

Abstract
By adulthood, sympathetic neurons have lost dependence on NGF and NT-3 and are able to survive in culture without added neurotrophic factors. To understand the molecular mechanisms that sustain adult neurons, we established low density, glial cell-free cultures of 12-wk rat superior cervical ganglion neurons and manipulated the function and/or expression of key proteins implicated in regulating cell survival. Pharmacological inhibition of PI 3-kinase with LY294002 or Wortmannin killed these neurons, as did dominant-negative Class IA PI 3-kinase, overexpression of Rukl (a natural inhibitor of Class IA PI 3-kinase), and dominant-negative Akt/PKB (a downstream effector of PI 3-kinase). Phospho-Akt was detectable in adult sympathetic neurons grown without neurotrophic factors and this was lost upon PI 3-kinase inhibition. The neurons died by a caspase-dependent mechanism after inhibition of PI 3-kinase, and were also killed by antisense Bcl-xL and antisense Bcl-2 or by overexpression of Bcl-xS, Bad, and Bax. These results demonstrate that PI 3-kinase/Akt signaling and the expression of antiapoptotic members of the Bcl-2 family are required to sustain the survival of adult sympathetic neurons.
Keywords: phosphoinositide 3-kinase; Akt kinase/protein kinase B; Bax; BcL-xL; signaling
Introduction
The molecular mechanisms that regulate neuronal survival and bring about apoptosis have been extensively studied in developing sympathetic neurons. In vivo studies using function-blocking anti-NGF and anti-NT3 antibodies (Levi-Montalcini, 1987; Zhou and Rush, 1995) together with detailed analysis of the timing of neuronal death in the sympathetic chain of mice lacking NGF or NT3 (Crowley et al., 1994; Wyatt et al., 1997; Francis et al., 1999) have shown that sympathetic neurons become dependent on the supply of these two neurotrophins shortly after they begin to innervate their targets. Because NGF and NT3 are normally produced in limiting amounts, only a proportion of the neurons initially generated survive into adulthood, with superfluous neurons being eliminated by apoptosis during the late fetal and early postnatal period. Sympathetic neurons cultured during this period of development die unless NGF or NT3 is added to the culture medium. However, these neurons lose dependence on neurotrophins with age, and, by adulthood, most sympathetic neurons are able to survive for at least a week in culture in defined medium without added neurotrophins (Orike et al., 2000).
Considerable progress has been made in delineating the molecular mechanisms that mediate the survival-enhancing effects of NGF and NT3 on embryonic and neonatal sympathetic neurons. Analysis of the Trk receptor tyrosine kinase isoforms expressed by sympathetic neurons (Wyatt et al., 1997) together with in vitro studies of TrkC-deficient neurons (Davies et al., 1995) and the effects of a mutant NT-3 protein that only signals via TrkA (Belliveau et al., 1997) have shown that both NGF and NT-3 promote sympathetic neuron survival by acting via TrkA. Ligand binding causes TrkA dimerization and autophosphorylation on specific tyrosine residues that form docking sites for several adapter proteins including Shc, PLCγ, and SHP. These proteins couple Trk to several intracellular signaling pathways that result in Shc/Grb2/Gab1-dependent activation of phosphoinositide (PI)* 3-kinase (Holgado-Madruga et al., 1997) and the production of 3′-phosphorylated phosphoinositides, Shc/Grb2/Sos-dependent activation of Ras, and PLC-γ–mediated generation of DAG and inositol trisphosphate (Kaplan and Miller, 1997).
Studies using function-blocking Ras antibodies or dominant-negative proteins have shown that Ras plays an important role in mediating the survival response of SCG neurons to NGF (Nobes et al., 1996; Markus et al., 1997; Mazzoni et al., 1999). Activated Ras stimulates several downstream signaling cascades including PI 3-kinase and the mitogen-activated protein kinase kinase (MEK)–mitogen-activated protein (MAP) kinase pathways (Shields et al., 2000). Studies using Ras effector mutants suggest that Ras exerts its effects on sympathetic neuron survival principally by activating the catalytic subunit of PI 3-kinase (Rodriguez-Viciana et al., 1994; Mazzoni et al., 1999; Xue et al., 2000), whereas MAP kinase activation plays only a minor role or no role at all (Creedon et al., 1996; Virdee and Tolkovsky, 1996; Klesse and Parada, 1998; Mazzoni et al., 1999; Xue et al., 2000). Although overexpression of P1 3-kinase prevents the death of sympathetic neurons after NGF withdrawal, suggesting that PI 3-kinase is sufficient for survival (Philpott et al., 1997; Crowder and Freeman, 1998), evidence for the involvement of PI 3-kinase in mediating the survival-promoting actions of NGF on embryonic and neonatal sympathetic neurons is conflicting. Whereas several studies have reported that pharmacological or dominant-negative inactivation of PI 3-kinase inhibits the NGF survival response (Crowder and Freeman, 1998; Mazzoni et al., 1999; Vaillant et al., 1999), other studies have reported only modest effects of PI 3-kinase inactivation on the survival of developing sympathetic neurons maintained with NGF (Philpott et al., 1997; Virdee et al., 1999; Tsui-Pierchala et al., 2000). Recently, it has been shown that there is a more critical dependence on PI 3-kinase for survival of sympathetic neurons supported by NGF acting on their distal axons as compared with neurons supported by NGF acting directly on their cell bodies (Kuruvilla et al., 2000).
The serine/threonine protein kinase Akt (protein kinase B), a downstream effector of PI 3-kinase, appears to play a key role in mediating the NGF survival response of developing sympathetic neurons since constitutively active Akt prevents the death of these neurons after NGF deprivation and dominant-negative Akt kills these neurons in the presence of NGF (Crowder and Freeman, 1998; Virdee et al., 1999). Akt influences the activity of several transcription factors implicated in regulating cell survival, including Forkhead 1 (Brunet et al., 1999), NF-κB (Kane et al., 1999; Ozes et al., 1999), and CREB (Du and Montminy, 1998). Phosphorylation of Forkhead 1 by Akt prevents it from inducing expression of the cytotoxic factor FasL whose synthesis after growth factor withdrawal plays a role in causing neuronal apoptosis by initiating a caspase cascade (Brunet et al., 1999; Le-Niculescu et al., 1999; Raoul et al., 1999). Induction of NF-κB and CREB transcriptional activity by Akt results in expression of their target genes that include those encoding Bcl-2 and inhibitor of apoptosis proteins, which are required for sympathetic neuron survival in the presence of NGF (Riccio et al., 1999; Wiese et al., 1999). Akt also phosphorylates Bad, a proapoptotic member of the Bcl-2 family, which may prevent it from interacting with and inactivating antiapoptotic members of this family (Datta et al., 1997; del Peso et al., 1997).
Proteins of the Bcl-2 family play a key role in controlling the activation of caspases, which are the proteases that dismantle the cell during apoptosis (Korsmeyer, 1999; Vaux and Korsmeyer, 1999). Bcl-2–related proteins fall into two groups that generally either repress apoptosis (e.g., Bcl-2 and Bcl-xL) or promote apoptosis (e.g., Bax, Bcl-xs, Bak, and Bad). These proteins influence caspase activation in part by controlling the release of cytochrome c from mitochondria that interacts with the adapter protein Apaf-1, which in turn activates procaspase-9 (Li et al., 1997; Qin et al., 1999). Proapoptotic members like Bax and Bak increase mitochondrial permeability allowing cytochrome c to pass into the cytosol, whereas antiapoptotic members like Bcl-2 and Bcl-xL prevent cytochrome c release (Kharbanda et al., 1997; Kluck et al., 1997; Yang et al., 1997; Shimizu et al., 1999). In addition, Bcl-2 is also able to regulate activation of membrane-associated procaspase-3 independently of cytochrome c (Krebs et al., 1999).
Although a good deal is known about the molecular mechanisms that mediate the survival effects of neurotrophins and induce apoptosis after their withdrawal in developing neurons, the mechanisms that sustain the survival of adult neurotrophic factor–independent neurons has received very little attention. One experimental paradigm used to address the intriguing issue of neurotrophic factor independence has involved growing embryonic sympathetic or sensory neurons in culture with NGF until they reach a point when they continue to survive after NGF is withdrawn. In sensory neurons grown under these conditions, inhibition of PI 3-kinase did not compromise survival after NGF withdrawal, suggesting that PI 3-kinase activation is not required for sustaining neuronal survival in this model of neurotrophin independence (Vogelbaum et al., 1998). In embryonic sympathetic neurons grown under these conditions, there is evidence for a block in the apoptotic pathway just upstream of caspase activation close to the point at which Bax acts (Greenlund et al., 1995; Easton et al., 1997). To investigate the molecular mechanisms that underlie neurotrophin independence in adult neurons, we purified and cultured sympathetic neurons from the superior cervical ganglion (SCG) of adult rats. We show that PI-3 kinase/Akt activation and the expression of antiapoptotic members of the Bcl-2 family are required for sustaining the survival of these neurons in the absence of added neurotrophic factors.
Results
Mature sympathetic neurons survive in isolation
Although it has been reported that mature SCG neurons survive without added neurotrophic factors in dissociated cultures enriched for neurons (Orike et al., 2000), it is possible that the small number of nonneuronal cells present in these cultures might produce sufficient neurotrophic factors to sustain neuronal survival. To demonstrate conclusively that mature sympathetic neurons are capable of surviving independently of factors produced by other cells, 12-wk adult rat SCG neurons were plated at single cell density in 96-well plates. The great majority of these neurons (>75%) survived for at least 5 d in defined, serum-free medium without added neurotrophic factors, and their survival was not enhanced by coculturing them with nonneuronal cells in these wells (Fig. 1) . Having established that mature sympathetic neurons can survive autonomously and independently of trophic support from other cells, we embarked upon a series of experiments to elucidate the molecular basis of their survival independence.
Figure 1.
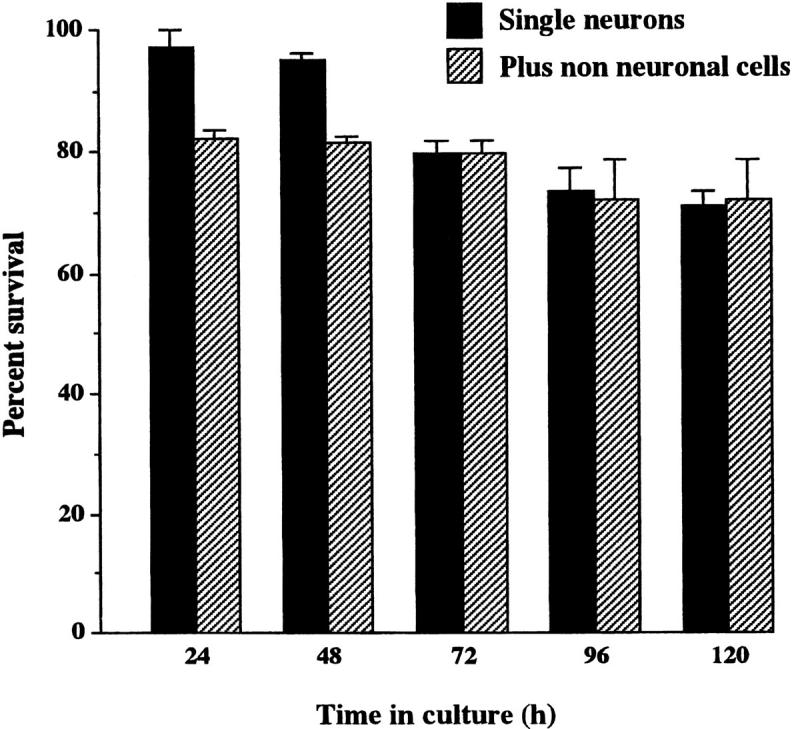
Survival of adult SCG neurons in culture. The neurons were grown in 96-well plates either as single cells in isolation (black bars) or single cells cocultured with one to four nonneuronal cells per well (hatched bars). The means and standard errors are shown for three experiments, with a total of 36 single neurons and 42 single neurons cocultured with nonneuronal cells.
PI 3-kinase activation is required for adult SCG neuron survival
We began our investigations of the survival independence of adult SCG neurons using pharmacological agents to inhibit signaling pathways that have been implicated in mediating the survival effects of growth factors. These and all subsequent experiments were carried out in low density–dissociated cultures of SCG neurons (<1,000 neurons per 60-mm petri dish) from which most nonneuronal cells were removed by differential sedimentation (>90% neurons). Addition of Wortmannin or LY294002 at concentrations shown previously to preferentially block PI 3-kinase activation (Okada et al., 1994, Vlahos et al., 1994) caused the rapid death of the majority of the neurons (Fig. 2) . Addition of PD98059 to the culture medium at concentrations shown previously to inhibit activation of MEK (Alessi et al., 1995) had little effect on neuronal survival, even when added repeatedly to the medium at daily intervals (Fig. 2). These results suggest that activation of PI 3-kinase is required for the neurotrophic factor–independent survival of adult SCG neurons, whereas MEK/MAP kinase signaling has no apparent role.
Figure 2.

Survival of adult SCG neurons cultured with inhibitors PI 3-kinase or MCK. The neurons were grown at low density in defined, serum-free medium alone (untreated) or containing 20 μM PD98059, 20 μM LY294002, or 20 μM Wortmannin. These reagents were added 3 h after plating when the neurons had become attached to the substratum. The number of neurons surviving at intervals after plating is expressed as a percentage of the initial number of neurons plated. The means and standard errors for the combined results of three separate experiments are shown.
To confirm the results of these pharmacological experiments, SCG neurons were injected with a plasmid that expresses Δp85, a dominant-negative form of the regulatory subunit of Class IA PI 3-kinase that is unable to bind to the catalytic p110 subunit, and thereby inhibits endogenous PI 3-kinase activity by sequestering upstream stimulatory proteins that bind to p85 (Dhand et al., 1994). Whereas neurons injected with an empty expression plasmid survived just as well as uninjected neurons (unpublished data), indicating that the injection procedure is not detrimental for the neurons, injection of the Δp85 expression plasmid caused the death of a substantial number of the neurons (Fig. 3) . Although the neurons did not die as fast as neurons treated with PI 3-kinase inhibitors (which may reflect the time taken to synthesize effective levels of Δp85), by 5 d, only 25% of the neurons injected with the Δp85 plasmid were surviving compared with >80% of control-injected neurons, suggesting that activation of Class IA PI 3-kinase is required for sustaining the survival of adult sympathetic neurons in defined medium lacking added neurotrophic factors.
Figure 3.

Survival of adult SCG neurons expressing inhibitors of PI 3-kinase. The neurons were injected with expression plasmids containing the Δp85 dominant-negative form of PI 3-kinase, Rukl, Rukm, a Ruk deletion mutant that lacks the proline-rich and cysteine-rich domains (RukΔ), or an empty plasmid (control plasmid). The neurons were initially grown for 12 h before injection, and the number of neurons surviving at intervals after injection is expressed as a percentage of the initial number of neurons injected. The means and standard errors for the combined results of three separate experiments are shown.
Further proof that PI 3-kinase activation is required for the survival of adult SCG neurons came from studying the effects of expressing a recently identified protein termed Rukl that interacts with the p85 regulatory subunit of Class IA PI 3-kinase and inhibits the lipid kinase activity of the holoenzyme (Gout et al., 2000). Injection of the Rukl expression plasmid into adult SCG neurons killed a substantial number of these neurons, leaving only 30% surviving 5 d after injection. In contrast, expression of either a shorter Ruk isoform (Rukm) that lacks two of the three SH3 domains found in Rukl (Gout et al., 2000) or a Rukl deletion mutant (RukΔ)that lacks the proline-rich and cysteine-rich domains, but retains all three SH3 domains, did not affect the survival of adult SCG neurons. 5 d after injection of the Rukm or RukΔ expression plasmids, the number of surviving neurons was no different from control-injected neurons (Fig. 3). These latter results are excellent controls for the microinjection experiments and for the Rukl expression studies as they show that expression of proteins that are closely related to a physiological inhibitor of Class IA PI 3-kinase has no detrimental effect on neuronal survival. Taken together, these results demonstrate that PI 3-kinase activation is required for the survival of adult SCG neurons.
Akt activation is required for adult SCG neuron survival
Because the downstream effector of PI 3-kinase, Akt, plays a key role in mediating the survival response of neurons to neurotrophins (Crowder and Freeman, 1998; Vaillant et al., 1999), we investigated whether Akt activation is required for the survival of adult SCG neurons. Injection of an expression plasmid that directs the synthesis of a kinase-inactive mutant of Akt that acts as a dominant-negative protein (Virdee et al., 1999) caused the death of a substantial number of adult SCG neurons. Over 70% of these neurons had died 5 d after injection with the dominant-negative Akt expression plasmid compared with 20% of control-injected neurons (Fig. 4) . The magnitude of neuronal death effected by expression of dominant-negative Akt was similar to that caused by expression of Δp85 or overexpression of Rukl. These results suggest that Akt is the main downstream effector of PI 3-kinase in sustaining the survival of adult SCG neurons in the absence of added neurotrophic factors.
Figure 4.

Survival of adult SCG neurons expressing dominant negative Akt. The neurons were injected with an expression plasmid containing the kinase-inactive mutant of PKB/Akt or with an empty plasmid (control plasmid). The neurons were initially grown for 12 h before injection, and the number of neurons surviving at intervals after injection is expressed as a percentage of the initial number of neurons injected. The means and standard errors for the combined results of three separate experiments are shown.
The demonstration that Akt plays a major role in mediating the PI 3-kinase survival signal in adult sympathetic neurons enabled us to carry out an additional control for the specificity of the pharmacological inhibition of PI 3-kinase by LY294002. Microinjecting a plasmid that expresses a constitutively active Akt protein (see Burgering and Coffer, 1995 for a description of the Gag-PKB protein) was able to rescue 73.2% of the neurons that would have otherwise died after 48-h incubation with 20 μM LY294002. The failure to rescue all neurons could be because Akt is not expressed at an optimal level.
Phospho-Akt is present in adult SCG neurons surviving without neurotrophic factors
To ascertain if the active, phosphorylated form of Akt is present in adult SCG neurons surviving without neurotrophic factors and whether this is dephosphorylated after inhibition of PI 3-kinase, we used immunocytochemistry to visualize Akt protein and phospho-Akt protein in SCG neurons grown with and without the PI 3-kinase inhibitor LY294002. Fig. 5 A shows that SCG neurons were clearly immunoreactive for phospho-Akt protein when grown at low density in defined medium alone. Although phospho-Akt immunoreactivity was clearly reduced by treatment with LY294002 for 45 min (Fig. 5G), expression of total Akt protein was little affected as revealed by the similar level of Akt immunofluorescence in the presence and absence of LY294002 (Fig. 5, B and C). There was no obvious change in the expression of either phospho-Akt immunoreactivity or total Akt protein in adult SCG neurons treated for 45 min with NGF (Fig. 5, A and E). No staining was observed when the primary antibodies were omitted from the staining protocol (Fig. 5 H), indicating that there was no detectable nonspecific staining. Taken together, these results suggest that Akt is constitutively active in adult SCG neurons surviving without neurotrophic factors and that PI 3-kinase is required for sustaining Akt activation.
Figure 5.

Micrographs of adult SCG neurons stained for Akt and phospho-Akt. The neurons were stained for Akt (A–C) or phospho-Akt (E–G) after being grown overnight and treated for 45 min with 10 ng/ml NGF (A and E), received no treatment (B and F) or were treated for 45 min with 20 μM LY294002 (C and G) before fixation. D and H show phase-contrast and epifluorescence micrographs of a neuron in a preparation from which the primary antiserum was omitted from the staining protocol, showing no detectable immunofluorescence. Bar, 10 μM.
Adult SCG neurons die by a caspase-dependent apoptotic mechanism after PI 3-kinase inhibition
Adult sympathetic neurons surviving without neurotrophic factors had smooth cell bodies, a smooth spherical nucleus, and unbroken short neurites (Fig. 6 A). Neurons grown without neurotrophins retained this appearance throughout the period of the study (up to 5 d in vitro). However, neurite outgrowth could be stimulated from these neurons after this prolonged period of culture without neurotrophins by adding NGF to the culture medium (unpublished data). This indicates that although NGF is not required for survival, it is capable of promoting neurite outgrowth from these neurons. A similar phenomenon has been observed for neonatal Bax-deficient sympathetic neurons that survive in culture without NGF, but display very poor neurite outgrowth unless NGF is added to the medium (Deckwerth et al., 1996).
Figure 6.

Micrographs of adult SCG neurons in culture. The neurons grown overnight and receiving no further treatment (A and C) or treated for 45 min with 20 μM LY294002 (B and D) are shown in phase-contrast optics (A and B) or stained with the fluorescent nuclear dye DAPI (C and D). (E). Bcl-xL staining in neurons injected 12 h earlier with an empty expression plasmid. (F). Bcl-xL staining in neurons injected 12 h earlier with a plasmid expressing antisense Bcl-xL RNA. Bar, 10 μM.
Neurons dying as a consequence of LY294002 treatment exhibited cell body condensation, neurite fragmentation, and chromatin condensation (Fig. 6, B and D) that is typical of neuronal apoptosis (Allsopp et al., 1993). To investigate whether caspase activation is required for neuronal death after LY294002 treatment, the caspase inhibitor Z-VAD-FMK (Fearnhead et al., 1995) was added to the culture medium to see if this would interfere with the cytotoxic effect of LY294002 and Wortmannin. Whereas Z-VAD-FMK did not affect the survival of neurons growing in defined medium alone (unpublished data), it largely prevented the cell death caused by LY294002 and Wortmannin (Fig. 7) , suggesting that adult SCG neurons die by a caspase-dependent mechanism after PI 3-kinase inhibition.
Figure 7.

Survival of adult sympathetic neurons following inhibition of PI 3-kinase and caspases. The neurons were grown at low density in defined, serum-free medium alone (untreated) or containing 20 μM LY294002, 20 μM Wortmannin, LY294002 plus 75 μM Z-VAD-FMK, or Wortmannin plus Z-VAD-FMK. The number of neurons surviving at intervals after plating is expressed as a percentage of the initial number of neurons plated. The means and standard errors for the combined results of three separate experiments are shown.
Bcl-xL and Bcl-2 expression is required for adult SCG neuron survival
Because members of the Bcl-2 family of proteins play key roles in controlling caspase activation, we carried out additional experiments to assess the importance of different members of this family in regulating adult sympathetic neuron survival. To ascertain the importance of antiapoptotic members of the Bcl-2 family in sustaining the survival of adult SCG neurons, we injected plasmids expressing specific antisense RNAs to interfere with the synthesis of these proteins. We also injected separate sets of neurons with an empty expression plasmid to control for the injection procedure and studied the effect of overexpressing antiapoptotic proteins. There was no significant difference in the survival of control-injected neurons compared with uninjected neurons (unpublished data), indicating that the microinjection procedure did not affect neuronal survival. Overexpression of the antiapoptotic proteins Bcl-xL or Bcl-2 had little effect on survival. Whereas neurons overexpressing Bcl-xL survived slightly better than control-injected neurons, the survival of Bcl-2–overexpressing neurons was not significantly different from control-injected neurons (Fig. 8) . In contrast, injection of plasmids containing Bcl-xL or Bcl-2 in the reverse orientation caused a marked reduction in neuronal survival. Expression of antisense Bcl-xL had the most dramatic effect, with over three quarters of the neurons dying within the first 24 h after injection, and by 5 d, <10% of the neurons were surviving. Expression of antisense Bcl-2 reduced the number of surviving neurons to ∼25% after 5 d (Fig. 8). To confirm that adult SCG neurons normally express Bcl-xL protein and that expression of this protein is reduced by antisense Bcl-xL RNA, we used immunocytochemistry to study Bcl-xL expression after injection of the antisense Bcl-xL plasmid. Whereas control-injected neurons exhibited intense Bcl-xL immunofluorescence (Fig. 6 E), neurons injected with the antisense Bcl-xL plasmid were either unlabeled by the Bcl-xL antiserum or exhibited negligible Bcl-xL immunofluorescence 12 h after injection (Fig. 6 F). Taken together, these results suggest that the endogenous expression of Bcl-xL and, to a lesser, extent Bcl-2 is required for the survival of adult SCG neurons.
Figure 8.

Survival of adult sympathetic neurons after manipulating expression of Bcl-xL and Bcl-2. The neurons were injected with expression plasmids containing Bcl-xL and Bcl-2 cDNAs in the sense or antisense orientations, Bcl-xS cDNA in the sense orientation or with an empty plasmid (control plasmid). The neurons were initially grown for 12 h before injection, and the number of neurons surviving at intervals after injection is expressed as a percentage of the initial number of neurons injected. The means and standard errors for the combined results of three separate experiments are shown.
In addition to using antisense Bcl-xL expression plasmid to interfere with the endogenous synthesis of the Bcl-xL protein, we also microinjected a plasmid that expresses a proapoptotic isoform of Bcl-x, Bcl-xS which lacks two of the four BH domains present in Bcl-xL (Boise et al., 1993). Expression of Bcl-xS also caused the rapid death of a large number of SCG neurons, leaving only 20% surviving 5 d after injection (Fig. 8).
Overexpression of Bad or Bax kills adult SCG neurons
In addition to interfering with the synthesis of antiapoptotic members of the Bcl-2 family to ascertain the importance of these proteins in sustaining the survival of adult SCG neurons, we also overexpressed proapoptotic members of this family to see if this would compromise the survival of these neurons. The most marked effect on survival was observed by overexpressing Bad. Injection of a Bad expression plasmid caused the rapid death of the majority of adult SCG neurons, leaving <20% surviving 5 d after injection (Fig. 9) . Injection of a Bax expression plasmid caused much less neuronal death, leaving 40% surviving 5 d after injection (Fig. 9).
Figure 9.

Survival of adult sympathetic neurons overexpressing Bad or Bax. The neurons were injected with Bad or Bax expression plasmids or with an empty plasmid (control plasmid). The neurons were initially grown for 12 h before injection, and the number of neurons surviving at intervals after injection is expressed as a percentage of the initial number of neurons injected. The means and standard errors for the combined results of three separate experiments are shown.
Discussion
We have shown that adult sympathetic neurons are able to survive for an extended period in culture in isolation without the need for trophic support from any other cells. Several complementary experimental observations have clearly demonstrated that the survival of these neurons depends on PI 3-kinase activity and on activation of the downstream effector of PI 3-kinase, the serine/threonine protein kinase Akt. These neurons are killed by pharmacological inhibition of PI 3-kinase with LY294002 or Wortmannin, by the expression of a dominant-negative Class IA PI 3-kinase p85 regulatory subunit, by a dominant-negative Akt protein, or by overexpressing Rukl, which is a natural inhibitor of Class IA PI 3-kinase. These neurons do not die as a nonspecific consequence of plasmid microinjection or the expression of new protein because overexpressing proteins that are very similar to Rukl (Rukm and a Rukl deletion mutant that retains all three SH3 domains) is not detrimental. Furthermore, phospho-Akt staining suggests that Akt is constitutively active in these neurons.
Although the role of PI 3-kinase in mediating the survival-promoting action of NGF on embryonic and neonatal sympathetic neurons is controversial (Philpott et al., 1997; Crowder and Freeman, 1998; Mazzoni et al., 1999; Vaillant et al., 1999; Virdee et al., 1999; Tsui-Pierchala et al., 2000), studies of several other kinds of neurons and many other cell types have shown that PI 3-kinase and serine/threonine kinase Akt/PKB play major roles in mediating the survival response of many different kinds of cells to a variety of growth factors (Datta et al., 1999). However, our study provides the first evidence that activation of PI 3-kinase and Akt is required to prevent the death of cells that survive independently of extracellular growth factors.
Even though adult SCG neurons survive as single cells in serum-free medium, nonetheless, it is possible that they could be sustained by neurotrophic factors that they produce themselves. Autocrine action of neurotrophic factors has been described previously for BDNF acting on embryonic and adult sensory neurons (Wright et al., 1992; Acheson et al., 1995) and for HGF acting on sympathetic neuroblasts (Maina et al., 1998). Although autocrine trophic support of adult sympathetic neurons is entirely possible, preliminary data suggest that several of the likely candidates are unlikely to be involved. Adult SCG neurons are not killed by the inhibitor of Trk receptor tyrosine kinases K252a (Orike et al., 2000) or by function-blocking antibodies to GFRα-3, a key component of the artemin receptor (Andres et al., 2001) or HGF (N. Orike, J. Thompson, and A.M. Davies, unpublished data). These findings suggest that neither NGF and NT-3, which promote the survival of developing sympathetic neurons (Levi-Montalcini, 1987; Crowley et al., 1994; Zhou and Rush, 1995; Wyatt et al., 1997; Francis et al., 1999), HGF, which enhances the survival of sympathetic neuroblasts (Maina et al., 1998), nor artemin, which is important for the survival of a SCG neurons during the postnatal period (Nishino et al., 1999; Andres et al., 2001), are required for the survival of adult SCG neurons. Furthermore, the activation of PI 3-kinase by NGF in SCG neurons is thought to occur predominantly by direct activation of the p110 catalytic subunit by Ras (Rodriguez-Viciana et al., 1994; Nobes et al., 1996; Markus et al., 1997; Mazzoni et al., 1999; Xue et al., 2000). Yet we have shown that inhibiting the function of the p85 regulatory subunit of Class IA PI 3-kinase by expressing a dominant-negative protein or overexpressing Rukl kills adult SCG neurons, indicating that this regulatory subunit is crucial for maintaining the activity of the catalytic subunit at the level required to sustain the survival of adult SCG neurons.
Our demonstration that PI 3-kinase activity is required for the survival of adult rat sympathetic neurons in the absence of neurotrophic factors differs from the findings of studies of embryonic sympathetic or sensory neurons that attained neurotrophic factor independence in culture after prolonged incubation with NGF. Dorsal root ganglion neurons from E15 rat embryos that were maintained for 3 wk in the presence of NGF survived after NGF withdrawal and were not killed by inhibiting PI 3-kinase activation with LY294002, but were killed by LY294002 treatment at earlier time points before the acquisition of NGF independence (Vogelbaum et al., 1998). E21 rat SCG neurons maintained for 3 wk with NGF also survived after NGF deprivation, but exhibited a rapid decrease in MAP kinase activation and early changes in gene expression after NGF deprivation that are typical of NGF withdrawal before the acquisition of NGF independence. However, like Bax-deficient embryonic neurons that survive after NGF deprivation, embryonic SCG neurons maintained in long-term culture with NGF do not exhibit c-fos induction after NGF deprivation. These observations, together with the finding that overexpression of Bax in these neurons restored their survival dependence on NGF, suggest that the apoptotic pathway was blocked at a point just upstream of caspase activation close to the point at which Bax acts (Greenlund et al., 1995; Easton et al., 1997). Thus, although embryonic sensory and sympathetic neurons lose dependence on NGF after being cultured with this factor for several weeks, the mechanism of neurotrophin independence appears to be different from that of adult sympathetic neurons that rely on PI 3-kinase activity for survival. However, one potential caveat of our study is that we have studied the role of PI 3-kinase shortly after plating when a stress response to axotomy might make them more susceptible to PI 3-kinase inhibition. In future work, it will be important to address the relevance of our in vitro observations to the role of PI 3-kinase in sustaining the survival of adult sympathetic neurons in vivo.
Another signaling pathway that has been implicated in neuronal survival under certain circumstances is the MEK/MAP kinase pathway. Although this Ras-activated pathway plays either no role or only a minor role in mediating the survival response of developing neurons to NGF (Creedon et al., 1996; Virdee and Tolkovsky, 1996; Klesse and Parada, 1998; Mazzoni et al., 1999; Xue et al., 2000), it plays a major role in BDNF neuroprotection against camptothecin (Hetman et al., 1999) and is capable of reducing p53-dependent apoptosis induced by cytosine arabinoside in sympathetic neurons (Anderson and Tolkovsky, 1999). Our demonstration that the MEK inhibitor PD98059 does not kill adult SCG neurons suggests that the MEK/MAP kinase activation is not required for sustaining the survival of these neurons.
Considerable evidence indicates that members of the Bcl-2 play a key role in regulating neuronal survival during development. Overexpression of Bcl-2 or Bcl-xL promote the survival of developing neurons in vitro and in vivo (Garcia et al., 1992; Allsopp et al., 1993; Martinou et al., 1994; Farlie et al., 1995; Gonzalez-Garcia et al., 1995; Middleton et al., 1996; Parsadanian et al., 1998), whereas reduction or elimination of endogenous Bcl-2 or Bcl-xL reduces neuronal survival (Allsopp et al., 1995; Motoyama et al., 1995; Michaelidis et al., 1996; Piñón et al., 1997). In contrast, Bax overexpression accelerates neuronal death after neurotrophin withdrawal (Vekrellis et al., 1997; Martinou et al., 1998), and reduction or elimination of endogenous Bax promotes the survival of neurons without neurotrophic factors in culture and prevents their death in vivo (Deckwerth et al., 1996; Gillardon et al., 1996; Miller et al., 1997; White et al., 1998; Bar-Peled et al., 1999). Our studies of manipulating the expression of Bcl-2 family proteins in adult sympathetic neurons have likewise shown that these proteins also play an important role in regulating the survival of mature sympathetic neurons. Expression of the proapoptotic proteins Bad, Bax, and Bcl-xS killed these neurons as did antisense RNA to the antiapoptotic proteins Bcl-xL and Bcl-2. A link between PI 3-kinase/Akt signaling and the function of these proteins has come from the demonstration that activated Akt phosphorylates Bad, resulting in its sequestration by cytosolic 14-3-3 proteins (Datta et al., 1997; del Peso et al., 1997). Because Bad brings about apoptosis by binding to and inhibiting the antiapoptotic actions of Bcl-xL, the sequestration of Bad in the cytosol by 14-3-3 proteins results in enhanced survival.
In summary, we have shown that the survival of adult sympathetic neurons is dependent on PI 3-kinase/Akt signaling and the expression of the antiapoptotic proteins Bcl-xL and Bcl-2. In future work, it will be important to ascertain which proteins interact with the p85 regulatory subunit of Class IA PI 3-kinase in adult sympathetic neurons to maintain the activity of the holoenzyme and understand what brings about the change in PI 3-kinase regulation between postnatal animals and the adult.
Materials and methods
Culture and microinjection of sympathetic neurons
Dissociated cultures of purified SCG neurons from 12-wk-old Sprague-Dawley rats were set were grown in defined, serum-free medium (Orike et al., 2000). In brief, SCG were cut into small pieces and incubated at 37°C for 30 min with 400 IU/ml collagenase class 2 (C9891; Sigma-Aldrich) followed by 40 min with 1 mg/ml trypsin (T2271; Sigma-Aldrich). The enzymes were dissolved in HBSS containing 6 mg/ml BSA (A4716; Sigma-Aldrich). The ganglion fragments were dissociated by gentle trituration using a fire-polished glass pipette, and the neurons were purified free of nonneuronal cells by differential sedimentation (Davies, 1986). For microinjection studies, the neurons (>90% pure) were plated in polyornithine/laminin-coated 60-mm-diam tissue culture petri dishes (Davies et al., 1995) at a density of 500 to 1,000 neurons per dish. For single cell cultures, the neurons were plated in 96-well dishes. The cultures were incubated at 37°C in a humidified 4.5% CO2 incubator in defined medium consisting of Ham's F14 supplemented with 2 mM glutamine, 0.35% BSA (Pathocyte-4; ICN Biomedicals), 60 ng/ml progesterone, 16 μg/ml putrescine, 400 ng/ml l-thyroxine, 38 ng/ml sodium selenite, and 340 ng/ml triiodothyronine.
The neurons were initially grown overnight and were microinjected intranuclearly with the aid of a manually operated Narishige micromanipulator (Allsopp et al., 1993) with expression plasmids encoding wild-type or mutated signaling proteins or members of the Bcl-2 family in the sense or antisense orientation. Expression plasmids without a cDNA insert were used to control for any nonspecific effects of the injection procedure. The expression plasmids were diluted in 100 mM potassium phosphate buffer, pH 7, to a concentration of 100 ng/ml and filtered through a 0.22-μm filter before injection. All neurons within a specific grid square marked on the inside of the culture dish were pressure-injected. Intranuclear injection resulted in >95% viability assessed 1 h after injection. The number of surviving neurons was counted at 12 or 24 hourly intervals after injection, and is expressed as a percentage of the number injected in the grid square. About 150 neurons were injected for each experimental condition and followed longitudinally in each experiment.
Live and dead neurons were clearly distinguished by the morphology when examined by phase-contrast microscopy. Live neurons had large, smooth, intact cell bodies and smooth, unbroken neurites. Dead neurons rapidly disintegrated, leaving cell body ghosts comprised of cellular debris and fragmented neurites, which are appearances that are typical of neuronal apoptosis in culture (Allsopp et al., 1993). In addition to these characteristic appearances, we also used DAPI to stain nuclear chromatin to determine if the chromatin condensation, which is typical of apoptosis, occurs in adult sympathetic neurons dying as a consequence of inhibiting PI-3 kinase.
Plasmids and pharmacological reagents
A variety of expression plasmids were used to express wild-type or mutated proteins or antisense RNA in cultured sympathetic neurons. cDNAs encoding members of the Bcl-2 family (Bcl-2, Bcl-xL, Bcl-xS, Bad, and Bax) were cloned into the pSFFV vector in the sense and antisense orientations. The pCMV vector was used to express the kinase-inactive mutant of PKB/Akt, Rukl and Rukm, and the pSG5 vector was used to express the Δp85 dominant-negative form of PI 3-kinase. The PI 3-kinase inhibitors LY294002 and Wortmannin, the MEK inhibitor PD98059, and the caspase inhibitor Z-VAD-FMK were obtained from Calbiochem.
Immunocytochemistry
Cultures of adult SCG neurons were fixed with fresh 4% paraformaldehyde for 15 min, washed in TBS with .1% Triton X-100 (TBT) and blocked with TBT plus 10% goat serum. They were incubated with primary antibody (rabbit polyclonals from Santa Cruz Biotechnology, Inc. and New England Biolabs, Inc.) at a dilution of 1:100 overnight at 4°C, washed in TBT, and incubated with FITC-labeled goat anti–rabbit antiserum (Sigma-Aldrich) at a dilution of 1:500 at room temperature for 1 h. The cultures were washed, mounted beneath coverslips, and viewed with an Axioskop fluorescence microscope.
Acknowledgments
We thank Julian Downward (Imperial Cancer Research Fund, London, UK) and Bart Vanhaesebroeck (Ludwig Institute for Cancer Research, London, UK) for the Δp85 and Gag-PKB cDNAs, Aviva Tolkovsky (Cambridge University, UK), and Brian Hemmings (Friedrich Miescher Institute, Basel, Switzerland) for the kinase-inactive mutant of PKB/Akt, Gabriel Nunez (University of Michigan Medical Center, Ann Arbor, MI) for the Bcl-2, Bcl-xL, Bcl-xS, Bad and Bax cDNAs, Jane Thompson for assistance in setting up cultures and David Kaplan for critical reading of the manuscript.
This work was supported by grants from the Wellcome Trust (to V. Buchman, T. Cowen, and A.M. Davies).
N. Orike and G. Middleton contributed equally to this work.
The present address of N. Orike is Montreal Neurological Institute, Montreal, Quebec, H3A 2B4, Canada.
Footnotes
Abbreviations used in this paper: MAP, mitogen-activated protein; MEK, mitogen-activated protein kinase kinase; PI, phosphoinositide; SCG, superior cervical ganglion.
References
- Acheson, A., J.C. Conover, J.P. Fandl, T.M. DeChlara, M. Russell, A. Thadani, S.P. Squinto, G.D. Yancopoulos, and R.M. Lindsay. 1995. A BDNF autocrine loop in adult sensory neurons prevents cell death. Nature. 374:450–453. [DOI] [PubMed] [Google Scholar]
- Alessi, D.R., A. Cuenda, P. Cohen, D.T. Dudley, and A.R. Saltiel. 1995. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J. Biol. Chem. 270:27489–27494. [DOI] [PubMed] [Google Scholar]
- Allsopp, T.E., S. Wyatt, H.F. Paterson, and A.M. Davies. 1993. The proto-oncogene bcl-2 can selectively rescue neurotrophic factor-dependent neurons from apoptosis. Cell. 73:295–307. [DOI] [PubMed] [Google Scholar]
- Allsopp, T.E., S. Kiselev, S. Wyatt, and A.M. Davies. 1995. Role of Bcl-2 expression in the BDNF survival response. Eur. J. Neurosci. 7:1266–1272. [DOI] [PubMed] [Google Scholar]
- Anderson, C.N.G., and A.M. Tolkovsky. 1999. A role for MAPK/ERK in sympathetic neuron survival: protection against a p53-dependent, JNK-independent induction of apoptosis by cytosine arabinoside. J. Neurosci. 19:664–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andres, R., A. Forgie, S. Wyatt, Q. Chen, F.J. de Sauvage, and A.M. Davies. 2001. Multiple effects of artemin on sympathetic neuron generation, survival and growth. Development. In press. [DOI] [PubMed] [Google Scholar]
- Bar-Peled, O., M. Knudson, S.J. Korsmeyer, and J.D. Rothstein. 1999. Motor neuron degeneration is attenuated in bax-deficient neurons in vitro. J. Neurosci. Res. 55:542–556. [DOI] [PubMed] [Google Scholar]
- Belliveau, D.J., I. Krivko, J. Kohn, C. Lachance, C. Pozniak, D. Rusakov, D. Kaplan, and F.D. Miller. 1997. NGF and neurotrophin-3 both activate TrkA on sympathetic neurons but differentially regulate survival and neuritogenesis. J. Cell Biol. 136:375–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boise, L.H., G.M. Gonzalez, C.E. Postema, L. Ding, T. Lindsten, L.A. Turka, X. Mao, G. Nunez, and C.B. Thompson. 1993. bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell. 74:597–608. [DOI] [PubMed] [Google Scholar]
- Brunet, A., A. Bonni, M.J. Zigmond, M.Z. Lin, P. Juo, L.S. Hu, M.J. Anderson, K.C. Arden, J. Blenis, and M.E. Greenberg. 1999. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 96:857–868. [DOI] [PubMed] [Google Scholar]
- Burgering, B.M., and P.J. Coffer. 1995. Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature. 376:599–602. [DOI] [PubMed] [Google Scholar]
- Creedon, D.J., E.M. Johnson, and J.C. Lawrence. 1996. Mitogen-activated protein kinase-independent pathways mediate the effects of nerve growth factor and cAMP on neuronal survival. J. Biol. Chem. 271:20713–20718. [DOI] [PubMed] [Google Scholar]
- Crowder, R.J., and R.S. Freeman. 1998. Phosphatidylinositol 3-kinase and Akt protein kinase are necessary and sufficient for the survival of nerve growth factor-dependent sympathetic neurons. J. Neurosci. 18:2933–2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley, C., S.D. Spencer, M.C. Nishimura, K.S. Chen, S. Pitts-Meek, M.P. Armanini, L.H. Ling, S.B. McMahon, D.L. Shelton, A.D. Levinson, and H.S. Phillips. 1994. Mice lacking nerve growth factor display perinatal loss of sensory and sympathetic neurons yet develop basal forebrain cholinergic neurons. Cell. 76:1001–1011. [DOI] [PubMed] [Google Scholar]
- Datta, S.R., A. Brunet, and M.E. Greenberg. 1999. Cellular survival: a play in three Akts. Genes Dev. 13:2905–2927. [DOI] [PubMed] [Google Scholar]
- Datta, S.R., H. Dudek, X. Tao, S. Masters, H. Fu, Y. Gotoh, and M.E. Greenberg. 1997. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 91:231–241. [DOI] [PubMed] [Google Scholar]
- Davies, A.M. 1986. The survival and growth of embryonic proprioceptive neurons is promoted by a factor present in skeletal muscle. Dev. Biol. 115:56–67. [DOI] [PubMed] [Google Scholar]
- Davies, A.M., L. Minichiello, and R. Klein. 1995. Developmental changes in NT3 signalling via TrkA and TrkB in embryonic neurons. EMBO J. 14:4482–4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deckwerth, T.L., J.L. Elliott, C.M. Knudson, E.M. Johnson, Jr., W.D. Snider, and S.J. Korsmeyer. 1996. BAX is required for neuronal death after trophic factor deprivation and during development. Neuron. 17:401–411. [DOI] [PubMed] [Google Scholar]
- del Peso, L., M. Gonzalez-Garcia, C. Page, R. Herrera, and G. Nunez. 1997. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 278:687–689. [DOI] [PubMed] [Google Scholar]
- Dhand, R., K. Hara, I. Hiles, B. Bax, I. Gout, G. Panayotou, M.J. Fry, K. Yonezawa, M. Kasuga, and M.D. Waterfield. 1994. PI 3-kinase: structural and functional analysis of intersubunit interactions. EMBO J. 13:511–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du, K., and M. Montminy. 1998. CREB is a regulatory target for the protein kinase Akt/PKB. J. Biol. Chem. 273:32377–32379. [DOI] [PubMed] [Google Scholar]
- Easton, R.M., T.L. Deckwerth, A.S. Parsadanian, and E.M. Johnson, Jr. 1997. Analysis of the mechanism of loss of trophic factor dependence associated with neuronal maturation: a phenotype indistinguishable from Bax deletion. J. Neurosci. 17:9656–9666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farlie, P.G., R. Dringen, S.M. Rees, G. Kannourakis, and O. Bernard. 1995. bcl-2 transgene expression can protect neurons against developmental and induced cell death. Proc. Natl. Acad. Sci. USA. 92:4397–4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearnhead, H.O., D. Dinsdale, and G.M. Cohen. 1995. An interleukin-1 beta-converting enzyme-like protease is a common mediator of apoptosis in thymocytes. FEBS Lett. 375:283–288. [DOI] [PubMed] [Google Scholar]
- Francis, N., I. Farinas, C. Brennan, K. Rivas-Plata, C. Backus, L. Reichardt, and S. Landis. 1999. NT-3, like NGF, is required for survival of sympathetic neurons, but not their precursors. Dev. Biol. 210:411–427. [DOI] [PubMed] [Google Scholar]
- Garcia, I., I. Martinou, Y. Tsujimoto, and J.C. Martinou. 1992. Prevention of programmed cell death of sympathetic neurons by the bcl-2 proto-oncogene. Science. 258:302–304. [DOI] [PubMed] [Google Scholar]
- Gillardon, F., M. Zimmermann, E. Uhlmann, S. Krajewski, J.C. Reed, and L. Klimaschewski. 1996. Antisense oligodeoxynucleotides to bax mRNA promote survival of rat sympathetic neurons in culture. J. Neurosci. Res. 43:726–734. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Garcia, M., I. Garcia, L. Ding, S. O'Shea, L.H. Boise, C.B. Thompson, and G. Nunez. 1995. bcl-x is expressed in embryonic and postnatal neural tissues and functions to prevent neuronal cell death. Proc. Natl. Acad. Sci. USA. 92:4304–4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gout, I., G. Middleton, J. Adu, N.N. Ninkina, L.B. Drobot, V. Filonenko, G. Matsuka, A.M. Davies, M. Waterfield, and V.L. Buchman. 2000. Negative regulation of PI 3-kinase by Ruk, a novel adaptor protein. EMBO J. 19:4015–4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenlund, L.J., S.J. Korsmeyer, and E.M. Johnson. 1995. Role of BCL-2 in the survival and function of developing and mature sympathetic neurons. Neuron. 15:649–661. [DOI] [PubMed] [Google Scholar]
- Hetman, M., K. Kanning, J.E. Cavanaugh, and Z. Xia. 1999. Neuroprotection by brain-derived neurotrophic factor is mediated by extracellular signal-regulated kinase and phosphatidylinositol 3-kinase. J. Biol. Chem. 274:22569–22580. [DOI] [PubMed] [Google Scholar]
- Holgado-Madruga, M., D.K. Moscatello, D.R. Emlet, R. Dieterich, and A.J. Wong. 1997. Grb2-associated binder-1 mediates phosphatidylinositol 3-kinase activation and the promotion of cell survival by nerve growth factor. Proc. Natl. Acad. Sci. USA. 94:12419–12424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane, L.P., V.S. Shapiro, D. Stokoe, and A. Weiss. 1999. Induction of NF-kappaB by the Akt/PKB kinase. Curr. Biol. 9:601–604. [DOI] [PubMed] [Google Scholar]
- Kaplan, D.R., and F.D. Miller. 1997. Signal transduction by the neurotrophin receptors. Curr. Opin. Cell Biol. 9:213–221. [DOI] [PubMed] [Google Scholar]
- Kharbanda, S., P. Pandey, L. Schofield, S. Israels, R. Roncinske, K. Yoshida, A. Bharti, Z.M. Yuan, S. Saxena, R. Weichselbaum, C. Nalin, and D. Kufe. 1997. Role for Bcl-xL as an inhibitor of cytosolic cytochrome c accumulation in DNA damage-induced apoptosis. Proc. Natl. Acad. Sci. USA. 94:6939–6942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klesse, L.J., and L.F. Parada. 1998. p21 ras and phosphatidylinositol-3 kinase are required for survival of wild-type and NF1 mutant sensory neurons. J. Neurosci. 18:10420–10428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluck, R.M., E. Bossy-Wetzel, D.R. Green, and D.D. Newmeyer. 1997. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 275:1132–1136. [DOI] [PubMed] [Google Scholar]
- Korsmeyer, S.J. 1999. BCL-2 gene family and the regulation of programmed cell death. Cancer Res. 59:1693s–1700s. [PubMed] [Google Scholar]
- Krebs, J.F., R.C. Armstrong, A. Srinivasan, T. Aja, A.M. Wong, A. Aboy, R. Sayers, B. Pham, T. Vu, K. Hoang, et al. 1999. Activation of membrane-associated procaspase-3 is regulated by Bcl-2. J. Cell Biol. 144:915–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuruvilla, R., H. Ye, and D.D. Ginty. 2000. Spatially and functionally distinct roles of the PI3-K effector pathway during NGF signaling in sympathetic neurons. Neuron. 27:499–512 [DOI] [PubMed] [Google Scholar]
- Le-Niculescu, H., E. Bonfoco, Y. Kasuya, F.X. Claret, D.R. Green, and M. Karin. 1999. Withdrawal of survival factors results in activation of the JNK pathway in neuronal cells leading to Fas ligand induction and cell death. Mol. Cell. Biol. 19:751–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi-Montalcini, R. 1987. The nerve growth factor 35 years later. Science. 237:1154–1162. [DOI] [PubMed] [Google Scholar]
- Li, P., D. Nijhawan, I. Budihardjo, S.M. Srinivasula, M. Ahmad, E.S. Alnemri, and X. Wang. 1997. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 91:479–489. [DOI] [PubMed] [Google Scholar]
- Maina, F., M.C. Hilton, R. Andres, S. Wyatt, R. Klein, and A.M. Davies. 1998. Multiple roles for hepatocyte growth factor in sympathetic neuron development. Neuron. 20:835–846. [DOI] [PubMed] [Google Scholar]
- Markus, A., A. von Holst, H. Rohrer, and R. Heumann. 1997. NGF-mediated survival depends on p21ras in chick sympathetic neurons from the superior cervical but not from lumbosacral ganglia. Dev. Biol. 191:306–310. [DOI] [PubMed] [Google Scholar]
- Martinou, I., M. Missotten, P.A. Fernandez, R. Sadoul, and J.C. Martinou. 1998. Bax and Bak proteins require caspase activity to trigger apoptosis in sympathetic neurons. Neuroreport. 9:15–19. [DOI] [PubMed] [Google Scholar]
- Martinou, J.C., M. Dubois-Dauphin, J.K. Staple, I. Rodriguez, H. Frankowsky, M. Missotten, P. Albertini, D. Talabot, S. Catsicas, C. Pietra, and J. Huarte. 1994. Overexpression of bcl-2 in transgenic mice protects neurons from naturally occurring cell death and experimental ischaemia. Neuron. 13:1017–1030. [DOI] [PubMed] [Google Scholar]
- Mazzoni, I.E., F.A. Said, R. Aloyz, F.D. Miller, and D. Kaplan. 1999. Ras regulates sympathetic neuron survival by suppressing the p53-mediated cell death pathway. J. Neurosci. 19:9716–9727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelidis, T.M., M. Sendtner, J.D. Cooper, M.S. Airaksinen, B. Holtmann, M. Meyer, and H. Thoenen. 1996. Inactivation of bcl-2 results in progressive degeneration of motoneurons, sympathetic neurons and sensory neurons during the early postnatal development. Neuron. 17:75–89. [DOI] [PubMed] [Google Scholar]
- Middleton, G., G. Nunez, and A.M. Davies. 1996. Bax promotes neuronal survival and antogonises the survival effects of trophic factors. Development. 122:695–701. [DOI] [PubMed] [Google Scholar]
- Miller, T.M., K.L. Moulder, C.M. Knudson, D.J. Creedon, M. Deshmukh, S.J. Korsmeyer, and E.M. Johnson, Jr. 1997. Bax deletion further orders the cell death pathway in cerebellar granule cells and suggests a caspase-independent pathway to cell death. J. Cell Biol. 139:205–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motoyama, N., F. Wang, K.A. Roth, H. Sawa, K. Nakayama, K. Nakayama, I. Negishi, S. Senju, Q. Zhang, S. Fujii, and D.Y. Loh. 1995. Massive cell death of immature hematopoietic cells and neurons in Bcl-x-deficient mice. Science. 267:1506–1510. [DOI] [PubMed] [Google Scholar]
- Nishino, J., K. Mochida, Y. Ohfuji, T. Shimazaki, C. Meno, S. Ohishi, Y. Matsuda, H. Fujii, Y. Saijoh, and H. Hamada. 1999. GFR alpha3, a component of the artemin receptor, is required for migration and survival of the superior cervical ganglion. Neuron. 23:725–736. [DOI] [PubMed] [Google Scholar]
- Nobes, C.D., J.B. Reppas, A. Markus, and A.M. Tolkovsky. 1996. Active p21Ras is sufficient for rescue of NGF-dependent rat sympathetic neurons. Neuroscience. 70:1067–1079. [DOI] [PubMed] [Google Scholar]
- Okada, T., Y. Kawano, T. Sakakibara, O. Hazeki, and M. Ui. 1994. Essential role of phosphatidylinositol 3-kinase in insulin-induced glucose transport and antilipolysis in rat adipocytes. Studies with a selective inhibitor wortmannin. J. Biol. Chem. 269:3568–3573. [PubMed] [Google Scholar]
- Orike, N., C. Thrasivoulou, A. Wrigley, and T. Cowen. 2000. Differential regulation of survival and growth in adult sympathetic neurons: an in vitro study of neurotrophin responsiveness. J. Neurobiol. 47:295–305. [DOI] [PubMed] [Google Scholar]
- Ozes, O.N., L.D. Mayo, J.A. Gustin, S.R. Pfeffer, L.M. Pfeffer, and D.B. Donner. 1999. NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature. 401:82–85. [DOI] [PubMed] [Google Scholar]
- Parsadanian, A.S., Y. Cheng, C.R. Keller-Peck, D.M. Holtzman, and W.D. Snider. 1998. Bcl-xL is an antiapoptotic regulator for postnatal CNS neurons. J. Neurosci. 18:1009–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philpott, K.L., M.J. McCarthy, A. Klippel, and L.L. Rubin. 1997. Activated phosphatidylinositol 3-kinase and Akt kinase promote survival of superior cervical neurons. J. Cell Biol. 139:809–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piñón, L.G.P., G. Middleton, and A.M. Davies. 1997. Bcl-2 is required for cranial sensory neuron survival at defined stages of embryonic development. Development. 124:4173–4178. [DOI] [PubMed] [Google Scholar]
- Qin, H., S.M. Srinivasula, G. Wu, T. Fernandes-Alnemri, E.S. Alnemri, and Y. Shi. 1999. Structural basis of procaspase-9 recruitment by the apoptotic protease-activating factor 1. Nature. 399:549–557. [DOI] [PubMed] [Google Scholar]
- Raoul, C., C.E. Henderson, and B. Pettmann. 1999. Programmed cell death of embryonic motoneurons triggered through the Fas death receptor. J. Cell Biol. 147:1049–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccio, A., S. Ahn, C.M. Davenport, J.A. Blendy, and D.D. Ginty. 1999. Mediation by a CREB family transcription factor of NGF-dependent survival of sympathetic neurons. Science. 286:2358–2361. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Viciana, P., P.H. Warne, R. Dhand, B. Vanhaesebroeck, I. Gout, M.J. Fry, M.D. Waterfield, and J. Downward. 1994. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature. 370:527–532. [DOI] [PubMed] [Google Scholar]
- Shields, J.M., K. Pruitt, A. McFall, A. Shaub, and C.J. Der. 2000. Understanding Ras: “it ain't over 'til it's over.” Trends Cell Biol. 10:147–154. [DOI] [PubMed] [Google Scholar]
- Shimizu, S., M. Narita, and Y. Tsujimoto. 1999. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature. 399:483–487. [DOI] [PubMed] [Google Scholar]
- Tsui-Pierchala, B.A., G.V. Putcha, E.M. Johnson, Jr. 2000. Phosphatidylinositol 3-kinase is required for the trophic, but not the survival-promoting, actions of NGF on sympathetic neurons. J. Neurosci. 20:7228–7237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaillant, A.R., I. Mazzoni, C. Tudan, M. Boudreau, D.R. Kaplan, and F.D. Miller. 1999. Depolarization and neurotrophins converge on the phosphatidylinositol 3-kinase-Akt pathway to synergistically regulate neuronal survival. J. Cell Biol. 146:955–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaux, D.L., and S.J. Korsmeyer. 1999. Cell death in development. Cell. 96:245–254. [DOI] [PubMed] [Google Scholar]
- Vekrellis, K., M.J. McCarthy, A. Watson, J. Whitfield, L.L. Rubin, and J. Ham. 1997. Bax promotes neuronal cell death and is downregulated during the development of the nervous system. Development. 124:1239–1249. [DOI] [PubMed] [Google Scholar]
- Virdee, K., and A.M. Tolkovsky. 1996. Inhibition of p42 and p44 mitogen-activated protein kinase activity by PD98059 does not suppress nerve growth factor-induced survival of sympathetic neurones. J. Neurochem. 67:1801–1805. [DOI] [PubMed] [Google Scholar]
- Virdee, K., L. Xue, B.A. Hemmings, C. Goemans, R. Heumann, and A.M. Tolkovsky. 1999. Nerve growth factor-induced PKB/Akt activity is sustained by phosphoinositide 3-kinase dependent and independent signals in sympathetic neurons. Brain Res. 837:127–142. [DOI] [PubMed] [Google Scholar]
- Vlahos, C.J., W.F. Matter, K.Y. Hui, and R.F. Brown. 1994. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J. Biol. Chem. 269:5241–5248. [PubMed] [Google Scholar]
- Vogelbaum, M.A., J.X. Tong, and K.M. Rich. 1998. Developmental regulation of apoptosis in dorsal root ganglion neurons. J. Neurosci. 18:8928–8935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White, F.A., C.R. Keller-Peck, C.M. Knudson, S.J. Korsmeyer, and W.D. Snider. 1998. Widespread elimination of naturally occurring neuronal death in Bax-deficient mice. J. Neurosci. 18:1428–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiese, S., M.R. Digby, J.M. Gunnersen, R. Gotz, G. Pei, B. Holtmann, J. Lowenthal, and M. Sendtner. 1999. The anti-apoptotic protein ITA is essential for NGF-mediated survival of embryonic chick neurons. Nat. Neurosci. 2:978–983. [DOI] [PubMed] [Google Scholar]
- Wright, E.M., K.S. Vogel, and A.M. Davies. 1992. Neurotrophic factors promote the maturation of developing sensory neurons before they become dependent on these factors for survival. Neuron. 9:139–150. [DOI] [PubMed] [Google Scholar]
- Wyatt, S., L.G.P. Piñón, P. Ernfors, and A.M. Davies. 1997. Sympathetic neuron survival and TrkA expression in NT3-deficient mouse embryos. EMBO J. 16:3115–3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue, L., J.H. Murray, and A.M. Tolkovsky. 2000. The Ras/phosphatidylinositol 3-kinase and Ras/ERK pathways function as independent survival modules each of which inhibits a distinct apoptotic signaling pathway in sympathetic neurons. J. Biol. Chem. 275:8817–8824. [DOI] [PubMed] [Google Scholar]
- Yang, J., X. Liu, K. Bhalla, C.N. Kim, A.M. Ibrado, J. Cai, T.I. Peng, D.P. Jones, and X. Wang. 1997. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 275:1129–1132. [DOI] [PubMed] [Google Scholar]
- Zhou, X., and R.A. Rush. 1995. Sympathetic neurons in neonatal rats require endogenous neurotrophin-3 for survival. J. Neurosci. 15:6521–6530. [DOI] [PMC free article] [PubMed] [Google Scholar]