

Abstract
The mitotic checkpoint prevents cells with unaligned chromosomes from prematurely exiting mitosis by inhibiting the anaphase-promoting complex/cyclosome (APC/C) from targeting key proteins for ubiquitin-mediated proteolysis. We have examined the mechanism by which the checkpoint inhibits the APC/C by purifying an APC/C inhibitory factor from HeLa cells. We call this factor the mitotic checkpoint complex (MCC) as it consists of hBUBR1, hBUB3, CDC20, and MAD2 checkpoint proteins in near equal stoichiometry. MCC inhibitory activity is 3,000-fold greater than that of recombinant MAD2, which has also been shown to inhibit APC/C in vitro. Surprisingly, MCC is not generated from kinetochores, as it is also present and active in interphase cells. However, only APC/C isolated from mitotic cells was sensitive to inhibition by MCC. We found that the majority of the APC/C in mitotic lysates is associated with the MCC, and this likely contributes to the lag in ubiquitin ligase activity. Importantly, chromosomes can suppress the reactivation of APC/C. Chromosomes did not affect the inhibitory activity of MCC or the stimulatory activity of CDC20. We propose that the preformed interphase pool of MCC allows for rapid inhibition of APC/C when cells enter mitosis. Unattached kinetochores then target the APC/C for sustained inhibition by the MCC.
Keywords: kinetochore; hBUBR1; anaphase promoting complex; MAD2; mitotic checkpoint
Introduction
The mitotic checkpoint ensures chromosomes are accurately segregated by preventing cells with unaligned chromosomes from exiting mitosis (Rieder and Salmon, 1998; Skibbens and Hieter, 1998; Zacharie and Nasmyth, 1999). Minimally, this pathway consists of three components: a sensor that detects the presence of unaligned chromosomes, an effector or transducer that relays or amplifies the signal generated by the sensor, and an inhibitor that is capable of stopping the cell cycle progression until after all chromosomes are properly aligned. This scheme is derived from early work that showed that a single unattached kinetochore can generate sufficient amounts of an inhibitory signal to prevent a cell from exiting from mitosis (Rieder et al., 1995). Molecular mechanisms that explain the different segments of this checkpoint pathway remain to be established, but recent work has identified many components of the system. The mitotic checkpoint pathway is specified by a group of evolutionarily conserved genes that include MAD1, MAD2, MAD3, BUB1, and BUB3 (Hoyt et al., 1991; Li and Murray, 1991; Roberts et al., 1994; Hardwick and Murray, 1995; Li and Benezra, 1996). These proteins preferentially associate with kinetochores of unaligned chromosomes where they are hypothesized to monitor and generate the inhibitory signal that delays the metaphase to anaphase transition (Chen et al., 1996, 1998; Taylor and McKeon, 1997; Taylor et al., 1998; Waters et al., 1998; Chan et al., 1999; Shah and Cleveland, 2000). This delay is achieved by inhibiting the anaphase-promoting complex/cyclosome (APC/C),* a multisubunit E3 ubiquitin ligase that targets key proteins whose destruction is necessary for sister chromatid separation and exit from mitosis (King et al., 1995, 1996; Sudakin et al., 1995; Hershko and Ciechanover, 1998).
Our current understanding of the mechanism by which the APC/C is inhibited by unaligned chromosomes has come primarily from studies of the MAD2 checkpoint protein. Consistent with the behavior of MAD2 mutants in yeast (Hoyt et al., 1991; Li and Murray, 1991; He et al., 1997), disruption of MAD2 function in mammalian cells and Xenopus extracts prevented the establishment of a checkpoint arrest in response to spindle damage (Chen et al., 1998; Gorbsky et al., 1998). The ability of MAD2 to selectively bind unattached kinetochores suggests that one of its functions is to monitor kinetochore–microtubule interactions. In mammalian cells, the evidence suggests that MAD2 may directly monitor the microtubule occupancy at kinetochores such that kinetochores fully saturated with microtubules exhibit no detectable MAD2, whereas MAD2 is prominently localized to unattached kinetochores (Waters et al., 1998). How this occurs remains unknown, but MAD2 is sensitive to microtubule interactions mediated by the kinetochore motor CENP-E. Depletion of CENP-E from kinetochores in mammalian cells disrupts chromosome alignment, and cells become arrested in mitosis with high levels of MAD2 at unattached kinetochores (Yao et al., 2000). In Xenopus extracts, depletion of CENP-E prevents MAD2 localization to kinetochores, and thus the extracts fail to arrest in mitosis when spindle assembly is inhibited (Abrieu et al., 2000). Despite the differences between how the checkpoint in these two experimental systems responds to loss of CENP-E function, it is clear that CENP-E activity at kinetochores is linked to MAD2 binding to kinetochores.
In addition to its role at kinetochores, MAD2 is thought to have a downstream role in directly blocking APC/C activity. MAD2 was found to bind to the APC/C in cytosol that was obtained from mitotically arrested HeLa cells (Li et al., 1997). Addition of excess MAD2 to mitotic egg extracts blocked exit from mitosis even in the absence of kinetochores (Chen et al., 1998). Furthermore, MAD2 was found to directly inhibit the ubiquitin ligase activity of purified APC/C in vitro (Li et al., 1997; Fang et al., 1998). Interestingly, bacterially expressed MAD2 was found to exist as either monomers or tetramers but only the tetrameric form was found to inhibit the APC/C (Fang et al., 1998). The possibility that there are inactive and active states of MAD2 provided a mechanistic explanation for an existing model in which unattached kinetochores are envisioned to convert MAD2 into a form that can inhibit APC/C activity (Gorbsky et al., 1998; Howell et al., 2000; Shah and Cleveland, 2000). One part of this model, whereby MAD2 is postulated to cycle on and off kinetochores, has been confirmed by FRAP experiments that measured the half-life of kinetochore-bound MAD2 (Howell et al., 2000). These studies estimated that the half-life of MAD2 at unattached kinetochores is ∼25 s. This rapid turnover rate was predicted to generate sufficient amounts of MAD2 to sustain a prolonged inhibition of the APC/C.
Although the collective studies have shed considerable light on MAD2, whether inhibition of APC/C is specified solely by MAD2 in vivo is unknown. In yeast and mammalian cells, MAD2 has been shown to interact with the APC/C through CDC20, a protein that specifies substrate selectivity by the APC/C (Dawson et al., 1995; Visintin et al., 1997; Fang et al., 1998; Hwang et al., 1998; Kallio et al., 1998; Kim et al., 1998). Furthermore, complexes consisting of MAD3, BUB3, CDC20, and MAD2 were identified in budding (Hardwick et al., 2000) and fission yeasts (Hardwick, K.G., personal communications). In mitotic HeLa cells, APC/C is associated with the hBUBR1 checkpoint kinase (Chan et al., 1999). This observation suggests that inhibition of APC/C in vivo might be achieved through more complex schemes. To obtain some insights into how APC/C is inhibited by the checkpoint in vivo, we set out to identify factors from mitotically arrested HeLa cells that inhibited APC/C. This search yielded a single stable complex named the mitotic checkpoint complex (MCC), consisting of the proteins hBUBR1, hBUB3, CDC20, and MAD2. We report here on the identification and characterization of MCC, and present evidence that this is a physiologically relevant inhibitor of the APC/C.
Results
Identification of an APC/C inhibitor that contains hBUBR1 kinase
We undertook a biochemical approach to identify factors in mitotically arrested cells that were responsible for inhibiting the APC/C in vivo. HeLa cells were chosen because they exhibit a robust checkpoint arrest in response to spindle or kinetochore defects. Lysates prepared from mitotically blocked cells were chromatographed through a gel filtration column and fractions were tested for their ability to inhibit APC/C in a standard in vitro assay (Sudakin et al., 1995). Our assay relied on APC/C that was partially purified from mitotic HeLa cells, and was based on a protocol used to purify the cyclosome from clam oocytes (Sudakin et al., 1995). Iodinated substrate consisting of protein A fused to the destruction box of cyclin B1 allowed quantitation of the APC/C activity (Glotzer et al., 1991). We found that fractions eluting from the 400-kD region of the gel filtration column strongly inhibited APC/C activity (Fig. 1 A, lane 4) whereas no inhibitory activity was detectable from other fractions from the column (unpublished data). The inhibitory factor eluted at a position that was very close to that described previously for the mitotic checkpoint kinase hBUBR1 (Chan et al., 1999). We attempted to separate the inhibitor away from hBUBR1 by using additional column chromatography (Fig. 1 B). At each of the three successive chromatographic steps, the peaks of APC/C inhibitory activity and hBUBR1 coincided. The results obtained from the final Superose 6 column show coelution of the APC/C inhibitor (MCC) and hBUBR1 (Fig. 1 C).
Figure 1.

Identification of an APC/C inhibitor that copurifies with hBUBR1 kinase. (A) Identification of APC/C inhibitory activity in mitotic lysates. Mitotic lysate (S100) was fractionated through a Superose 6 gel filtration column by FPLC and proteins eluting at approximately the 300–400-kD range were found to inhibit APC/C activity. To maintain some physiological relevance to the APC/C inhibition assays, equal cell equivalents of APC/C and the various column fractions were used. Purified mitotic APC/C was incubated for 30 min with either buffer B alone (lane 2) or fractions from the 300–400-kD range of the Superose 6 column (lane 4) and then assayed for ubiquitin ligase activity. The same column fractions were assayed without addition of APC/C to monitor contaminating APC/C activity (lane 3). A reaction containing only substrate served as a negative control (lane 1). Asterisk denotes an iodinated contaminant bacterial protein that is not a substrate for APC/C and was excluded from the quantitation. (B) Flow chart of the purification of the APC/C inhibitory complex from HeLa cells. (C) Inhibitor of APC/C cofractionates with the hBUBR1 kinase complex. The elution profiles of hBUBR1 (inset) and APC/C inhibitory activity from the final Superose 6 column is shown. Arrows point to thyroglobulin (670 kD), γ-globulin (158 kD), and ovalbumin (44 kD), which served as migration standards.
To test whether hBUBR1 is an essential component of MCC, fractions containing high levels of MCC activity were immunodepleted of hBUBR1 and the remaining supernatant was tested for APC/C inhibitory activity. Incubation of MCC with nonimmune IgG did not deplete hBUBR1 or reduce its inhibitory activity relative to untreated MCC (Fig. 2 A, compare lanes 2 and 3). In contrast, MCC fractions that were immunodepleted of hBUBR1 did not block APC/C activity to the same extent as mock-depleted or untreated MCC (Fig. 2 A, compare lane 4 with lanes 2 and 3). Because hBUBR1 was not completely depleted from this MCC fraction (∼25% of the input hBUBR1 was still detected by Western analysis, Fig. 2 A, upper panel), some residual APC/C inhibitory activity was detected when compared with APC/C that was incubated with buffer alone (Fig. 2 A, compare lanes 1 and 4, Fig. 2 B). These results show that hBUBR1 or an associated factor is essential for MCC activity.
Figure 2.
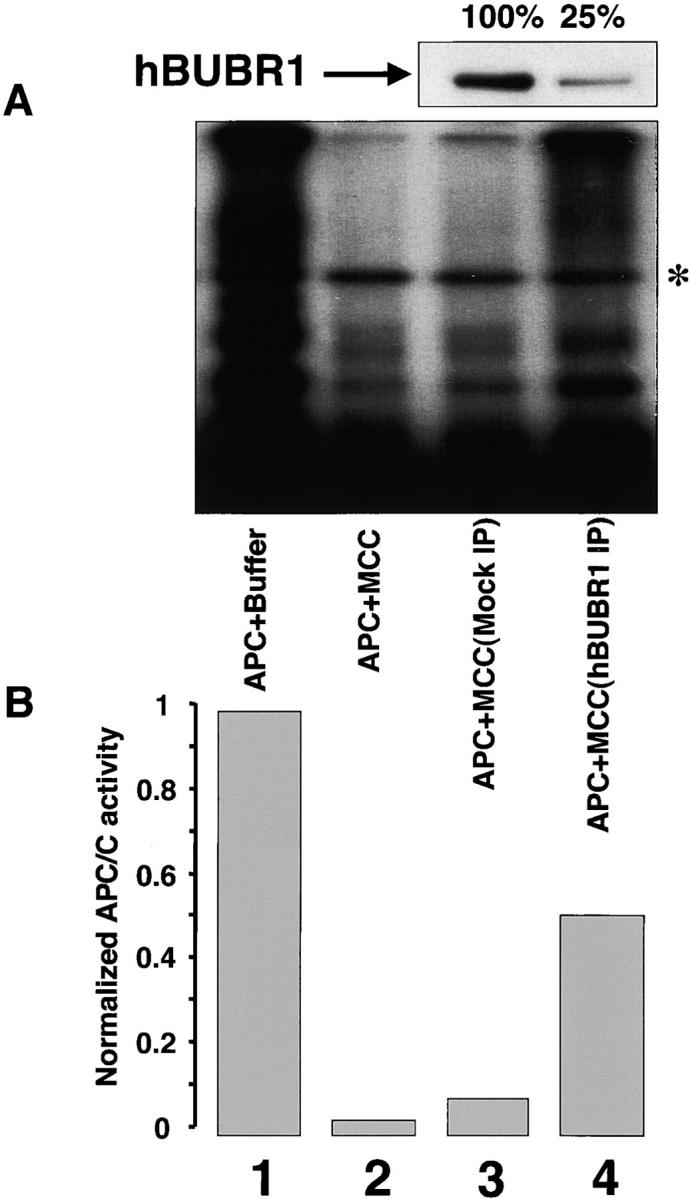
MCC inhibition of the APC/C requires hBUBR1. Purified mitotic APC/C was incubated in the presence of buffer (lane 1), MCC (lane 2), MCC that was mock depleted with nonimmune IgG (lane 3) or immunodepleted with hBUBR1 antibodies (lane 4), and then assayed for ubiquitin ligase activity. (A) Ubiquitination assay. Mock- and hBUBR1-immunodepleted supernatants were probed for hBUBR1 (top panel). Asterisk denotes the contaminant iodinated protein. (B) Quantification of the ubiquitination assay on phosphorimager.
MCC consists of multiple checkpoint proteins
To further characterize the composition of MCC, we probed the complex for MAD2. Fractionation of crude mitotic lysates through a Superose 6 column showed that the vast majority of MAD2 was found as monomers. However, some MAD2 (<5% of total) comigrated with the MCC and the APC/C (Fig. 3 A). MAD2 was determined to be part of MCC, as it was detected in hBUBR1 immunoprecipitates obtained from fractions containing MCC (Fig. 3 B). Furthermore, MCC was found to contain CDC20, an activator of the APC/C that has been shown to target MAD2 to the APC/C (Fang et al., 1998; Wassmann and Benezra, 1998). Consistent with the previous data that showed that hBUB3 was associated with hBUBR1 (Chan et al., 1999, Taylor et al., 1998), hBUB3 was also present in the MCC (Fig. 3 B). The presence of MAD2 in MCC was verified in immunoprecipitation experiments using anti-MAD2 antibodies. The MAD2 immunoprecipitates contained all four MCC subunits (Fig. 3 B). Importantly, we did not detect hBUB1 or MAD1 in the MCC (unpublished data) despite the fact that they have been shown to interact with some of the MCC subunits (hBUB1 with hBUB3, and MAD1with MAD2). Our recently published data indicate that MAD1 binds MAD2 but not CDC20 (Campbell et al., 2001). This suggests that hBUB1 and MAD1 form complexes distinct from the MCC.
Figure 3.

APC/C inhibitor is a complex of mitotic checkpoint proteins. (A) MAD2 comigrates with the hBUBR1 kinase complex. Mitotic HeLa extracts were separated through a Superose 6 column and the column fractions were probed for MAD2, hBUBR1, and the APC7. Fractions exhibiting APC/C inhibitory activity are denoted as MCC. (B) MCC consists of hBUBR1, hBUB3, MAD2, and CDC20. Fractions eluting from the 300–400-kD region of the Superose 6 column in A that exhibited APC/C inhibitory activity were pooled and immunoprecipitated with nonimmune IgG, anti-hBUBR1, and anti-MAD2 antibodies, washed, and probed with for hBUBR1, hBUB3, MAD2, and CDC20. (C) MCC can exist independently of APC/C. Fractions 19–28 from the Superose 6 column shown in A were combined and rechromatographed through a MonoQ anion exchange column by FPLC. Fractions were assayed for APC/C activity (solid line) and probed for hBUBR1, hBUB3, CDC20, MAD2, and CDC27 by Western blots. (D) Separation of active and inactive APC/C. Fractions 40–42 from the MonoQ column shown in C that exhibited APC/C activity and MCC were immunodepleted successively with anti-hBUBR1 and anti-CDC20 antibodies. The ubiquitin ligase activity was tested in the supernatants and immunoprecipitates. (Lane 1) Input APC/C activity; (lanes 2 and 3, respectively) APC/C activity in the supernatants after depletion of hBUBR1 and CDC20; (lanes 4 and 5, respectively) APC/C activity associated with corresponding hBUBR1 and CDC20 immunoprecipitates; and (lane 6) immunoprecipitate performed with nonimmune IgG. hBUBR1, CDC20, and nonimmune IgG immunoprecipitates were probed for CDC27 to estimate relative amount of APC/C (bottom panel). Asterisk denotes the contaminant iodinated protein.
The MCC and APC/C migrated closely to each other through the gel filtration column so it was critical to obtain additional evidence that the interaction between hBUBR1–hBUB3 and CDC20–MAD2 did not occur through their mutual association with the APC/C. MCC and APC/C fractions were pooled from the Superose 6 column and rechromatographed through a MonoQ column. Western blots of the eluted fractions showed that the four MCC subunits could be separated from the APC/C as determined by lack of CDC27 immunoreactivity and ubiquitin ligase activity (Fig. 3 C, fractions 27 and 28). Only fractions containing the four MCC subunits exhibited APC inhibitory activity (unpublished data). We found that some of the MCC subunits did not comigrate with each other, most likely because they had dissociated. Paradoxically, fractions containing active APC/C were found to also contain MCC (Fig. 3 C, fractions 40–42). We reasoned that the ability of APC/C to remain active in the presence of MCC could be explained by the existence of both active and inactive populations of APC/C in these fractions. To test this hypothesis, we immunoprecipitated the MCC from these fractions with hBUBR1 antibodies, and found that a small fraction of the input APC/C activity was detected in the hBUBR1 immunoprecipitates as nearly all of the input APC/C activity remained in the supernatant (Fig. 3 D, lanes 2 and 4). The majority of the APC/C activity in this supernatant was significantly depleted after immunoprecipitation with CDC20 antibodies (Fig. 3 D, lane 3), as it was mostly recovered in the CDC20 immunoprecipitates (Fig. 3 D, lane 5). When the APC/C activities associated with the hBUBR1 and CDC20 immunoprecipitates were normalized to the amount of CDC27 that coimmunoprecipitated with each antibody, we estimate that the APC/C activity that was associated with hBUBR1 (MCC) was approximately sixfold weaker than the APC/C that was not associated with hBUBR1. This is likely an underestimate, as the APC/C activity detected in the hBUBR1 beads may have been due to dissociation of the MCC. Regardless, these data provide further evidence that MCC is likely a potent inhibitor of the APC/C in vivo.
Next, we sought evidence to directly support our finding that hBUBR1 is part of the inhibitory complex that blocks APC/C activity. Initial attempts to purify MCC with the hBUBR1 antibodies were unsuccessful because the harsh conditions required to elute hBUBR1 from the antibody-linked beads destroyed the complex. An alternative approach was to express a glutathione S-transferase (GST)-tagged hBUBR1 in human embryonic kidney cell line HEK293T and then use glutathione beads to affinity purify it along with the appropriate subunits from extracts of transfected cells (Fig. 4, A and B) . Western blot analysis confirmed that the affinity-purified GST–hBUBR1 formed the MCC, as it contained endogenous hBUBR1, hBUB3, CDC20, and MAD2. As shown previously, only a small fraction of the total pool of MAD2 was associated with GST–hBUBR1:MCC (Fig. 4 A, lane 3). All of these associations are specific to hBUBR1, as these proteins did not associate with GST alone (Fig. 4 A, lane 1). Purified GST–hBUBR1:MCC was found to inhibit ubiquitin ligase activity of the APC/C (Fig. 4 B, lane 5) as was seen with conventionally purified MCC (Fig. 4 B, lane 4). GST purified from transfected cells did not exhibit significant inhibitory activity (Fig. 4 B, lane 3).
Figure 4.


Affinity-purified GST–hBUBR1:MCC inhibits APC/C. (A) GST–hBUBR1 is associated with the MCC. A GST–hBUBR1 construct was transfected into cells and the protein was affinity purified using glutathione beads. Affinity-purified GST–hBUBR1 (lane 3) and the remaining supernatant (lane 4) were probed for hBUBR1, hBUB3, CDC20, and MAD2. In parallel, a GST construct was transfected and the affinity-purified GST (lane 1) and the remaining supernatant (lane 2) was probed with the same antibodies and a GST antibody to confirm expression of GST in transfected cells. The black arrowhead points to endogenous hBUBR1 and the open arrowheads point to GST–hBUBR1 and its degradation product (as confirmed by GST Western blot, unpublished data). (B) Affinity-purified GST–hBUBR1:MCC inhibits APC/C. Mitotic APC/C (lane 2) was incubated with GST (lane 3), conventionally purified MCC (lane 4), and GST-MCC (lane 5) and assayed for ubiquitin ligase activity. A control reaction lacking APC/C (lane 1) provided the level of input substrate that was used for calculating APC/C activity in the various reactions.
MCC is a more potent inhibitor than recombinant MAD2
Recombinant MAD2 purified from bacteria can exist as monomers and tetramers, but only tetrameric MAD2 was found to inhibit APC/C in vitro (Fang et al., 1998). Gel filtration of mitotic HeLa extracts cells showed that >90% of MAD2 is not associated with MCC or APC/C (Fig. 3 A) and these fractions did not inhibit APC/C (unpublished data). To understand why recombinant MAD2 can inhibit APC/C whereas MAD2 in HeLa cells cannot, we compared the inhibitory activities between MCC and recombinant MAD2. Optimally, this comparison should be made based on protein concentration. However, we could not estimate the amount of MCC as our preparations were not purified to homogeneity. Therefore, we standardized the inhibitory activities using MAD2 levels. First, we estimated the amount of MAD2 that was present in MCC by quantitative immunoblots using known amounts of recombinant MAD2 (Fig. 5 A). We then compared the inhibitory activities obtained with different amounts of MCC or recombinant tetrameric MAD2 and determined the amount of each inhibitor that gave comparable activities (Fig. 5 B). Based on this analysis, we estimated that the inhibitory activity of the MCC is >3,000-fold greater than recombinant MAD2 oligomers.
Figure 5.

Characterization of the MCC. (A) Estimation of MAD2 content in MCC. Quantitative immunoblot of purified MCC with known amounts of recombinant MAD2. Recombinant (His) 6-tagged MAD2 migrated slower than native MAD2. (B) Comparison of APC/C inhibitory activity between the recombinant oligomeric MAD2 and purified MCC. Increasing amounts of oligomeric MAD2 and purified MCC were added to the APC/C assay to compare inhibitory activities. Phosphorimage of the APC/C reactions (upper panel) was quantified to determine the amount of recombinant MAD2 and MCC required to achieve equivalent levels of inhibition (lower panel). (C) MCC subunits exist in near equal stoichiometry. HeLa cells were labeled in 35S-Trans label for 6 h and mitotic and interphase cells were separated and then incubated with nonimmune IgG or hBUBR1 antibodies. Phosphorimage of the immunoprecipitate obtained from mitotic cells shows the radiolabeled hBUBR1, hBUB3, MAD2, and CDC20. The counts from each subunit were normalized to their cysteine and methionine contents (without initiating Met) to estimate their stoichiometry.
Next, we estimated the stoichiometry of the MCC subunits to see if there was any evidence for the existence of MAD2 oligomers. MCC was immunoprecipitated from lysates prepared from 35S-Trans–labeled HeLa cells and the radioactivity in each subunit was counted (Fig. 5 C). When the counts were normalized to the methionine and cysteine contents in hBUBR1, hBUB3, CDC20, and MAD2 (40, 13, 14, and 4, respectively, without initiating Met), we obtained a ratio of 1.4 ± 0.3:1 ± 0.3:1 ± 0.2:1.2 ± 0.2. A complex with such a ratio would have a molecular mass of ∼300 kD. Therefore, it is unlikely that MAD2 oligomers exist in the MCC.
It is noteworthy that our radiolabeled immunoprecipitate contained additional prominent bands. As hBUBR1 is known to form a separate complex with CENP-E (which elutes in the void of Superose 6 columns), these complexes were likely to be present in the immunoprecipitates. These additional hBUBR1 complexes explain the higher stoichiometry of hBUBR1 in our calculations. If this is taken into account, we estimate that the MCC subunits exist in near equal stoichiometry. Regardless, the results obtained from the radiolabeling experiment do not support the presence of oligomeric forms of MAD2 in the MCC. Our data show that hBUBR1 forms additional uncharacterized complexes in HeLa cells (Fig. 3 C, fractions 32–35); the complexes possessed no inhibitory activity (unpublished data). The identity and functions of these complexes remain to be elucidated. We suggest that the existence of the additional complexes contributed to an increased amount of hBUBR1 in the calculated MCC ratio presented above. Another possibility is that the MCC contains yet unidentified subunits. The complex migrates at 400 kD, whereas its molecular mass estimated by our radiolabeling analysis gives a molecular mass of ∼300 kD.
MCC is expressed throughout the cell cycle but only targets mitotic APC/C
We showed previously that the 400-kD hBUBR1 complex was present in both interphase and mitotic cells (Chan et al., 1999). Given that hBUBR1 was found to associate with the APC/C only in mitosis (Chan et al., 1999), we expected that only the mitotic form of the MCC would inhibit the APC/C. Surprisingly, hBUBR1 complex isolated from interphase HeLa cells (synchronized in the G1/S boundary) inhibited APC/C activity and contained the same subunits found in mitotic MCC (unpublished data). Furthermore, both interphase and mitotic forms of MCC exhibited similar activities when equivalent amounts of hBUBR1 were tested (Fig. 6 A). Although this MCC preparation was obtained from cells arrested at the G1/S boundary, analysis of synchronized cells obtained by centrifugal elutriation showed that MCC is present in all stages of the cell cycle (unpublished data).
Figure 6.

Regulation of the MCC. (A) MCC is present and active in interphase HeLa cells. hBUBR1 complex was purified from HeLa cells that were arrested at the G1/S boundary by a double thymidine block. The hBUBR1 and APC/C inhibitor profiles (tested by APC/C partially purified on gel filtration column) from the final Superose 6 column are shown. (B) Only APC/C from mitotic cells is sensitive to inhibition by MCC. APC/C purified from mitotic (M) and interphase (I) cells were incubated in the absence and presence of the MCC that was purified from interphase HeLa cells. Relative APC/C activity denotes the ratio of the ubiquitin ligase activity in reactions performed in the presence and absence of MCC. (C) hBUBR1 binds preferentially APC/C that contains hyperphosphorylated CDC27. Mitotic HeLa lysates were fractionated through a Superose 6 column as described and the portion containing intact APC was immunoprecipitated with hBUBR1 antibodies. The immunoprecipitate (IP) and remaining supernatant (Sup) were separated on SDS-PAGE and probed with hBUBR1, CDC27, CDC16, and APC7 antibodies.
We reasoned that if MCC activity is not subject to cell cycle regulation, perhaps the sensitivity of the APC/C to MCC inhibition differed between interphase and mitosis. This possibility was confirmed as only APC/C that was obtained from mitotic cells was efficiently inhibited by MCC, whereas APC/C that was purified from interphase cells was only modestly inhibited by MCC (Fig. 6 B). This modest level of inhibition is likely contributed by the APC/C present in the small fraction of mitotic cells in the asynchronous cell population. As several subunits of the APC/C are hyperphosphorylated during mitosis (King et al., 1995; Lahav-Baratz et al., 1995; Descombes and Nigg, 1998; Kotani et al., 1999), we examined whether these modifications might specify interactions between MCC and the mitotic APC/C. Mitotic lysates were fractionated through a Superose 6 column and the portion containing intact APC was immunoprecipitated with hBUBR1 antibodies. As shown previously, hBUBR1 was associated with the APC/C as determined by the presence of the APC subunits CDC27, CDC16, and APC7 (Fig. 6 C). However, hBUBR1 was found to associate preferentially with APC that contained hyperphosphorylated CDC27 despite the presence of APC that contained unphosphorylated CDC27 (Fig. 6 C). Thus, mitotic phosphorylation of APC/C might specify its inhibition by the MCC.
Sorting out the interactions between chromosomes, APC/C, and MCC
The ability of MCC from interphase cells to block APC/C shows that its activity does not require the presence of unattached kinetochores, as mature kinetochores are not found until mitosis. To clarify the interactions between MCC, APC/C, and kinetochores, we set out to reconstitute the checkpoint inhibition of the APC/C in lysates prepared from HeLa cells. Kinetic studies showed that the APC/C activity in crude mitotic lysates exhibited a reproducible lag of ∼15 min, compared with the APC/C activity in lysates prepared from asynchronous cells that were >90% interphase (Fig. 7 A, compare ⋄ and ○). The initial lag seen in the mitotic lysates may have been due to checkpoint inhibition, as lysates were prepared from mitotically arrested cells whose APC/C is associated with MCC. In the absence of chromosomes, the checkpoint inhibition cannot be sustained and the APC/C eventually recovers its activity. This was confirmed when addition of purified chromosomes to the mitotic lysate suppressed the reactivation of ACP/C (Fig. 7 A, ♦). Chromosomes do not permanently inactivate the APC/C, as APC/C was reactivated when chromosomes were removed from during the incubation (unpublished data). Chromosomes were only effective in mitotic lysates as they modestly reduced APC/C activity in asynchronous lysates (Fig. 7 A, •). This modest reduction can be attributed to the small fraction of mitotic cells that were present in the asynchronous population.
Figure 7.
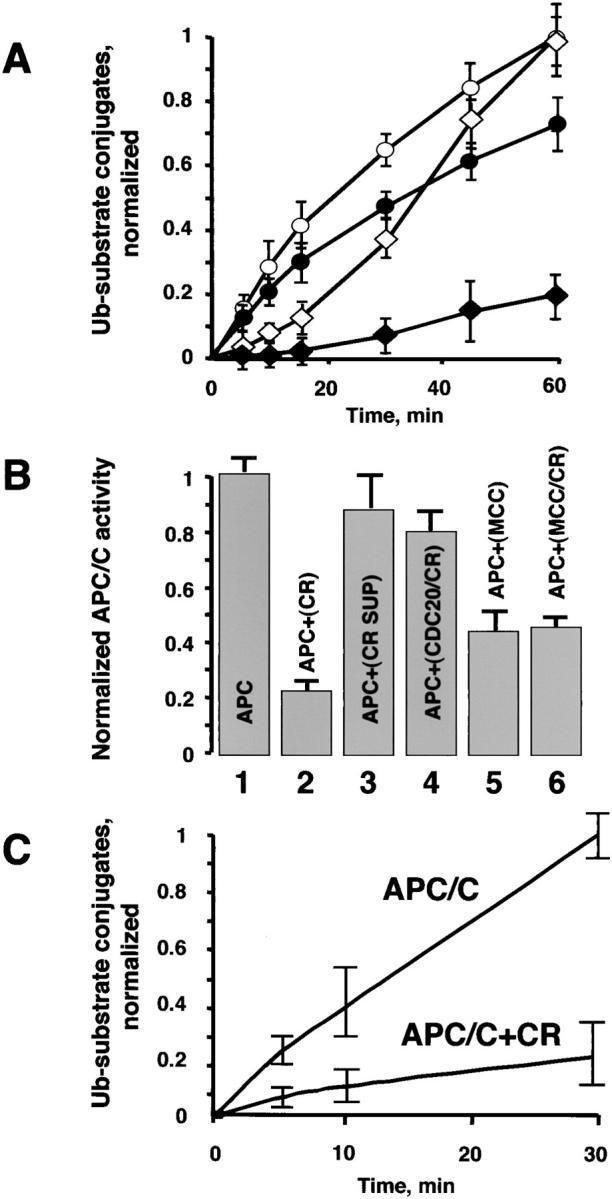
Chromosomes inhibit APC/C. (A) Chromosomes prolong the inhibition of the APC/C activity in mitotic lysates. APC/C activity in lysates prepared from interphase (○) and mitotic (⋄) cells were monitored for up to 60 min. The same lysates were assayed for ACP/C activity in the presence of chromosomes (• and ♦). (B) Chromosomes inhibit APC/C fraction directly. Partially purified mitotic APC/C (gel filtration form of the APC/C was prepared as described above), MCC and in vitro–translated CDC20 were preincubated, respectively, in the presence or absence of chromosomes at 300°C. Upon completion of the preincubation, the APC/C inhibitory assays were assembled as described below. After 30 min, E1, E2-C, and radio- labeled substrate were added to initiate the ubiquitin ligase reaction and APC/C activity was determined over the course of 30 min. (1) APC. APC/C was preincubated alone; (2) APC+CR. APC/C was preincubated with chromosomes; (3) APC+CR SUP. Chromosomes were preincubated, the mix was centrifuged for 10 min at 14,000 rpm to remove chromosomes, and the supernatant was tested for the APC/C inhibitory activity; (4) APC+CDC20/CR. In vitro–translated CDC20 was preincubated with chromosomes, the mix was centrifuged for 10 min at 14.000 rpm to remove chromosomes, and the supernatant was tested for the APC/C inhibitory activity; (5) APC+MCC. MCC was preincubated alone; (6) APC+MCC/CR. MCC was preincubated with chromosomes, the mix was centrifuged for 10 min at 14,000 rpm to remove chromosomes, and the supernatant was tested for the APC/C inhibitory activity. All reactions were supplemented with CDC20 to maintain the same concentration with the samples where preincubation of CDC20 with chromosomes has been tested. (C) Chromosomes inhibit highly purified APC/C. Gel filtration fraction of the APC/C was further purified on MonoQ anion exchange FPLC column as described above. The APC/C was preincubated with or without chromosomes and the ubiquitination reaction was initiated upon addition of all necessary ingredients as described above. 0, 5, 10, and 30 min time points were taken to determine the APC/C activity. In all experiments presented in the figure the Ub–substrate conjugates were visualized and quantified as described above.
The ability of APC/C to recover its activity in the absence of chromosomes suggested that MCC interactions with APC/C are likely labile, as might be expected for a component of the checkpoint. To learn more about the role of chromosomes in suppressing the reactivation of APC/C, we tested whether chromosomes enhanced the inhibitory activity of MCC or inhibited the stimulatory activity of CDC20. APC/C that is purified from cells is depleted of its activator CDC20, as addition of recombinant CDC20 significantly stimulates APC/C activity (Fang et al., 1998). Preincubation of the APC/C with chromosomes led to significant inhibition of the ubiquitination activity (Fig. 7 B, compare lanes 1 and 2). To test whether chromosomes blocked APC/C through CDC20, in vitro–translated CDC20 was preincubated with chromosomes and then assayed for its ability to stimulate APC/C that was partially purified from mitotic cells. No difference in stimulatory effect was observed between untreated and treated CDC20 (Fig. 7 B, compare lanes 1 and 4). We next preincubated chromosomes with purified MCC to test whether its inhibitory activity might be enhanced. No enhancement of MCC inhibitory activity was observed compared with untreated MCC (for this assay, we predetermined the amount of MCC that produced a 50–60% inhibition of APC/C activity) (Fig. 7 B, compare lanes 5 and 6). To address the question of whether a soluble factor emitting from chromosomes is responsible for the inhibition of the APC/C, the chromosome sample was preincubated for 30 min at 30°C. The chromosomes were removed by centrifugation and we found no significant inhibitory activity in the remaining supernatant (Fig. 7 B, compare 1 and 3). All reactions were supplemented with CDC20 to maintain consistency with the samples where preincubation of CDC20 with chromosomes has been tested.
We next attempted to reconstitute the suppression of APC/C activity observed in crude lysates by using purified components. It was important to verify that the chromosome-induced inhibition of the APC/C activity was not caused by unidentified factors in the crude preparation. As in crude mitotic lysates, we found that chromosomes are able to effectively sustain the APC/C inhibition in a highly purified sample (Fig. 7 C). Given that chromosomes did not exert an effect on MCC or CDC20, its ability to suppress the activation of APC/C maybe mediated through the APC/C.
Discussion
MCC is likely the APC/C inhibitor in vivo
We have isolated from HeLa cells a complex consisting of hBUBR1, hBUB3, MAD2, and CDC20 that is responsible for inhibiting the APC/C in mitotically arrested cells. MCC obtained by conventional biochemical purification or by affinity purification blocked APC/C in an in vitro assay. In addition, we found that the majority of the APC/C in a mitotic cell is bound to MCC. However, this complex exhibited very little activity (∼5% of the total) when compared with APC/C that lacked MCC. Although many reports show that recombinant MAD2 can directly inhibit APC/C, we determined that the inhibitory activity was over three orders of magnitude (>3,000-fold) weaker than the MCC. This difference in activity accounts for why the population of free MAD2 in cells, which is present at an ∼20–50-fold higher level than the amount of MAD2 in MCC, cannot block APC/C in vivo. We have determined that the concentration of MAD2 in mitotic lysates prepared from HeLa cells is ∼400 nM (unpublished data), but this is about fivefold lower than the concentration of recombinant MAD2 that is required to inhibit APC/C in vitro. Thus, HeLa cells simply do not express MAD2 at the levels that are required to inhibit APC/C in vitro.
Although oligomerization of recombinant MAD2 was found to enhance its ability to inhibit APC/C activity (Fang et al., 1998), it cannot account for the inhibitory activity of the MCC. Based on the near equal stoichiometry of the MCC subunits, it is unlikely that MAD2 oligomers are present in the MCC despite its ability to oligomerize in bacteria (Fang et al., 1998). It is unclear whether the inhibitory activity of MAD2 is enhanced through its association with the other MCC subunits, or whether the other subunits of MCC also directly block APC/C activity. Taken together, our data strongly suggest that MCC is the physiologically relevant inhibitor of the APC/C in HeLa cells. Interestingly, a similar complex consisting of MAD3, CDC20, and MAD2 has been identified in yeast but its role in regulating APC/C has not been elucidated (Hardwick et al., 2000). It appears that the MCC may be conserved throughout evolution.
It is noteworthy that we did not detect MAD1 in MCC (unpublished data) even though MAD1 and MAD2 form a complex in vivo (Chen et al., 1998, 1999; Jin et al., 1998; Campbell et al., 2001). Studies in Xenopus extracts suggest that MAD1 specifies kinetochore binding by MAD2 but that is not directly involved in inhibiting the APC/C (Chen et al., 1998). We have also not detected the presence of checkpoint proteins such as hBUB1, hZW10, or hROD in the MCC. Therefore, these components are likely to provide functions that are distinct from the MCC.
We believe that MCC inhibits APC/C by directly binding to it rather than via catalytic inactivation. This conclusion is based on the finding that the intracellular concentration of hBUBR1 and APC/C are roughly the same (∼52 and 65 nM for hBUBR1 and CDC27 in crude lysate, respectively; ratio 1:1 in anti-hBUBR1 immunoprecipitates from whole mitotic lysate; unpublished data). Our unpublished data indicate that there is ∼400 nM of MAD2 in mitotic HeLa lysates. The gel filtration profiles show that only 5–10% of the total MAD2 is a part of high molecular mass complexes and comigrates with APC/C and MCC (Fig. 3 A). Given the hBUBR1 and CDC27 concentrations we have a roughly equal ratio between hBUBR1, CDC27, and “active” MAD2 in our extracts (∼52, 65, and 20–40 nM, respectively). This ratio of hBUBR1 to active MAD2 is similar to our estimate using radiolabeled proteins.
Examination of the MCC and APC/C profiles in a MonoQ column showed that MCC was associated with ∼75% of the APC/C (Fig. 3 D). However, the remaining 25% of the APC/C most likely lost its association with its MCC, as we found MCC in fractions that did not contain any APC/C (Fig. 3 C). We had relied previously on immunoprecipitation to estimate that ∼25% of the APC/C was bound to hBUBR1 (Chan et al., 1999). This is likely to be an underestimate, as we now know that the conditions used to wash the immunoprecipitate dissociated much of the APC/C from hBUBR1. The identification of the molecular interactions among the various subunits within APC/C and MCC will be crucial for understanding the mechanism of inhibition.
Roles of MCC and kinetochores in inhibition of APC/C
The finding that MCC is present during interphase and can inhibit the APC/C was unexpected because kinetochores (which are not present during interphase) are thought to generate the inhibitor of the APC/C. However, we found that only APC/C from mitotic cells was sensitive to MCC inhibition. These findings have changed our view of the checkpoint pathway in two important ways (Fig. 8) . First, our data suggest that the formation and activity of the APC/C inhibitor can be uncoupled from kinetochores. This may be an important feature considering that APC/C is known to be activated at the onset of mitosis. The existence of a preformed pool of inhibitor would rapidly block precocious ubiquitination activity by the APC/C at the onset of mitosis. It is noteworthy that only the mitotically modified form of the APC/C can be inhibited (Fig. 6, B and C; Fig. 7 A), which can explain why the preformed pool of MCC doesn't inhibit APC before mitosis. By necessity, the inhibition must be reversible so that APC/C can be activated once cells are ready to exit mitosis. We believe that in vivo, the interaction between APC/C and MCC is quite labile in the absence of unattached kinetochores. This would explain why APC/C that is purified from mitotic cells exhibits a lag before it becomes reactivated.
Figure 8.

Model for MCC function and regulation. MCC is present throughout the cell cycle but is incapable of inhibiting APC/C until it has undergone mitotic modifications. APC/C is normally rapidly activated in mitosis by phosphorylations and association with CDC20. These modifications may also be recognized by the MCC so that APC/C is prevented from ubiquitinating target proteins until all chromosomes are aligned. We speculate that the interaction between MCC and APC/C is not stable so APC/C is not permanently inhibited. In the presence of unattached kinetochores (•), signals initiated at kinetochores modify the APC/C or APC-bound MCC to enhance their interactions and thus prolong its inhibition. The signal from the kinetochore can either be MAD2 as proposed or a kinase cascade that is initiated at the kinetochore by hBUBR1, hBUB1, or other kinases. After chromosomes align, the signal from kinetochores decays along with the modifications. The MCC then dissociates and APC/C becomes active to drive cells out of mitosis. We believe that MCC may not inhibit APC/C during interphase because it either lacks the appropriate phosphorylations or the APC/C is bound to a different substrate specificity factor, CDH1.
We also propose that the role of unattached kinetochores is to sustain the inhibition of APC/C by MCC. We found that APC/C activity in mitotic lysates and purified mitotic APC/C was suppressed upon addition of chromosomes. We propose that the “wait anaphase” signal that is produced by unattached kinetochores enhances or prolongs the inhibition of the APC/C mediated by the MCC. Given that APC/C copurifies with MCC (Fig. 3, C and D), we cannot as yet clearly distinguish whether the target is the MCC or the APC/C. We examined the effect of chromosomes on purified MCC and CDC20 to address this issue and found that neither MCC inhibitory activity nor CDC20 stimulatory activity was affected by chromosomes. This leaves open the possibility that chromosomes target subunits of the APC/C to sensitize it to prolonged inhibition by MCC.
The role of the kinetochore in generating wait anaphase signal
The biochemical nature of the wait anaphase signal that is generated at kinetochores remains to be elucidated but it is likely to involve many of the checkpoint proteins that reside there. FRAP analysis of MAD2 showed that its half-life at kinetochores is ∼25 s, and that ∼1,300 molecules are present on kinetochores at any given time (Howell et al., 2000). The rapid turnover rate supports a model whereby unattached kinetochores catalytically convert MAD2 into a form that inhibits the APC/C upon its release from kinetochores (Chen et al., 1998; Gorbsky et al., 1998; Howell et al., 2000). Our data cannot eliminate this possibility despite the fact that kinetochores are not required for MCC formation or activity. In light of our findings, we propose that MAD2 that is derived from kinetochores may enhance the sensitivity of the APC/C to the inhibitory action of MCC.
As the fate of MAD2 after it has dissociated from kinetochores is not known, other possibilities remain open. An alternative view is that the APC/C inhibition is mediated by checkpoint kinases such as BUBR1 and BUB1 that reside at kinetochores. In this scenario, MAD2 could regulate these kinetochore-associated kinases such that its presence at unattached kinetochores is required to maintain a threshold level of kinase activity. It is noteworthy that hBUBR1 that is associated with kinetochores becomes quantitatively hyperphosphorylated within 15 min after disruption of microtubule attachments (Chan et al., 1999). This may contribute to MAD2 binding, as in vitro studies show that MAD2 will only bind to kinetochores that are phosphorylated (Waters et al., 1998). We speculate that hyperphosphorylation of hBUBR1 stimulates its kinase activity that is required for generating the wait anaphase signal. In vivo, efficient phosphorylation of APC/C by kinetochores may be achieved through a kinase cascade. Our attempt at reconstituting the checkpoint inhibition of APC/C by adding just chromosomes probably does not reflect the in vivo situation. In our experiments, we believe that chromosomes are directly modifying the APC/C while in vivo, an occurrence probably accomplished by a signaling cascade amplifying the signal that originates from a single unattached kinetochore. Whatever the nature of the modification that is introduced to the APC/C by kinetochores, it must be labile in order to relieve MCC inhibitory activity upon chromosome alignment.
MCC interactions with APC/C
Regardless of whether kinetochores modify APC/C to prolonged inhibition by the MCC, our data show that APC/C must undergo mitotic modifications in order for it to be recognized by the MCC. This is supported by the observation that interphase APC/C was insensitive to inhibition by MCC. The insensitivity cannot be due simply to the lack of modifications introduced by kinetochores, as APC/C activity in interphase lysates was unaffected in the presence of chromosomes. We believe that the mitotic modifications that target the MCC to APC/C include phosphorylations of specific APC/C subunits (King et al., 1995; Lahav-Baratz et al., 1995; Charles et al., 1998; Descombes and Nigg, 1998; Kotani et al., 1999; Shirayama et al., 1998) and its association with CDC20 (Fang et al., 1998; Shirayama et al., 1998; Chan et al., 1999; Shteinberg et al., 1999). The importance of CDC20 in checkpoint control is highlighted by the finding that certain CDC20 mutants in fission (Kim et al., 1998) and budding yeast (Hwang et al., 1998; Schott and Hoyt, 1998) exhibit checkpoint defects. Whether this mutation affects formation of an MCC-like complex (Hardwick et al., 2000; Millband, D., and K.G. Hardwick, personal communications) or alters the sensitivity of the APC/C to inhibition by the checkpoint remains to be clarified. How modifications to the APC/C contribute to recognition by MCC remains to be determined, but it has been reported that MAD2, CDC20, and hBUBR1 bind preferentially to mitotically phosphorylated APC/C (Fang et al., 1998; Kallio et al., 1998; Kotani et al., 1999; Wu et al., 2000). We suggest that these previously reported findings reflect the MCC–APC/C association during mitosis. It remains to be seen whether the modifications that are required for APC/C activity are also used by the MCC to inhibit the APC/C.
Materials and methods
Preparation of HeLa extracts
Frozen HeLa cell pellets were obtained from the National Cell Culture Center (Cellex Biosciences). Mitotic extracts were prepared from HeLa cells exposed to 60 ng/ml nocodazole for 18 h. Interphase extracts were prepared from cells arrested at the G1/S boundary after double thymidine block. Cells were washed twice in ice cold PBS, diluted 1:1 with the disruption buffer (40 mM Hepes, pH 7.4, 10 mM EDTA, 2 mM DTT, and protease inhibitors), and lysed using a high-pressure nitrogen bomb (Parr Instruments). 10,000 g (S10) supernatants were supplemented with glycerol (10% final concentration) and frozen at −80°C. To prepare high-speed extracts, S10 extracts were spun in the ultracentrifuge (Beckman Coulter) at 100,000 g for 30 min. Final protein concentration of the extracts was 15–18 mg/ml. All procedures were carried out at 4°C.
Purification of mitotic chromosomes
Chromosomes were purified from mitotically blocked HeLa cells as described (Yen et al., 1991). In brief, HeLa cells were blocked with nocodazole for 18 h, pelleted and hypotonically swollen in RSB (10 mM Tris-HCl, pH 8.0, 10 mM MgCl2, 10 mM NaCl), and lysed in digitonin containing buffer A (50 mM Tris-HCl, pH 7.5, 80 mM KCl, 2 mM EGTA, 3 mM spermine, 7.5 mM spermidine, 0.1% digitonin supplemented with 1 μM pepstatin, 1 μg/ml leupeptin, 1 mM PMSF, 2 μg/ml aprotinin). Cells were lysed using a dounce (10–12 times using a pestle B) and centrifuged at 500 g for 5 min to remove nuclei and cell debris. Chromosomes in the supernatant were pelleted at 1,600 g for 10 min, washed three times in buffer 3 (50 mM Tris-HCl, pH 7.5, 40 mM KCl, 3 mM spermine, 3.75 mM spermidine with protease inhibitors), resuspended in buffer 4 (50 mM Tris-HCl, pH 7.5, 20 mM KCl, 3 mM spermine, 3.75 mM spermidine, 15% glycerol with protease inhibitors), and used immediately or snap frozen. Chromosomes were examined by DAPI staining and kinetochore integrity was monitored by staining for CENP-E and various mitotic checkpoint proteins.
Ubiquitination assay and APC/C isolation
Crude extracts or purified factors were supplemented with purified E1, E2, ATP regenerating system, and iodinated protein A–cyclin B substrate (Glotzer et al., 1991), and the APC/C activity was determined as described (Sudakin et al., 1995) in final volume of 10 μl. Standard reactions were performed for 30 min at 30°C but kinetic studies relied on sampling the reaction at various times. Reaction products were separated by SDS-PAGE and the dried gel was analyzed with a phosphorimager (Fujix) to quantify the amount of ubiquitinated substrate. This amount was compared with the amount of input substrate to obtain the percent of substrate that was ubiquitinated in each reaction. In all experiments, the percent of substrate that is conjugated in a positive control reaction that contained APC/C was set at 1 and served as a reference for the activities obtained in other reactions in the same experiment. Most of the figures show normalized activities. APC/C was partially purified from HeLa lysates (15 mg total protein) by fast protein liquid chromatography (FPLC) (Amersham Pharmacia Biotech) using Superose 6 or Superdex 200 gel filtration columns. Gel filtration columns were eluted with buffer A (50 mM Tris-Cl, pH 7.2, 250 mM NaCl, 1 mM DTT, 0.2 mg/ml BSA) and fractions exhibiting APC/C activity were pooled, desalted, and concentrated in buffer B (50 mM Tris-Cl, pH 7.2, 1 mM DTT, 10% glycerol). Further purification of APC/C was achieved by chromatography through a MonoQ column. Proteins were eluted with a salt gradient (0.15–0.5 M NaCl) in buffer containing 50 mM Hepes, pH 7.4, and 1 mM DTT. BSA was added to the fraction collector tubes (0.2 mg/ml final concentration in collected fractions) and active fractions were washed with buffer B and concentrated. For immunoprecipitation experiments, affinity-purified hBUBR1 or CDC20 antibodies were incubated with APC/C-containing fractions and complexes recovered with 10 μl protein A–Sepharose beads. The beads were washed five times in ice cold PBS containing 0.1% NP-40 and 10% glycerol, and three times in ice cold buffer containing 50 mM Tris, pH 7.6, 1 mM DTT, 1 mg/ml BSA, and 5% glycerol. After the last wash, the buffer was aspirated completely and the beads were resuspended in 10 μl of the ubiquitination reaction mix as described above. Samples were incubated at 30°C for 30 min and mixed gently every 5 min. The reactions were terminated by SDS-PAGE sample buffer, boiled, and separated by SDS-PAGE. Ub–substrate conjugates were quantified as described above.
The pET28 plasmid encoding the CDC20 protein was a gift from Dr. M. Brandeis (Hebrew University, Jerusalem, Israel). The CDC20 protein was expressed in vitro in a TNT Quick reticulocyte system (Promega).
Purification of the MCC and of recombinant MAD2
APC/C inhibitory activity was recovered in the 20–40% ammonium sulfate cut of an S100 extract. This was subjected to FPLC separation through Superdex 200 gel filtration, MonoQ anion exchange, and Superose 6 gel filtration columns. Gel filtration columns were eluted with buffer A and fractions were desalted and concentrated in buffer B. MonoQ column was eluted with shallow salt gradient (0.2–0.5 M NaCl) in buffer B without glycerol. To show that MCC can exist independently of APC/C, the Superose 6 fractions of MCC and APC/C were pooled and refractionated on MonoQ anion exchange column by a shallow salt gradient (0.2–0.4 M NaCl in buffer B). Column fractions were concentrated, desalted into buffer B, and tested for inhibitory activity in the APC/C assay. During the initial purification of the inhibitory factor, equal cell equivalents of column fractions and APC/C were used for the inhibitor assay.
Human MAD2 cDNA was amplified by PCR from HeLa cell cDNA (CLONTECH Laboratories, Inc.) and cloned into pET28A expression vector (Novagen). Recombinant protein was expressed in JM109(DE3) and purified with a nickel–agarose column (QIAGEN). Tetrameric recombinant hMAD2 was purified by Superdex 75 gel filtration chromatography as described (Fang, et al. 1998).
Affinity purification of MCC with GST–hBUBR1
Full-length hBUBR1 cDNA was obtained by digestion of gfp-hBUBR1 (Chan et al., 1998) with BamH1 and KpnI, and transferred to the mammalian GST vector pEBG (gift from N. Grammatikakis, Lankenau Institute for Medical Research, Wynnewood, PA). Transient transfections were performed by lipofection using Lipofectamine Plus (Life Technologies) as per manufacturer's instruction. Transfected HeLa cells were lysed in 50 mM Tris, pH 7.2, 150 mM NaCl, 1 mM DTT, and protease inhibitors (10 μg/ml AEBSF, 10 μg/ml leupeptin, 5 μg/ml pepstatin, 5 μg/ml chymostatin, 10 μg/ml aprotinin) by repeated freeze and thaw cycles. Lysates were clarified by spinning at 10,000 g at 4°C. GST–hBUBR1 was affinity purified from the 10,000 g supernatant by glutathione Sepharose 4B beads (Amersham Pharmacia Biotech), washed five times with wash buffer (50 mM Tris, pH7.2, 250 mM NaCl, 1 mM DTT, and protease inhibitors), and eluted with 10 mM glutathione in lysis buffer. Eluted GST–hBUBR1 was dialyzed and concentrated using Centricon10 (Millipore) into 50 mM Tris, pH 7.2, and 1 mM DTT for APC/C assay.
Stoichiometry of MCC
Synchronous populations of HeLa cells released from a G1/S boundary were labeled in 35S-Trans label for 6 h. Mitotic and interphase cells were mechanically separated, lysed in the NP-40 lysis buffer (0.1% NP-40, 10% glycerol in PBS), and the extracts were then incubated with nonimmune IgG or hBUBR1 antibodies. Immunoprecipitates were washed five times in ice cold lysis buffer and separated on SDS-PAGE. Radiolabeled proteins were visualized and quantified with the phosphorimager (Fujix). Cys + Met amounts in the MCC proteins (without initiating Met) have been used to calculate the ratio of the MCC subunits.
Acknowledgments
The authors are grateful to B. Conner and J. Hittle for excellent technical assistance, A. Hershko for ubiquitin reagents, and M.S. Campbell for radiolabeling experiments. We would also like to thank A. Hershko, A. Hoyt, E.D. Salmon, and S.A. Jablonski for helpful comments and discussion during the course of the work and in reading our manuscript.
This work was supported by the National Institutes of Health (POI CA 75138; Core Grant CA06927), March of Dimes Foundation, and an appropriation from the Commonwealth of Pennsylvania. V. Sudakin was supported by a fellowship from the Fulbright and Human Frontiers Science Foundations.
Footnotes
Abbreviations used in this paper: APC/C, anaphase-promoting complex/cyclosome; FPLC, fast protein liquid chromatography; GST, glutathione S-transferase; MCC, mitotic checkpoint complex.
References
- Abrieu, A., J.A. Kahana, K.W. Wood, and D.W. Cleveland. 2000. CENP-E as an essential component of the mitotic checkpoint in vitro. Cell. 102:817–826. [DOI] [PubMed] [Google Scholar]
- Campbell, M.S., G.K.T. Chan, and T.J. Yen. 2001. Mitotic checkpoint proteins HsMAD1 and HsMAD2 are associated with nuclear pore complexes in interphase. J. Cell Sci. 114:953–963. [DOI] [PubMed] [Google Scholar]
- Chan, G.K.T., B.T. Schaar, and T.J. Yen. 1998. Characterization of the kinetochore binding domain of CENP-E reveals interactions with the kinetochore proteins CENP-F and hBUBR1. J. Cell Biol. 143:49–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, G.K.T., S.A. Jablonski, V. Sudakin, J.C. Hittle, and T.J. Yen. 1999. Human BUBR1 is a mitotic checkpoint kinase that monitors CENP-E functions at kinetochores and binds the cyclosome/APC. J. Cell Biol. 146:941–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles, J.F., S.L. Jaspersen, R.L. Tinker-Kulberg, L. Hwang, A. Szidon, and D.O. Morgan. 1998. The Polo-related kinase Cdc5 activates and is destroyed by the mitotic cyclin destruction machinery in S. cerevisiae. Curr. Biol. 8:497–507. [DOI] [PubMed] [Google Scholar]
- Chen, R.H., J.C. Waters, E.D. Salmon, and A.W. Murray. 1996. Association of spindle assembly checkpoint component XMAD2 with unattached kinetochores. Science. 274:242–246. [DOI] [PubMed] [Google Scholar]
- Chen, R.H., A. Shevchenko, M. Mann, and A.W. Murray. 1998. Spindle checkpoint protein Xmad1 recruits Xmad2 to unattached kinetochores. J. Cell Biol. 143:283–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, R.H., D.M. Brady, D. Smith, A.W. Murray, and K.G. Hardwick. 1999. The spindle checkpoint of budding yeast depends on a tight complex between the Mad1 and Mad2 proteins. Mol. Biol. Cell. 10:2607–2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson, I.A., S. Roth, and S. Artavanis-Tsakonas. 1995. The Drosophilla cell cycle gene fizzy is required for normal degradation of cyclins A and B during mitosis and has homology to the CDC20 gene of Saccharomyces cerevisiae. J. Cell Biol. 129:725–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Descombes, P., and E.A. Nigg. 1998. The polo-like kinase Plx1 is required for M phase exit and destruction of mitotic regulators in Xenopus egg extracts. EMBO J. 17:1328–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang, G., H. Yu, and M.W. Kirschner. 1998. The checkpoint protein MAD2 and the mitotic regulator CDC20 form a ternary complex with the anaphase-promoting complex to control anaphase initiation. Genes Dev. 12:1871–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glotzer, M., A.W. Murray, and M.W. Kirschner. 1991. Cyclin is degraded by the ubiquitin pathway. Nature. 349:132–138. [DOI] [PubMed] [Google Scholar]
- Gorbsky, G.J., R.H. Chen, and A.W. Murray. 1998. Microinjection of antibody to Mad2 protein into mammalian cells in mitosis induces premature anaphase. J. Cell Biol. 141:1193–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick, K.G., and A.W. Murray. 1995. Mad1p, a phosphoprotein component of the spindle assembly checkpoint in budding yeast. J. Cell Biol. 131:709–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick, K.G., R.C. Johnston, D.L. Smith, and A.W. Murray. 2000. MAD3 encodes a novel component of the spindle checkpoint which interacts with Bub3p, Cdc20p, and Mad2p. J. Cell Biol. 148:871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, X., T.E. Patterson, and S. Sazer. 1997. The Saccharomyces pombe spindle checkpoint protein Mad2p blocks anaphase and genetically interacts with the anaphase-promoting complex. Proc. Natl. Acad. Sci. USA. 94:7965–7970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershko, A., and A. Ciechanover. 1998. The ubiquitin system. Annu. Rev. Biochem. 67:425–479. [DOI] [PubMed] [Google Scholar]
- Howell, B.J., D.B. Hoffman, G. Fang, A.W. Murray, and E.D. Salmon. 2000. Visualization of Mad2 dynamics at kinetochores, along spindle fibers, and at spindle poles in living cells. J. Cell Biol. 150:1233–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyt, M.A., L. Totis, and B.T. Roberts. 1991. S. cerevisiae genes required for cell cycle arrest in response to loss of microtubule function. Cell. 66:507–517. [DOI] [PubMed] [Google Scholar]
- Hwang, L.H., L.F. Lau, D.L. Smith, C.A. Mistrot, K.G. Hardwick, E.S. Hwang, A. Amon, and A.W. Murray. 1998. Budding yeast Cdc20: a target of the spindle checkpoint. Science. 279:1041–1044. [DOI] [PubMed] [Google Scholar]
- Jin, D.Y., F. Spencer, and K.T. Jeang. 1998. Human T cell leukemia virus type 1 oncoprotein Tax targets the human mitotic checkpoint protein MAD1. Cell. 93:81–91. [DOI] [PubMed] [Google Scholar]
- Kallio, M., J. Weinstein, J.R. Daum, D.J. Burke, and G.J. Gorbsky. 1998. Mammalian p55CDC mediates association of the spindle checkpoint protein Mad2 with the cyclosome/anaphase-promoting complex, and is involved in regulating anaphase onset and late mitotic events. J. Cell Biol. 141:1393–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, S.H., D.P. Lin, S. Matsumoto, A. Kitazono, and T. Matsumoto. 1998. Fission yeast Slp1: an effector of the Mad2-dependent spindle checkpoint. Science. 279:1045–1047. [DOI] [PubMed] [Google Scholar]
- King, R.W., J.-M. Peters, S. Tugendreich, M. Rolfe, P. Hieter, and M.W. Kirschner. 1995. A 20S complex containing CDC27 and CDC16 catalyses the mitosis-specific conjugation of ubiquitin to cyclin B. Cell. 81:279–288. [DOI] [PubMed] [Google Scholar]
- King, R.W., R.J. Deshaies, J.M. Peters, and M.W. Kirschner. 1996. How proteolysis drives the cell cycle. Science. 274:1652–1659. [DOI] [PubMed] [Google Scholar]
- Kotani, S., H. Tanaka, H. Yasuda, and K. Todokoro. 1999. Regulation of APC activity by phosphorylation and regulatory factors. J. Cell Biol. 146:791–800. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Lahav-Baratz, S., V. Sudakin, J.V. Ruderman, and A. Hershko. 1995. Reversible phosphorylation controls the activity of cyclosome-associated cyclin-ubiquitin ligase. Proc. Natl. Acad. Sci. USA. 92:9303–9307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, R., and A.W. Murray. 1991. Feedback control of mitosis in budding yeast. Cell. 66:519–531. [DOI] [PubMed] [Google Scholar]
- Li, Y., and R. Benezra. 1996. Identification of a human mitotic checkpoint gene: hsMAD2. Science. 274:246–248. [DOI] [PubMed] [Google Scholar]
- Li, Y., C. Gorbea, D. Mahaffey, M. Rechsteiner, and R. Benezra. 1997. MAD2 associates with the cyclosome/anaphase-promoting complex and inhibits its activity. Proc. Natl. Acad. Sci. USA. 94:12431–12436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder, C.L., and E.D. Salmon. 1998. The vertebrate cell kinetochore and its roles during mitosis. Trends Cell Biol. 8:310–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder, C.L., R.W. Cole, A. Khodjakov, and G. Sluder. 1995. The checkpoint delaying anaphase in response to chromosome monoorientation is mediated by an inhibitory signal produced by unattached kinetochores. J. Cell Biol. 130:941–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts, B.T., K.A. Farr, and M.A. Hoyt. 1994. The Saccharomyces cerevisiae checkpoint gene BUB1 encodes a novel protein kinase. Mol. Cell. Biol. 14:8282–8291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schott, E.J., and A.M. Hoyt. 1998. Dominant alleles of Saccharomyces cerevisiae CDC20 reveal its role in promoting anaphase. Genetics. 148:599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah, J.V., and D.W. Cleveland. 2000. Waiting for anaphase: Mad2 and the spindle assembly checkpoint. Cell. 103:997–1000. [DOI] [PubMed] [Google Scholar]
- Shirayama, M., W. Zachariae, R. Ciosk, and K. Nasmyth. 1998. The polo-like kinase Cdc5p and the WD-repeat protein Cdc20p/fizzy are regulators and substrates of the anaphase promoting complex in Saccharomyces cerevisiae. EMBO J. 17:1336–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shteinberg, M., Y. Protopopov, T. Listovsky, M. Brandeies, and A. Hershko. 1999. Phosphorylation of the cyclosome is required for its stimulation by fizzy/cdc20 Biochem Biophys. Res. Commun. 260:193–198. [DOI] [PubMed] [Google Scholar]
- Skibbens, R.V., and P. Hieter. 1998. Kinetochores and the checkpoint mechanism that monitors for defects in the chromosome segregation machinery. Annu. Rev. Genet. 32:307–337. [DOI] [PubMed] [Google Scholar]
- Sudakin, V., D. Ganoth, A. Dahan, H. Heller, J. Hershko, F.C. Luca, J.V. Ruderman, and A. Hershko. 1995. The cyclosome, a large complex containing cyclin-selective ubiquitin lygase activity, targets cyclins for destruction at the end of mitosis. Mol. Biol. Cell. 6:185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, S.S., and F. McKeon. 1997. Kinetochore localization of murine Bub1 is required for normal mitotic timing and checkpoint response to spindle damage. Cell. 89:727–735. [DOI] [PubMed] [Google Scholar]
- Taylor, S.S., E. Ha, and F. McKeon. 1998. The human homologue of Bub3 is required for kinetochore localization of Bub1 and a Mad3/Bub1-related protein kinase. J. Cell Biol. 142:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visintin, R., S. Prinz, and A. Amon. 1997. CDC20 and CDH1: a family of substrate-specific activators of APC-dependent proteolysis. Science. 278:460–463. [DOI] [PubMed] [Google Scholar]
- Wassmann, K., and R. Benezra. 1998. Mad2 transiently associates with an APC/p55Cdc complex during mitosis. Proc. Natl. Acad. Sci. USA. 95:11193–11198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters, J.C., R.H. Chen, A.W. Murray, and E.D. Salmon. 1998. Localization of Mad2 to kinatochores depends on microtubule attachment, not tension. J. Cell Biol. 141:1181–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, H., Z. Lan, W. Li, S. Wu, J. Weinstein, K.M. Sakamoto, and W. Dai. 2000. p55CDC/hCDC20 is associated with BUBR1 and may be a downstream target of the spindle checkpoint kinase. Oncogene. 19:4557–4562. [DOI] [PubMed] [Google Scholar]
- Yen, T.J., D.A. Compton, D. Wise, R.P. Zinkowski, B.R. Brinkley, W.C. Earnshaw, and D.W. Cleveland. 1991. CENP-E, a novel human centromere-associated protein required for progression from metaphase to anaphase. EMBO J. 10:1245–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao, X., A. Abrieu, Y. Zheng, K.F. Sullivan, and D.W. Cleveland. 2000. CENP-E forms a link between attachment of spindle microtubules to kinetochores and the mitotic checkpoint. Nat. Cell Biol. 2:484–491. [DOI] [PubMed] [Google Scholar]
- Zacharie, W., and K. Nasmyth. 1999. Whose end is destruction: cell division and the anaphase-promoting complex. Genes Dev. 13:2039–2058. [DOI] [PubMed] [Google Scholar]