG recording, respectively. The rhythmic EGM recordings of suckling-like responses were detected in 87 out of 116 trials in wild-type mice (n = 5), and 53 out of 69 trials in JP-1 knockout mice (n = 3).
The direct cause of death in the JP-1 knockout mice is unclear, but could be the result of undernourishment caused by the absence of milk suckling. It has been reported that mutant mice bearing suckling failure die shortly after birth but could grow by bottle feeding for several days (see, for example, Yagi et al., 1993; Kutsuwada et al., 1996). We tried to feed the JP-1 knockout neonates manually, providing artificial milk every several hours through a tube inserted into the stomach. Control mice could be maintained at least for several days, but under the same conditions we could not prolong the survival of the JP-1 knockout mice. All of the mutants seemed to suffocate due to milk regurgitation and/or infirm breathing. Thus, it is possible that the perinatal death of the JP-1 knockout mice is caused not only by suckling failure, but also other defects such as dysfunction of pharyngoesophageal or diaphragm muscles.
Morphological abnormalities in skeletal muscle lacking JP-1
Because JP-1 is predominantly expressed in skeletal muscle, we surveyed morphological defects in skeletal muscles from the JP-1 knockout neonates. Cell density, diameter, and the shape of mutant muscle fibers seemed to be normal, and photomicroscopic examination detected no obvious abnormalities. However, the electron microscopic examination detected ultrastructural abnormalities in the membrane systems of the mutant muscles (Fig. 4) . In muscles from newborn mice, formation of the junctional complexes between the T-tubule and the SR are not yet completed, and both diads and triads partially occupy A-I junctions. In contrast to flattened T-tubular structures in mature triads, the T-tubules pinched from both sides by the SR are often elliptical in shape in immature muscles from neonates. No obvious ultrastructural abnormalities in the membrane system were observed in mutant muscles from JP-1 knockout neonates immediately after birth. However, mutant muscles from the knockout neonates that were near death (15–20 h after birth) showed the following abnormal SR features: the terminal regions of the SR were frequently swollen, longitudinal SR regions were partially vacuolated, and the orientation of SR networks was irregular. Therefore, junctional membrane complexes of both diads and triads were structurally abnormal in mutant muscles. These morphological abnormalities were detected in all muscle types examined in neonates 15–20 h after birth, and jaw muscle cells showed the most severe defects. On the other hand, mitochondria and myofibrils retained normal structures in JP-1 knockout muscles.
Figure 4.
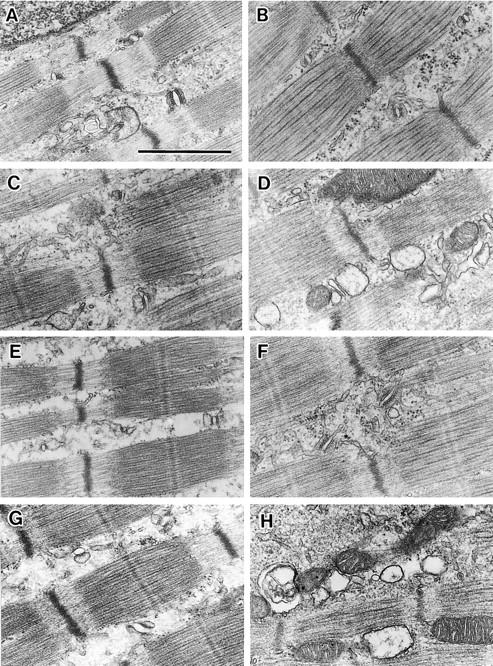
Morphological abnormalities of membrane system in JP-1 knockout skeletal muscle. Longitudinal ultrathin sections of hindlimb (A–D) and jaw (E–H) muscles were analyzed using electron microscopy. Muscle preparations were derived from wild-type (A and E) and JP-1 knockout (C and G) neonates immediately after birth, and wild-type (B and F) and JP-1 knockout (D and H) neonates ∼15 h after birth. In JP-1 knockout muscles, morphology of the SR turned worsened after birth, although mitochondria and myofibril retained normal structures. Bar, 1.0 μm.
In mutant muscle cells prepared immediately after birth, both diads and triads were apparently normal in morphology. The T-SR gap size in the junctional membrane structures was 13 ± 1 nm in both wild-type and mutant muscles. However, statistical analysis demonstrated that the number of triad junctions was significantly reduced in all examined muscles from the JP-1 knockout neonates immediately after birth (Fig. 5) . These observations clearly suggest that the loss of JP-1 affects the triad formation in skeletal muscle.
Figure 5.

Quantitative analysis of junctional membrane structures between wild-type and JP-1 knockout skeletal muscles. Skeletal muscles were prepared from tongue, jaw (digastric muscle), diaphragm, and hindlimb (thigh region) of newborn mice immediately after birth. The junctional membrane structures at the A-I junctions were analyzed by electron microscopic observations, and the results of random samples were compared between the genotypes. The data were obtained from at least 3,000 A-I junctions of three neonates and are shown as the mean ± SEM. Statistical differences between the genotypes are indicated by asterisks (t test, *P < 0.05 and **P < 0.01).
As shown in the electron microscopic observations, JP-1 knockout muscle contained few triad junctions at birth, and the SR structures worsened after birth in the mutant muscle. The observations may suggest that the triad deficiency affects SR structures in a stimulation activity–dependent manner. In striated muscle, disruption of the RyR genes produces abnormal SR and mitochondria showing vacuolated and swollen structures, probably due to SR Ca2+ overloading (Ikemoto et al., 1997; Takeshima et al., 1998). The RyR and JP-1 knockout muscles show similar structural abnormalities in the SR, although no obvious defects were developed in the mitochondria of JP-1 knockout muscle. Frequency of stimulation from motor nerves should increase after birth, and resulting Ca2+ signaling during excitation–contraction (E–C) coupling might produce the structurally abnormal SR in JP-1 knockout muscle. As in the case of the RyR-deficient muscle, SR Ca2+ overloading might underlie abnormalities in JP-1 knockout muscle because the reduced formation of triad junctions could inhibit DHPR-mediated activation of RyR.
In mutant jaw muscle, formation of both triads and diads was weak, and therefore the number of total junctional membrane complexes was significantly reduced (Fig. 5). In contrast, normal levels of diads were retained in other muscle types examined. The extreme reduction of junctional complexes in jaw muscle may cause the disturbance of mouth opening in the mutant mice. The jaw muscle–specific defect on junctional membranes does not reflect different expressions of JP subtypes among muscle types. Our Western blot experiments did not find a significant difference in JP expression levels among jaw, tongue, and hindlimb muscles in neonatal mice (unpublished data). The mechanism underlying the jaw muscle–specific abnormality is unknown.
Physiological abnormalities in skeletal muscle lacking JP-1
To survey functional abnormalities of the JP-1 knockout muscle, we examined the contraction profiles of a hindlimb muscle bundle (musculus vastus lateralis) from the mutant neonates. Fig. 6 shows significant differences in the force–frequency relationship between JP-1 knockout and wild-type muscles in normal bathing solution. Electrical stimuli at low frequencies developed less contractile forces in the mutant muscle compared with wild-type muscle. Twitch tension developed in the mutant muscle was reduced to about half the value of that in control muscle, although there is no significant difference in the maximum force between the genotypes (Table I). Thus, the force–frequency curve of the mutant muscle was shifted downward, indicating that the loss of JP-1 reduced the efficiency of E–C coupling. The low efficiency in the mutant muscle is likely due to the deficiency of triad junctions where functional coupling occurs between DHPR and RyR.
Figure 6.

Force–frequency relationship in skeletal muscles from wild-type and JP-1 knockout neonates. Isometric tension of muscle preparations at several frequencies was determined in modified Krebs-Ringer solution, and typical recordings from wild-type and mutant muscles are shown in A. Force–frequency relationships in wild-type and mutant muscles are shown in B. Each value from at least n =13 from seven mice was normalized to the maximum force and represents the mean ± SEM. Statistical differences between the genotypes are indicated by asterisks (t test, *P < 0.05 and ** P < 0.01).
Table I. Twitch and tetanic tension of hindlimb muscles from wild-type and JP-1 knockout neonatal mice.
| Twitch | Tetanus | |
|---|---|---|
| mg/mm2 | ||
| Wild-type muscle | 22.4 ± 3.5 | 52.0 ± 8.3 |
| JP-1–deficient muscle | 11.2 ± 1.8a | 49.4 ± 7.0 |
Data (mean ± SEM) were derived from at least eight muscle preparations from six mice and each value was normalized by cross-sectional surface area of the muscle bundle. Twitch and tetanic responses were evoked by electrical stimuli at 0.33 and 30 Hz, respectively.
Indicates statistical difference (P , 0.01 in t test).
We then examined the effects of extracellular Ca2+ levels in the JP-1 knockout muscle. In mature muscle, an increase in extracellular Ca2+ does not obviously affect the twitch force, because contraction is produced by Ca2+ release from the SR by functional-coupling DHPR and RyR during E–C coupling. On the other hand, when the extracellular Ca2+ level was increased from 2.5 mM to 5 mM, twitch tension was gradually enhanced in muscle from neonatal mice (Fig. 7 A). The enhancement of twitch amplitude by high Ca2+ in the mutant muscle was significantly higher than that of control muscle. In accordance with a previous report (Beam and Knudson, 1988), the results suggest that immature skeletal muscle cells contain a detectable population of Ca2+ channels that are uncoupled with RyR and devoted to voltage-dependent Ca2+ influx. The highly increased twitch by high Ca2+, together with weak contractile responses at low-frequency stimuli (Fig. 6), might suggest an increased population of Ca2+ channels uncoupled with RyR in the JP-1 knockout muscle. Alternatively, the abnormal response to extracellular Ca2+ might require dysregulation of SR Ca2+ loading in the mutant muscle.
Figure 7.

Effects of extracellular Ca2+ concentrations on twitch tension in wild-type and JP-1 knockout skeletal muscles. Control twitch tension was measured in modified Krebs-Ringer solution containing 2.5 mM CaCl2. The bathing solution was then changed to a high-Ca2+ solution containing 5 mM CaCl2 (A) or a Ca2+-free solution containing 0.1 mM EGTA (B). Twitches evoked by 0.33-Hz stimuli were monitored continuously after the replacement, and the data (at least n = 6 from four neonates in each plot) were normalized and represent the mean ± SEM. Statistical differences between the genotypes are indicated by asterisks (t test, *P < 0.05 and **P < 0.01).
Contraction of mature skeletal muscle is highly resistant under Ca2+-free conditions, and it is difficult to deplete the SR as the intracellular Ca2+ store. However, the high resistance is not fully established in immature muscles from neonatal mice. We examined JP-1 knockout muscle under Ca2+-free conditions (Fig. 7 B). The mutant muscle showed a faster decrease of twitch tension than control muscle. The apparent time constants of the decay in wild-type and JP-1 knockout muscles were 13.7 and 7.4 min, respectively. A similar result has been observed in mitsugumin29 knockout muscle bearing structurally irregular membrane systems around the triad junction (Nishi et al., 1999). The observations on the mutant muscles lacking mitsugumin29 or JP-1 suggest that the formation of refined triad junctions is important for the Ca2+ preservation mechanism in skeletal muscle.
Proposed physiological roles of JP subtypes during triad formation
JP subtypes are composed of a COOH-terminal hydrophobic segment spanning the ER–SR membrane and the remaining cytoplasmic region interacting with the PM. Based on the biochemical properties, JP can produce junctional membrane structures between the ER and the PM in a heterologous expression system. The heart predominantly contains JP-2, and the deficiency of peripheral coupling was observed in embryonic cardiac myocytes from the JP-2 knockout mice (Takeshima et al., 2000). Despite the fact that both JP-1 and JP-2 were expressed in skeletal muscle (Fig. 1) and that JP-1 deficiency did not affect the subcellular localization of JP-2 (Fig. 2), the JP-1 knockout muscle exhibited both ultrastructural and functional abnormalities. The reduction in total JP protein level might cause the abnormalities, although JP-1 and JP-2 are functionally similar. Alternatively, JP-1 and JP-2 might be functionally different, and JP-1 is essential for the formation and/or maintenance of triad junctions. The latter theory is likely supported by several observations: (1) the formation of triad junctions was specifically reduced in muscles other than the jaw muscle from the JP-1 knockout mice; (2) the expression of JP-1 and the formation of triad junctions significantly increase during muscle maturation after birth; and (3) only a few triad-like structures are detected in cardiac myocytes predominantly expressing JP-2. However, future studies will be needed to examine whether JP-1 and JP-2 are functionally and qualitatively different during the formation of skeletal muscle triad junctions. Although the molecular basis of the triad formation is largely unknown, recent gene knockout experiments pointed out protein components involved in the construction (Fig. 8) .
Figure 8.

Protein components involved in formation of triad junction in skeletal muscle. JP subtypes specifically interact with the cell membrane through the cytoplasmic region and span the ER–SR membrane in the COOH-terminal end. Therefore, JP subtypes can contribute to the formation of junctional membrane complexes. JP-1 knockout mice and other observations likely suggest that JP-1 is essential for the construction and/or maintenance of the triad junction in skeletal muscle (see text). In the triad junction, DHPR and RyR function as the cell surface voltage sensor and SR Ca2+ release channel, respectively, and proposed direct coupling between them converts depolarization into intracellular Ca2+ signaling. Furthermore, our previous results indicated that mitsugumin29 (MG29) is important for structural refinement of the triad junction, such as T-tubular vending and correct localization of the T-tubule (Nishi et al., 1999).
Why did the JP-1 knockout mice show a suckling defect and fail to raise squeaky voices in response to pain? Both the suckling response and vocalization mainly depend on reflexes, and the EMG recording indicated that the mutant mice retain a normal neural circuit for suckling response (Fig. 3). Furthermore, the tissue distribution of JP-1 indicates that primarily damaged tissues are skeletal muscle systems in the mutant mice. Therefore, one possibility is that in the mutant mice, apparently normal stimuli from motor neurons cannot control mouth movements for suckling or vocalization due to a low efficiency of E–C coupling in jaw muscles. Because the mutant thigh muscles (containing weak triads but retaining normal level of diads) showed several functional abnormalities in tension measurements, the significant reduction of both triads and diads might produce fatal damage in E–C coupling of mutant jaw muscles. Apparently normal respiration and limb movements in the mutant mice suggest that diaphragmatic and limb muscle motor neurons evoke excess levels of stimuli to overcome the impaired efficiencies of E–C coupling. The muscle type–specific defect proposed in the JP-1 knockout mice could be explained by differences in stimulation amplitude among innervated motor neurons, and also in reduced efficiency of E–C coupling.
Materials and methods
Immunochemical analyses
The mouse JP-1 cDNA fragment encoding the divergent region (amino acid residues 425–633) was cloned into pGEX4T-2 (Amersham Pharmacia Biotech) for production of a bacterial glutathione S-transferase fusion protein. Rabbits were repeatedly immunized with the fusion protein purified using glutathione-Sepharose (Amersham Pharmacia Biotech) to yield antiserum. The antibody specific to JP-1 was affinity purified using protein A–Sepharose and N-hydroxysuccinimide–Sepharose (Amersham Pharmacia Biotech) conjugated with the fusion protein. The preparation of rat antibody to mouse JP-2 was described previously (Takeshima et al., 2000). Both antibodies were specific to corresponding JP subtypes and did not show cross-reactivity in the bacterial fusion proteins and mouse membrane preparations. For immunoblot analysis, total microsomal proteins were prepared from mouse tissues and analyzed as described previously (Takeshima et al., 1994). For immunohistochemical analysis, cryosections were prepared from skeletal muscles fixed in acetone. The sections were immunolabeled with the JP subtype–specific antibodies and Alexa 546– and 488–labeled secondary antibodies (Molecular Probes).
Generation of JP-1 knockout mice
A mouse genomic DNA library was screened with a JP-1 cDNA fragment as a hybridization probe to yield a phage clone carrying the 5′-terminal region of the JP-1 gene. Chromosomal mapping was carried out using fluorescence-labeled genomic fragments as probes as described previously (Shimuta et al., 1998). Our basic methods for the gene targeting were described previously (Takeshima et al., 1994). The targeting vector was constructed with the genomic DNA fragments, the neomycinresistance gene from pMC1 Neo polyA (Stratagene), the diphtheria toxin gene from pMC1-DT-A (Yagi et al., 1990), and pBluescript SK(−) (Stratagene) as shown in Fig. 2. The vector linearized with NotI was transfected into J1 embryonic stem (ES) cells (Li et al., 1992) and of the resulting G418-resistant ES cells, several clones were shown to carry the expected homologous mutation by Southern blot analysis using several restriction enzymes and hybridization probes. Chimeric mice produced with the positive ES clone numbered 397 were crossed with C57BL/6J mice and could transmit the mutant gene to the pups. The JP-1 knockout and wild-type mice obtained by crossing the heterozygous mutants were used for the analyses in this report. To determine the mouse genotypes, PCR was carried out using the synthetic primers P72-2 (27mer, CCAACATCCTACTTATGGGGTTCTGGC), P72-3 (26mer, TCCCCATTTTCGAGGGAGACCACTTGG), and PneoS (27mer, CGCTATCAGGACATAGCGTTGGCTACC), as shown in Fig. 2.
EMG recording during suckling response
The suckling behavior consists of nipple attachment, rhythmic suckling, and stretch response during milk letdown. Low-frequency and rhythmic movements of the jaw and tongue are observed in rhythmic suckling, and similar responses can be produced by mechanical stimulation to the lips (Westneat and Hall, 1992). EMG was recorded from a jaw muscle (musculus masseter), essentially as described previously (Kutsuwada et al., 1996). The suckling-like responses were induced by gentle stimulation of the area around the lips or tongue using a soft polyethylene tube, and the stimulation site and induced jaw movements were observed under a binocular microscope. The EMG signals evoked were picked up with bipolar electrodes (0.13-mm-diam enamel-coated copper wire) inserted into the jaw muscle, and were monitored using the electric amplifier (AB6212G; Nihon Koden) and polygraph system (PM-6000; Nihon Koden).
Morphological analyses
Skeletal muscle preparations were treated with a prefixative solution (3% paraformaldehyde, 2.5% glutaraldehyde, and 0.1 M sodium cacodylate, pH 7.4) and postfixed with a buffer containing 1% OsO4 and 0.1 M sodium cacodilate, pH 7.4. The fixed muscles were washed, dehydrated with alcohol and acetone, and embedded in Epon. For histological analysis, sections were prepared, stained with Toluidine blue, and observed under an optic microscope. For ultrastructural examination, thin sections were prepared, stained with uranyl acetate and lead citrate, and observed under an electron microscope (JEM-200CX; JEOL).
Muscle contraction measurements
Skeletal muscle preparations (musculus vastus lateralis) were dissected from hindlimbs of newborn mice and were mounted on a force transducer in a chamber containing modified Krebs-Ringer solution (121.9 mM NaCl, 4.7 mM KCl, 2.5 mM CaCl2, 1.2 mM MgCl2, 1.2 mM NaH2PO4, 15.5 mM NaHCO3, and 11.5 mM glucose) constantly bubbled with 95% O2 and 5% CO2. To induce contraction, field stimulation (10 ms duration) with supramaximal voltage was given at various frequencies for 10 s, and developed force was recorded online using Chart (v. 3.5; ADInstruments). The effects of extracellular Ca2+ concentrations were examined on twitches evoked at 0.33 Hz. After the experiments, the muscle preparations were fixed with 3% paraformaldehide and were subjected to size measurements. Cross sectional surface area was determined using an Mac scope (v. 2.3; Mitani Sangyo Co. Ltd.) based on the observations under a binocular microscope.
Acknowledgments
We thank Miyuki Kameyama and Hiromi Sawamizu for maintaining the mutant mice.
This work was supported in part by grants from the Ministry of Education, Science, Sports and Culture of Japan, the Ministry of Health and Welfare of Japan, the Takeda Science Foundation, the Mochida Memorial Foundation, the Japan Heart Foundation, the Japanese Foundation of Metabolism and Disease, the Kimura Memorial Heart Foundation, the Kaibara Morikazu Medical Science Promotion Foundation, the Toray Science Foundation, and the Inamori Foundation.
Footnotes
Abbreviations used in this paper: DHPR, dihydropyridine receptor; E–C, excitation–contraction; EMG, electromyogram; ES, embryonic stem; JP, junctophilin; PM, plasma membrane; RyR, ryanodine receptor; SR, sarcoplasmic reticulum; T-tubule, transverse-tubule.
References
- Beam, K.G., and M.C. Knudson. 1988. Calcium currents in embryonic and neonatal mammalian skeletal muscle. J. Gen. Physiol. 91:781–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge, M.J. 1998. Neuronal calcium signaling. Neuron. 21:13–26. [DOI] [PubMed] [Google Scholar]
- Fan, H., R.N. Brandt, and H.A. Caswell. 1995. Disulfide bonds, N-glycosylation and transmembrane topology of skeletal muscle triadin. Biochemistry. 34:14902–14908. [DOI] [PubMed] [Google Scholar]
- Flucher, B.E. 1992. Structural analysis of muscle development: transverse tubules, sarcoplasmic reticulum, and the triad. Dev. Biol. 154:245–260. [DOI] [PubMed] [Google Scholar]
- Ikemoto, T., S. Komazaki, H. Takeshima, M. Nishi, T. Noda, M. Iino, and M. Endo. 1997. Functional and morphological features of skeletal muscle from mutant mice lacking both type 1 and type 3 ryanodine receptors. J. Physiol. 501:305–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, L.R., L. Zhang, K. Sanborn, O.A. Jorgensen, and J. Kelley. 1995. Purification, primary structure, and immunological characterization of the 26-kDa calsequestrin binding protein (junctin) from cardiac junctional sarcoplasmic reticulum. J. Biol. Chem. 270:30787–30796. [DOI] [PubMed] [Google Scholar]
- Kutsuwada, T., K. Sakimura, T. Manabe, C. Takayama, N. Katakura, E. Kushiya, R. Natsume, M. Watanabe, Y. Inoue, and T. Yagi, et al. 1996. Impairment of suckling response, trigeminal neuronal pattern formation, and hippocampal LTD in NMDA receptor ε2 subunit mutant mice. Neuron. 16:333–344. [DOI] [PubMed] [Google Scholar]
- Li, E., H.T. Bestor, and R. Jaenisch. 1992. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 69:915–926. [DOI] [PubMed] [Google Scholar]
- Nakamura, Y., and N. Katakura. 1995. Generation of masticatory rhythm in the brainstem. Neurosci. Res. 23:1–19. [PubMed] [Google Scholar]
- Nishi, M., S. Komazaki, M. Iino, K. Kangawa, and H. Takeshima. 1998. Mitsugumin23, a novel transmembrane protein on endoplasmic reticulum and nuclear membranes. FEBS Lett. 432:191–196. [DOI] [PubMed] [Google Scholar]
- Nishi, M., S. Komazaki, N. Kurebayashi, Y. Ogawa, T. Noda, M. Iino, and H. Takeshima. 1999. Abnormal features in skeletal muscle from mice lacking mitsugumin29. J. Cell Biol. 147:1473–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishi, M., A. Mizushima, K. Nakagawara, and H. Takeshima. 2000. Characterization of human junctophilin subtype genes. Biochem. Biophys. Res. Commun. 273:920–927. [DOI] [PubMed] [Google Scholar]
- Pozzan, T., R. Rizzuto, P. Volpe, and J. Meldolesi. 1994. Molecular and cellular physiology of intracellular calcium stores. Physiol. Rev. 74:595–636. [DOI] [PubMed] [Google Scholar]
- Schneider, M.F. 1994. Control of calcium release in functioning skeletal muscle fibers. Annu. Rev. Physiol. 56:463–484. [DOI] [PubMed] [Google Scholar]
- Shimuta, M., S. Komazaki, M. Nishi, M. Iino, K. Nakagawara, and H. Takeshima. 1998. Structure and expression of mitsugumin29 gene. FEBS Lett. 431:263–267. [DOI] [PubMed] [Google Scholar]
- Takekura, H., H. Takeshima, S. Nishimura, M. Takahashi, T. Tanabe, V. Flockerzi, F. Hofmann, and C. Franzini-Armstrong. 1995. Co-expression in CHO cells of two muscle proteins involved in excitation-contraction coupling. J. Muscle Res. Cell Motil. 16:465–480. [DOI] [PubMed] [Google Scholar]
- Takeshima, H., M. Iino, H. Takekura, M. Nishi, J. Kuno, O. Minowa, H. Takano, and T. Noda. 1994. Excitation-contraction uncoupling and muscular degeneration in mice lacking functional skeletal muscle ryanodine-receptor gene. Nature. 369:556–559. [DOI] [PubMed] [Google Scholar]
- Takeshima, H., M. Shimuta, S. Komazaki, S. Ohmi, M. Nishi, M. Iino, A. Miyata, and K. Kangawa. 1998. Mitsugumin29, a novel synaptophysin family member from the triad junction in skeletal muscle. Biochem. J. 331:317–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeshima, H., S. Komazaki, M. Nishi, M. Iino, and K. Kangawa. 2000. Junctophilins: a novel family of junctional membrane complex proteins. Mol. Cell. 6:11–22. [DOI] [PubMed] [Google Scholar]
- Tanabe, T., G.K. Beam, A.J. Powell, and S. Numa. 1988. Restoration of excitation-contraction coupling and slow calcium current in dysgenic muscle by dihydropyridine receptor complementary DNA. Nature. 336:134–139. [DOI] [PubMed] [Google Scholar]
- Westneat, M.W., and G.W. Hall. 1992. Ontogeny of feeding motor patterns in infant rats: an electromyographic analysis of suckling and chewing. Behav. Neurosci. 106:539–554. [DOI] [PubMed] [Google Scholar]
- Yagi, T., Y. Ikawa, K. Yoshida, Y. Shigetani, N. Takeda, I. Mabuchi, T. Yamamoto, and S. Aizawa. 1990. Homologous recombination at c-fyn locus of mouse embryonic stem cells with use of diphtheria toxin A-fragment gene in negative selection. Proc. Natl. Acad. Sci. USA. 87:9918–9922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi, T., S. Aizawa, T. Tokunaga, Y. Shigetani, N. Takeda, and Y. Ikawa. 1993. A role for Fyn tyrosine kinase in the suckling behaviour of neonatal mice. Nature. 366:742–745. [DOI] [PubMed] [Google Scholar]