

Abstract
Fibronectin (FN) assembly into a fibrillar extracellular matrix is a stepwise process requiring participation from multiple FN domains. Fibril formation is regulated in part by segments within the first seven type III repeats (III1–7). To define the specific function(s) of this region, recombinant FNs (recFNs) containing an overlapping set of deletions were tested for the ability to assemble into fibrils. Surprisingly, recFN lacking type III repeat III1 (FNΔIII1), which contains a cryptic FN binding site and has been suggested to be essential for fibril assembly, formed a matrix identical in all respects to a native FN matrix. Similarly, displacement of the cell binding domain in repeats III9–10 to a position close to the NH2-terminal assembly domain, as well as a large deletion spanning repeats III4–7, had no effect on assembly. In contrast, two deletions that included repeat III2, ΔIII1–2 and ΔIII2–5, caused significant reductions in fibril elongation, although binding of FN to the cell surface and initiation of assembly still proceeded. Using individual repeats in binding assays, we show that III2 but not III1 contains an FN binding site. Thus, these results pinpoint repeat III2 as an important module for FN–FN interactions during fibril growth.
Keywords: fibronectin; matrix assembly; type III repeats; RGD sequence; self-association
Introduction
Fibronectin (FN)* functions from within a fibrillar matrix, and proper formation of matrix fibrils is crucial for controlling tissue structure and cell motility, growth, and differentiation (Mosher, 1989; Hynes, 1990; Schwarzbauer and Sechler, 1999). Multiple FN domains have been implicated in intermolecular interactions required for the assembly process, including FN's dimer structure and NH2-terminal assembly domain (McKeown-Longo and Mosher, 1985; McDonald et al., 1987; Schwarzbauer, 1991). Integrin binding to the arg-gly-asp (RGD) cell binding sequence within the cell binding domain is necessary for initiation of fibril formation, but not for fibril elongation (Sechler et al., 1996). In the absence of the synergy site, α5β1-mediated assembly is stalled, suggesting that fibril growth requires strong interactions between FN and integrins (Sechler et al., 1997). This is further supported by the demonstration that activation of α4β1, αvβ3, and αIIbβ3 integrins can promote FN assembly (Wu et al., 1995, 1996; Wennerberg et al., 1996; Sechler et al., 2000).
FN–FN interactions are also important for fibril formation. The major site of interaction is the NH2-terminal assembly domain which consists of repeats I1–5 and binds FN and many other molecules (Mosher, 1989; Hynes, 1990; Schwarzbauer, 1991). Other FN binding sites have been localized to the first one or two type III repeats (Morla and Ruoslahti, 1992; Aguirre et al., 1994; Hocking et al., 1994; Ingham et al., 1997), the cell binding repeat III10 (Hocking et al., 1996), and the COOH-terminal heparin binding domain (III12–14) (Bultmann et al., 1998). Each of these sites interacts with the NH2-terminal assembly domain.
Results from binding, inhibition, and matrix assembly studies show that FN fibrils form via a multistep process (McKeown-Longo and Mosher, 1983; Schwarzbauer and Sechler, 1999). During the initiation stage of assembly, integrin binding immobilizes dimeric FN and promotes formation of deoxycholate (DOC)-soluble fibrils in a process that depends on the NH2-terminal assembly domain. Mutations that affect the RGD cell binding sequence or the NH2-terminal domain ablate fibril formation (Schwarzbauer, 1991; Sottile et al., 1991; Sechler et al., 1996; Sottile and Mosher, 1997). Assembly then progresses into a growth phase that involves incorporation of additional FN dimers into nascent fibrils, fibril elongation, and conversion of fibrils into a DOC-insoluble form. The matrix is further stabilized as DOC-insoluble FN is formed into high molecular mass multimers.
We have shown previously that a recombinant FN (recFN) lacking the first seven type III repeats (FNΔIII1–7) is able to form a fibrillar matrix, albeit at an altered rate (Sechler et al., 1996). It appears that this set of seven repeats, or a subset of them, has a regulatory role in FN assembly. In this study, recFNs containing overlapping deletions across this region were tested for the ability to form fibrils, DOC-insoluble matrices, and high molecular mass multimers. Surprisingly, deletion of repeat III1, a site proposed to be essential for assembly, had no detrimental effects on assembly. Similarly, relatively large deletions of up to four type III repeats, as well as displacement of the cell binding domain toward the NH2 terminus, caused no deficiencies in matrix formation. However, deletions that included repeat III2 reduced the assembly of a DOC-insoluble matrix and blocked fibril elongation. Binding studies using recombinant fragments showed that III2, but not III1, has FN binding activity. Our results indicate that repeat III2 is a key element in the regulation of FN–FN interactions during matrix assembly.
Results
Repeat III1 is not essential for FN assembly
To delineate the elements involved in the regulation of FN matrix assembly, a set of recFNs containing in-frame deletions within the first seven type III repeats was prepared (Fig. 1) . Of particular interest was the first type III repeat, which has FN binding activity when in denatured form and has been implicated in FN matrix formation (Morla and Ruoslahti, 1992; Aguirre et al., 1994; Hocking et al., 1994; Ingham et al., 1997). Using the baculovirus expression system, FNΔIII1 was expressed from a rat FN cDNA mutated by PCR to eliminate the entire segment encoding repeat III1. Assembly was tested using CHO K1 cells transfected with human α5 integrin cDNA (CHOα5) that do not assemble an endogenous matrix (Sechler et al., 1996). FNΔIII1 was assembled into a fibrillar matrix morphologically identical to that formed by native FN (Fig. 2 , A–D) or full-length recFNA-B- (unpublished data and Sechler et al., 1996). No differences in native and FNΔIII1 matrices were detected at any of the time points studied. AtT-20 mouse pituitary cells transfected with human α5 integrin cDNA (AtT-20α5), a cell line that does not express any endogenous FN (Sechler et al., 1996), also assembled morphologically identical native and FNΔIII1 matrices (Fig. 2, E and F). Therefore, in two independent cell lines, FN matrix assembly was not altered by the deletion of repeat III1.
Figure 1.

Schematic representation of FN and recFNs. The structural organization of FN consisting of type I (rectangles), type II (triangles), and type III (ovals) repeats is shown at top. Darkened ovals represent alternatively spliced EIIIA and EIIIB repeats that were not included in any recFNs. All recFNs contain the V120 variant of the alternatively spliced V region (cross-hatched box) as well as the COOH-terminal cysteine pair (S-S). The inverted triangle over FNrIII4–5/9–10 indicates a deleted RGD sequence; white ovals are III9–10 in place of III4–5. All six recFNs were constructed from rat FN cDNA.
Figure 2.
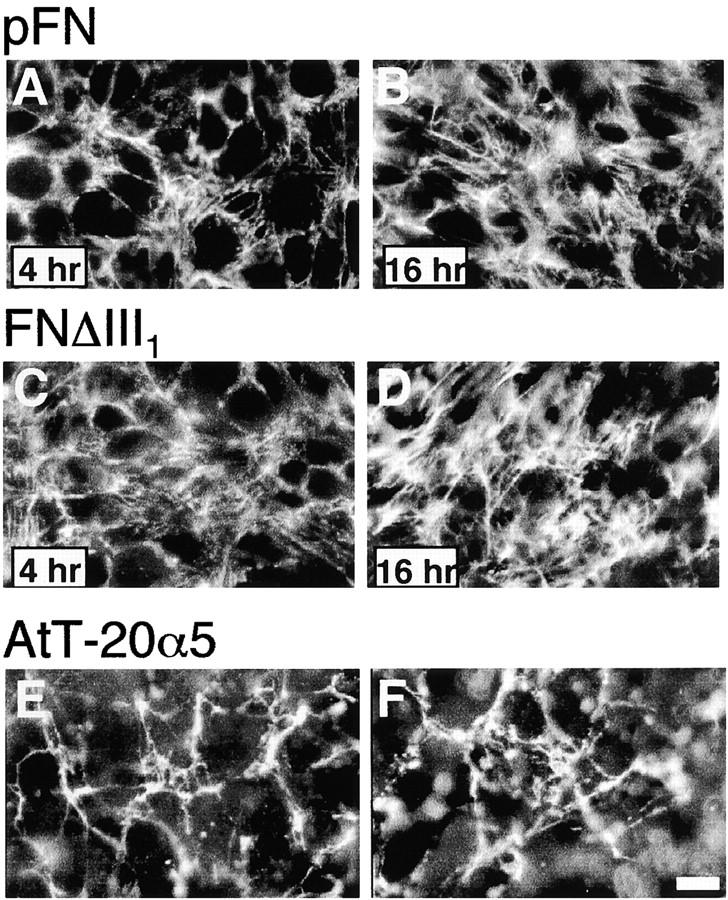
Assembly of FNΔIII1. CHOα5 cells were cultured in the presence of 50 μg/ml pFN (A and B) or 50 μg/ml FNΔIII1 (C and D) for 4 (A and C) or 16 (B and D) h. AtT-20α5 cells were incubated with 25 μg/ml pFN (E) or FNΔIII1 (F) for 16 h. Cells were then fixed and FN fibrils detected by indirect immunofluorescence with monoclonal anti–rat FN antibody IC3. Bar, 10 μm.
Biochemically, FNΔIII1 and native FN matrices were indistinguishable. Equivalent amounts of both proteins were associated with CHOα5 cells in the DOC-soluble fractions (Fig. 3 A). Similar proportions of DOC-insoluble material were formed from FN and FNΔIII1 during a 16-h incubation (Fig. 3 B). As has been shown previously for FN (McKeown-Longo and Mosher, 1983; Sechler et al., 1996), there was continued incorporation of FNΔIII1 into DOC-insoluble matrices and high molecular mass aggregates (Fig. 3 B, 48 h). These data show that III1 is neither required for the formation of fibrils, nor responsible for the altered rate of assembly observed with FNΔIII1–7.
Figure 3.

DOC-soluble and -insoluble FNΔIII1. CHOα5 cells were incubated with 50 μg/ml pFN or FNΔIII1 for the indicated times and lysed in buffered DOC. DOC-soluble (A) and -insoluble (B) fractions were separated in 5% polyacrylamide-SDS gels without reduction and transferred to nitrocellulose. FN was detected on immunoblots with monoclonal anti-FN antibody IC3 and chemiluminescence reagents. Dimeric pFN and FNΔIII1 are present (arrowhead) as well as high molecular mass multimers at the top of the stacking (bracket) and at the interface of the stacking and separating gels (arrow).
Reduced matrix accumulation in the absence of repeats III2–5
In contrast to the normal matrix formed with FNΔIII1, deletion of III2–5 decreased matrix accumulation. Some fibril assembly was initiated, but the amount of FNΔIII2–5 matrix assembled by CHOα5 cells was less than that of native FN, and the distribution of fibrils was more sparse than for native FN (Fig. 4 A). Furthermore, DOC-insoluble matrix was at least threefold less for FNΔIII2–5 than for native FN (Fig. 4 B). Limited conversion of DOC-soluble FNΔIII2–5 into DOC-insoluble matrix suggests that this recFN is impaired in its ability to participate in fibril growth.
Figure 4.

Assembly of FNΔIII2–5. (A) Native pFN and FNΔIII2–5 were added to CHOα5 cells at a concentration of 50 μg/ml and cultured for the indicated times. Fibrils were visualized by indirect immunofluorescence as in Fig. 2. (B) DOC-insoluble material isolated at the indicated times was analyzed under reducing conditions by immunoblotting with IC3 monoclonal antibody. Bar, 10 μm.
Analyses of the assembly of two other deletion mutants, FNΔIII4–5 and FNΔIII4–7, showed formation of characteristic fibrils at all time points (unpublished data). Furthermore, biochemical analyses did not reveal any defects in incorporation into DOC-insoluble matrix or the formation of high molecular mass multimers, results similar to those observed for FNΔIII1. FNΔIII2–5 and FNΔIII4–7 both lack four type III repeats but differ in their capacity to be assembled. This indicates that alterations in the ability to assemble matrices cannot be attributed solely to the size of the deletion. Instead, there appear to be specific roles for individual type III repeats and normal assembly of FNΔIII4–5 compared with FNΔIII2–5 indicates that III2 and/or III3 may be important.
Fibril growth depends on repeats III1–2
Because III1–2 has FN binding activity (Aguirre et al., 1994) and a proteolytic fragment containing III1 plus part of III2 inhibits incorporation of FN into matrix (Chernousov et al., 1991; Morla and Ruoslahti, 1992), we generated FNΔIII1–2 to test whether these two repeats together comprise a matrix regulatory region. Immunofluorescence analysis of FN assembled by CHOα5 cells showed that early during assembly, FNΔIII1–2 formed aggregates on the cell surface (Fig. 5 A) similar to, but much smaller than, those formed by FNΔIII1–7 (Sechler et al., 1996). As assembly progressed, FNΔIII1–2 formed mainly short fibrils between cells although occasional long thin fibrils were also visible. An extensive fibrillar network was never observed even after prolonged incubations. Levels of DOC-insoluble material were significantly reduced, especially at later times of assembly, and no high molecular mass multimers were observed (Fig. 5 B). Incubations with higher concentrations of FNΔIII1–2 did not increase the amount of DOC-insoluble matrix (unpublished data). Unlike FNs lacking the RGD sequence, FNΔIII1–2 was not deficient in binding to cells. In fact, substantially more FNΔIII1–2 than native FN was isolated as cell-associated DOC-soluble material. This indicates that FNΔIII1–2 can efficiently bind to the cell surface to initiate assembly, but is defective in the growth phase when conversion from DOC-soluble to -insoluble matrix occurs.
Figure 5.

Assembly of FNΔIII1–2. (A) CHOα5 cells were incubated for either 4 or 24 h in the presence of 50 μg/ml FNΔIII1–2. Monoclonal antibody IC3 was used to detect recFN matrix by immunofluorescence. (B) DOC-soluble and -insoluble cell lysates were isolated from CHOα5 cells incubated in the presence of 50 μg/ml FNΔIII1–2 or pFN for 0.5, 4, 7, 16, 24, and 48 h. DOC-soluble and -insoluble material was analyzed as described in Fig. 3. Arrow and bracket indicate locations of high molecular mass multimers. Dash indicates location of 180-kD molecular mass standard. Bar, 10 μm.
FN lacking the RGD sequence is unable to initiate matrix assembly but can be incorporated once assembly has been primed by native FN (Sechler et al., 1996). However, preinitiation by native FN would not be expected to rescue FNΔIII1–2 assembly, as this protein shows a defect in fibril growth. In fact, neither FNΔIII1–2 nor FNΔIII2–5 was able to efficiently incorporate into a preformed human FN matrix. A very few short fibrils were formed by FNΔIII1–2 (Fig. 6 B) and FNΔIII2–5 fibrils were found in small patches distributed unevenly throughout the matrix (Fig. 6 C). In contrast, an extensive fibrillar network was assembled by native FN (Fig. 6 A). In all cases, a fibrillar human plasma FN (pFN) matrix could be readily detected with a human FN-specific monoclonal antibody (unpublished data). Thus, the presence of repeat III2 is required for the continued growth of FN fibrils.
Figure 6.
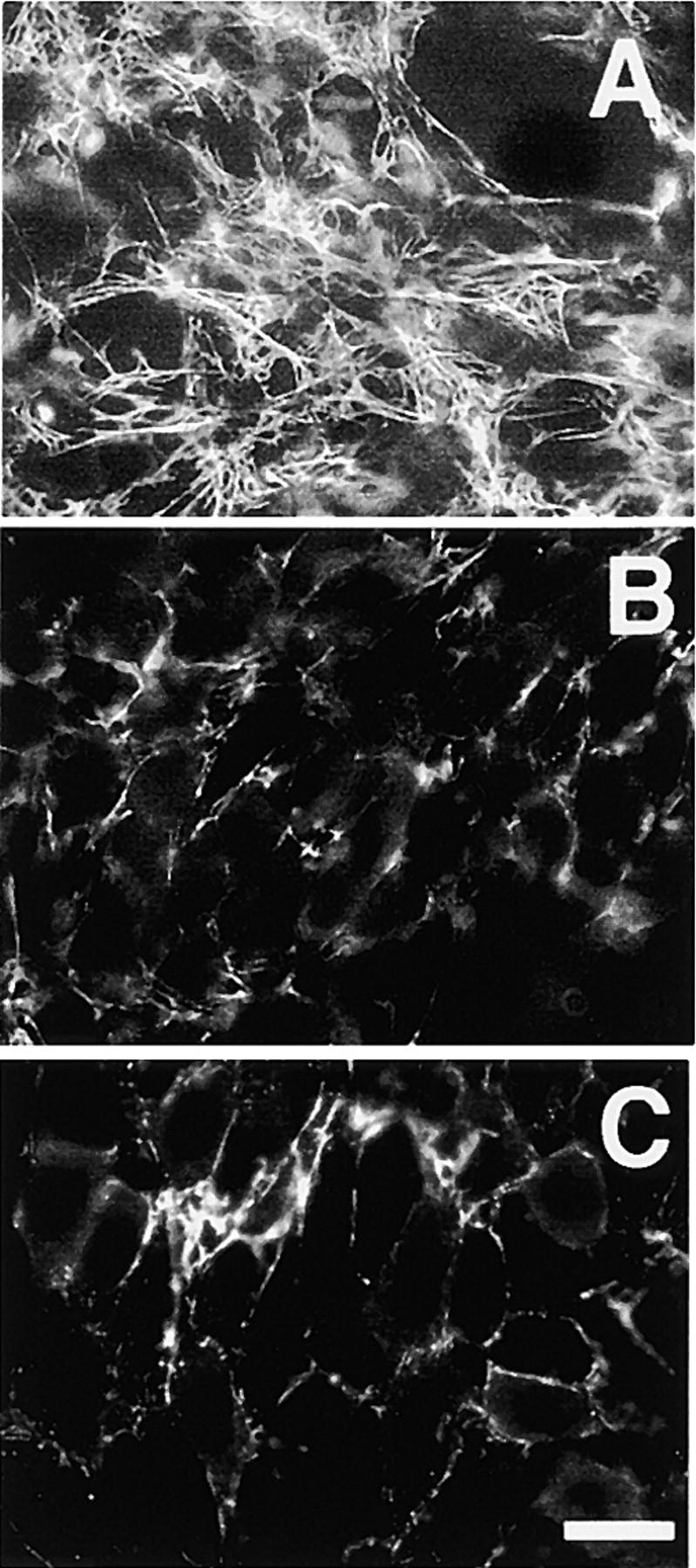
RecFN assembly in the presence of human FN matrix. CHOα5 cells were incubated with 50 μg/ml human pFN for 8 h. Medium was then removed, cell layers were washed to remove unbound FN, and cells were fed with fresh medium containing 25 μg/ml full-length recFN (A), FNΔIII1–2 (B), or FNΔIII2–5 (C). After incubation for an additional 16 h, cells were fixed and recFN fibrils detected with an anti–rat FN-specific monoclonal antibody. Bar, 20 μm.
Repeat III2 contains an FN binding site
To demonstrate a requirement for III2 in FN assembly, attempts were made to generate a recFN lacking repeat III2 or with another homologous repeat in its place. Unlike the recFNs reported here, secretion of mutant recFNs lacking III2 from infected insect cells was very inefficient, suggesting that in the absence of this repeat the proteins were not properly folded. We have shown previously that III1–2 purified in soluble form from bacterial lysates is able to bind FN (Aguirre et al., 1994). To identify the repeat responsible for FN binding activity, maltose-binding protein (MBP) fusion proteins containing either III1 or III2 were generated and tested for binding in solid phase binding assays. MBP-III2, but not MBP-III1, showed significant FN binding activity (Fig. 7 A) that was reversed by treatment with buffered SDS (unpublished data). The lack of MBP-III1 binding confirms the results of Ingham et al. (1997) who showed that native III1 does not bind to FN or its fragments. FN binding was concentration dependent and a complementary binding site was localized to the 70-kD region (Fig. 7 B). Apparent dissociation constants for FN and 70 kD binding to III2 differ by only 3.5-fold, 28 nM, and 8 nM, respectively. The higher dissociation constant for FN may reflect a difference in affinity. However, it may also be due to molecular differences between the relatively small 70-kD fragment and dimeric pFN, which has a compact conformation that may reduce accessibility to III2 binding sites.
Figure 7.
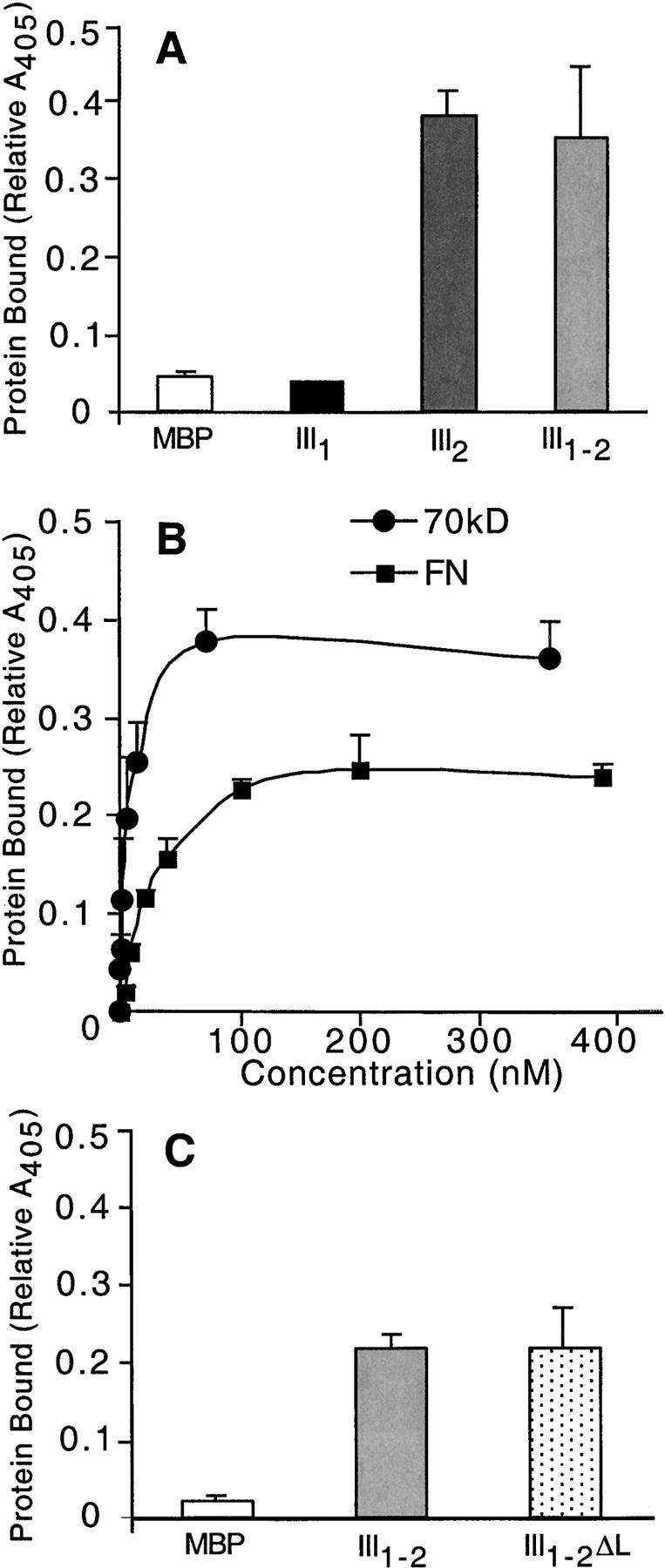
Repeat III2 contains a FN binding site. Solid phase binding assays were used to examine binding of FN (A–C) and NH2- terminal 70-kD fragment (B) to immobilized fusion proteins. (A and C) MBP and fusion proteins were coated at 2 μg/ml and incubated with 50 μg/ml rat pFN. (B) MBP-III2 coated at 4 μg/ml was incubated with rat pFN or 70-kD fragment at increasing concentrations. Background binding to MBP was subtracted from these data. Binding was detected with 5G4 anti-FN monoclonal antibody for FN (A and C) or R457 anti–70-kD polyclonal antiserum (B). Results are presented as the average of normalized values from two to three experiments.
Between III1 and III2 is a 17–amino acid “linker” segment that was present in the MBP-III2 protein. To determine whether the binding site was located in the type III repeat or the linker, an MBP protein was generated containing III1–2 but lacking the linker (MBP-III1–2ΔL) and tested for FN binding. MBP-III1–2ΔL bound to FN (Fig. 7 C) and the 70-kD fragment (unpublished data) to an extent identical to MBP-III1–2. Similar III1–2 and III1–2ΔL fragments prepared from human FN also showed concentration-dependent binding to 70 kD (unpublished data). Therefore, the site resides in III2 itself and is present in both rat and human FNs. These results identify repeat III2 as a major FN binding site and suggest that the lack of fibril formation by FNΔIII1–2 is due to the absence of this site.
Repositioning of III9–10 cell binding domain
The cell binding domain consisting of an RGD sequence and synergy site in repeats III9–10 is essential for initiation of FN matrix assembly by α5β1 integrin (Sechler et al., 1996, 1997). Deletions within repeats III1–7 position the cell binding domain closer to the NH2-terminal assembly domain and could affect recFN fibril formation. To eliminate the possibility that changes in the domain organization can alter FN assembly, we created FNrIII4–5/9–10. Repeats III4–5 were replaced with III9–10 and the RGD sequence was deleted from its native position within the cell binding domain (Fig. 1). Therefore, the only functional III9–10 pair is in the position normally occupied by III4–5. CHOα5 cells efficiently assembled FNrIII4–5/9–10 into a DOC-insoluble fibrillar matrix identical to that of native FN at all time points (Fig. 8 , A–C). Cell cycle progression by CHOα5 cells assembling either FN or FNrIII4–5/9–10 was identical, as were the levels of focal adhesion kinase phosphorylation (unpublished data). These results demonstrate that the location of the cell binding domain is not restricted to the center of the molecule and that displacement toward the NH2 terminus does not reduce FN function in matrix assembly or its ability to influence cell cycle progression.
Figure 8.

Assembly of FNrIII4–5/9–10. CHOα5 cells were cultured in the presence of 50 μg/ml FNrIII4–5/9–10 for 4 (A) and 16 (B) h. (C) DOC-insoluble material was isolated from CHOα5 cells 4 and 16 h after incubation with 50 μg/ml FNrIII4–5/9–10 and analyzed as in Fig. 3. Locations of high molecular mass multimers are indicated with arrow and bracket. 180-kD molecular mass standard is indicated by dash. Bar, 10 μm.
Discussion
FN fibril assembly is initiated by binding to integrin receptors and propagated by FN–FN interactions. The progression of FN conversion from soluble dimer into insoluble matrix fibrils is regulated in part by sequences within repeats III1–7 (Sechler et al., 1996). Using a set of overlapping deletions within this region, we have identified repeat III2 as an important FN binding site involved in fibril elongation. Deletions spanning this repeat severely limit fibril assembly and block formation of DOC-insoluble material. In addition to implicating III2, our results also eliminate several candidate sites as important contributors to this process. Specifically, we have shown that: (a) the III1 repeat and its cryptic FN binding site are not needed for fibril formation; (b) relatively large deletions of as many as four repeats do not affect fibril assembly; and (c) there is considerable pliability in the location of the cell binding domain. None of the deletions tested here had an obvious effect on FN binding to integrins or initiation of fibril assembly, indicating that reported interactions between III1 and the cell binding domain (Hocking et al., 1996) are not required in the early stages of FN matrix assembly.
Our data with FNΔIII1–2 and FNΔIII2–5 show that repeat III2 participates in the growth phase of FN assembly. Assembly was initiated by both of these recFNs but DOC-insoluble matrix formation was blocked. Together with data showing a direct interaction between III2 and the NH2-terminal assembly domain of FN, our matrix assembly results indicate that this FN binding site plays a key role in FN–FN interactions during fibril assembly. Repeats III2–3 have been shown previously to interact with repeats III12–14, an interaction that appears to contribute to the formation of the compact conformation of soluble FN (Johnson et al., 1999). Thus, the III2 module may participate in matrix assembly through interactions with several sites on FN. These interactions may promote elongation by aligning fibrils into a stable, uniform structure that can then be converted into a DOC-insoluble form. In the absence of this repeat, fibrils begin to form but become stalled during elongation and are inefficiently converted into the DOC-insoluble matrix. DOC-insolubility of FN fibrils appears to occur through hydrophobic protein–protein interactions that resist SDS denaturation (Chen and Mosher, 1996). Perhaps III2 participates in the formation of that hydrophobic interface.
Others have shown the presence of a cryptic FN binding site in III1 that is exposed by denaturation (Hocking et al., 1994; Ingham et al., 1997). The apparent affinity of 70 kD for III2 is higher than that reported for 70 kD binding to heat-denatured III1 (Hocking et al., 1994). The identification of two distinct sites indicates that the III1–2 segment contains more than one FN binding site, the site in III2 that is critical for fibril assembly and a cryptic site in III1 that is dispensable for this process. It is also possible that repeats III1–2 act as a functional unit to regulate fibril assembly and promote elongation. For example, interactions between III1 and III2 could regulate the accessibility of an FN binding site. Previous studies lend support to the idea of cooperation between these repeats. Both repeats were required for formation of an in vitro ternary complex with heat-denatured III10 and the NH2-terminal 70-kD fragment (Hocking et al., 1996). III2 has also been proposed to contribute to interactions between III1 and the COOH-terminal heparin binding domain (Bultmann et al., 1998). Furthermore, the reduced secretion of recFNs lacking III2 or carrying another type III repeat in place of III2 indicates that interactions between adjacent repeats contribute to domain structure and stability. III1-specific inhibitory peptides and antibodies have been described (Chernousov et al., 1987, 1991; Morla and Ruoslahti, 1992). If III1 and III2 do indeed function together, these inhibitory reagents may exert their effects indirectly through disruption of activities mediated by the adjacent III2 module.
Regulated assembly depends in part on conformational changes in the FN molecule. Accumulating evidence indicates that soluble FN dimers must be converted from a compact inactive form into an “unfolded” activated form in order for assembly to proceed (Alexander et al., 1979; Williams et al., 1982; Erickson and Carrell, 1983; Rocco et al., 1983; Ugarova et al., 1995; Schwarzbauer and Sechler, 1999). In vivo, integrin binding induces FN activation and this may expose the III2 binding site, allowing intermolecular interactions between cell surface–bound FNs. Whereas recFNs lacking III2 are able to initiate assembly and form short fibrils, the significantly reduced levels of DOC-insoluble FNΔIII1–2 suggest that in the absence of the III2 binding site, this recFN cannot effectively participate in the essential FN–FN interactions needed for fibrillogenesis. FN molecules can also be induced to associate in solution by the addition of a peptide corresponding to part of the III1 module (Morla et al., 1994). This treatment may expose the III2 binding site by local perturbation of intramolecular interactions involving this region of the molecule.
Comparison of the progression of assembly by FNΔIII1–2 with FNΔIII1–7 shows that both initially form short stitches around cell peripheries and connect to adjacent cells. FNΔIII1–7 then forms aggregates that prematurely become insoluble in DOC (Sechler et al., 1996). These aggregates can apparently be remodeled by binding to adjacent cells and getting stretched into fibrils. In this way, FNΔIII1–7 forms a relatively normal-appearing fibrillar matrix. On the other hand, FNΔIII1–2 forms only a few small, DOC-soluble aggregates that can be converted into predominantly short fibrils. FNΔIII1–2 does not accumulate in DOC-insoluble material, nor does it form an extensive fibrillar matrix. The differences between assembly of FNΔIII1–2 and FNΔIII1–7 suggests that repeats III3–7 contribute to the progression of FN fibril formation. A few activities have been mapped to the III3–7 region. Repeats III4–6 can bind to heparin and DNA under low salt conditions (Hynes, 1990). Cryptic binding sites within repeat III5 have been reported for activated α4β1 and α4β7 integrins (Moyano et al., 1997) as well as for repeat III1 (Hocking et al., 1996). Repeat III1 can also bind to repeat III7 (Ingham et al., 1997). Thus, it is possible that during assembly this region of FN interacts with cell surface or matrix proteins or glycosaminoglycans, and that these interactions may help to control fibril formation.
FNΔIII1–2 and FNΔIII1–7 probably also differ in the alignment of FN dimers into fibrils. For example, binding of the NH2-terminal assembly domain of one FNΔIII1–7 dimer to the COOH-terminal heparin domain of another (Bultmann et al., 1998) would align their cell binding domains relatively close to each other. This juxtaposition could result in increased clustering of integrins and more stable contacts between matrix and cytoskeleton, giving the strong connections needed to remodel aggregates into fibrils and form DOC-insoluble material. Tension applied to FN fibrils has been predicted to cause slight unfolding of type III repeats (Erickson, 1994; Krammer et al., 1999), and this might allow the formation of SDS-resistant protein–protein interactions (Chen and Mosher, 1996). On the other hand, the inclusion of III3–7 may yield a potentially different organization of both cell surface receptors and cytoskeletal elements, thus precluding the formation of a stable matrix.
Clearly, multiple options exist for establishing FN–FN interactions during matrix assembly. For example, the NH2-terminal assembly domain is required throughout the assembly process, whereas the III2 module participates after initiation during a phase of fibril growth. This indicates that different FN binding sites have distinct temporal and spatial roles, and suggests that control of domain-specific FN interactions may play an important role in regulating the structural and functional organization of the FN matrix.
Materials and methods
Cell culture
CHOα5, clone 17, and AtT-20α5, clone 11, transfected with a cDNA to the human α5 integrin subunit have been described previously (Sechler et al., 1996). For all experiments, CHOα5 cells were cultured in DME supplemented with 2 mM glutamine, 1% nonessential amino acids, 100 μg/ml Geneticin (Life Technologies/GIBCO BRL), and 10% fetal calf serum (Hyclone Labs) depleted of FN. AtT-20α5 cells were cultured in a 50:50 mixture of Ham's F12 and DME, plus 20 mM Hepes, pH 7.4, 4 mM glutamine, 0.25 mg/ml Geneticin (Life Technologies/GIBCO BRL), and 10% fetal calf serum (Hyclone Labs) and 10% Nu-serum both depleted of FN.
FN cDNA constructions and recombinant protein production
All recFNs were expressed with baculovirus vector pVL1392. Restriction enzymes and T4 DNA ligase were purchased from New England Biolabs, Inc. Oligonucleotide primers were prepared by the Synthesis and Sequencing Facility (Princeton University, Princeton, NJ).
All deletions were created by PCR amplification of rat FN cDNA. A KpnI site was engineered into each oligonucleotide to join the regions spanning each deletion. PCR amplification for 25 cycles was performed for all constructions under the following conditions: 95°C, 30 s; 60°C, 60 s; and 72°C, 60 s. PCR products were digested with flanking enzymes and inserted into the FN cDNA using convenient restriction sites. The following 5′ and 3′ primers were used to generate the indicated deletions (base positions of the primers within the FN cDNA are in parentheses and base changes to introduce the Kpn I sites are underlined): ΔIII1: GGGGTACCTGTGCCTGGGTA (1831-1812), CTGGTACCAGCAACACAGTG (2116-2130); ΔIII4–7: CGGGTACCTCATCGGATCGT (2721-2702), GCGGTACCTCCTCCCACGGA (4065-4084); ΔIII2–5: TGTTGCTGGGTACCGGTGTG (2124-2105), CCTGGTACCTCTGCGCTCCA (3248-3267). pVL1392 FNΔIII1–2 was prepared by ligating a fragment from FNΔIII1 with a PCR-amplified fragment made using the primer CTGGTACCGCACCTGATGCGCCTCCAG (2424-2442). pVL1392 FNΔIII4–5 was created by ligating a 5′ fragment from pVL1392 FNΔIII4–7 with a 3′ fragment from pVL1392 FNΔIII2–5. To generate pVL1392 FNrIII4–5/9–10, a segment spanning repeats III9–10 was amplified using 5′ primer GAGGTACCGGACTCCCCAACTGGTTTTG (4552-4570) and 3′ primer GAGGTACCGCTGTTTGATAATTGATGGAAACTGGC (5098-5073) containing KpnI sites (underlined). The resulting KpnI fragment was then inserted at the engineered KpnI site in pVL1392 FNΔIII4–5 and a segment encoding an RGD deletion was inserted into the cell binding domain (Schwarzbauer, 1991). All regions obtained from PCR products were verified by DNA sequence analysis.
Recombinant baculoviruses were created and recombinant proteins and rat pFN were purified as described (Sechler et al., 1996, 1997). Yields of recFNs were at least 0.8 mg/100 ml of culture supernatant. The identity of FNΔIII1 and FNΔIII1–2 recombinant baculoviruses was further confirmed using DNA isolated from infected High Five insect cells as template for PCR amplification with flanking primers. Correctly sized PCR products were obtained as analyzed by PAGE before and after restriction with KpnI.
Immunofluorescence
As described in Sechler et al. (1996), CHOα5 and AtT-20α5 cells were seeded in medium containing FN-depleted serum and plated onto glass coverslips in a 24-well dish or four-well chamber slides at a concentration of 1.5 × 105 and 4 × 105, respectively. After an overnight incubation, fresh medium was added along with either pFN or purified recFN and incubated for the times indicated. Cell layers were fixed with 3.7% formaldehyde and stained with a 1:1,000 dilution of IC3 ascites in PBS with 2% ovalbumin followed by a 1:400 dilution of fluorescein-conjugated goat anti–mouse secondary antibody. Stained cells were mounted with FluoroGuard (Bio-Rad Laboratories), and fibrils were visualized with a Nikon Optiphot-2 microscope. Images were collected with a DEI-750 cooled CCD camera (Optronics Engineering) and transferred to a Macintosh G3 computer with an LG3 frame grabber (Scion Corp.) and Adobe Photoshop v. 5.0.
Isolation and detection of DOC-soluble and -insoluble matrix
DOC-soluble and -insoluble material was isolated from CHOα5 cells cultured in a 24-well dish with pFN or recFNs as described above. After the indicated time periods, cells were washed with serum-free DME and lysed with 200 μl of DOC lysis buffer (2% deoxycholate, 0.02 M Tris-HCl, pH 8.8, 2 mM phenylmethylsulfonyl fluoride, 2 mM EDTA, 2 mM iodoacetic acid, and 2 mM N-ethylmaleimide) per well. Lysates were separated into DOC-soluble and -insoluble fractions that were analyzed by SDS-PAGE. Immunodetection was performed as described (Sechler et al., 1996) using ascites fluid from rat FN–specific monoclonal antibody IC3 at a dilution of 1:1,000. Immunoblots were developed with Super Signal chemiluminescence reagents (Pierce Chemical Co.). Band intensities were quantified at two exposure times using IPLab software (Mac v. 3.5; Scanalytics, Inc.).
Expression of bacterial fusion proteins and FN binding assays
Rat FN cDNA fragments encoding repeat III1 or III2 were inserted into pMAL-cRI (New England Biolabs, Inc.) for expression as MBP fusion proteins. III1 spanned amino acid positions 604–700 (TYP … TTS) and III2 extended from residue 701 to 808 (AST … QTT). BamHI sites and XbaI sites were engineered at the 5′ and 3′ ends, respectively. PCR amplification using primers homologous to sequences flanking the 17–amino acid linker (701–717) was used to replace these residues (AST … APF) with a KpnI site to generate III1–2ΔL. Escherichia coli TB1 cells expressing individual MBP fusion proteins were lysed with B-PER (Pierce Chemical Co.) and proteins were purified by amylose resin affinity chromatography following the manufacturer's recommendations. Rat pFN, recombinant 70-kD fragment, MBP, and MBP-III1–2 were purified as described previously (Aguirre et al., 1994).
Solid phase binding assays were performed essentially as described by Aguirre et al. (1994). Fusion proteins containing III1, III2, III1–2, and III1–2ΔL were immobilized on Nunc Maxisorp microtiter plates by overnight incubation at the indicated concentrations. Relative amounts of immobilized proteins were determined by ELISA with an anti-MBP antiserum. After washing and blocking with 1% BSA in PBS, wells were incubated with rat pFN at 50 μg/ml or NH2-terminal 70-kD fragment of FN at 30 μg/ml for 2 h at room temperature. Bound FN and 70 kD were detected and quantified by ELISA using anti–rat FN monoclonal antibody 5G4 or anti–70-kD polyclonal antiserum R457.
Acknowledgments
The authors would like to thank Jennifer Luczak, Jennifer Podesta, and Nedra Guckert for technical assistance, Dr. Siobhan Corbett for helpful discussions, and Chris Hoyte for production of MBP-III1.
This work was supported by a grant from the National Institutes of Health (CA44627 to J.E. Schwarzbauer). J.L. Sechler was a recipient of a New Jersey Commission on Cancer Research postdoctoral fellowship. T. Murata was supported by the Japan Society for the Promotion of Science.
J.L. Sechler's present address is R.W. Johnson Research Pharmaceutical Institute, Welsh and McKean Roads, Spring House, PA 19477.
T. Murata's present address is School of Dentistry, Niigata University, 2-5274 Gakkou-Machi Dori, Niigata-Si, 951-8514 Japan.
Footnotes
Abbreviations used in this paper: DOC, deoxycholate; FN, fibronectin; pFN, plasma FN; recFN, recombinant FN; MBP, maltose-binding protein; RGD, arg-gly-asp.
References
- Aguirre, K.M., R.J. McCormick, and J.E. Schwarzbauer. 1994. Fibronectin self-association is mediated by complementary sites within the amino-terminal one-third of the molecule. J. Biol. Chem. 269:27863–27868. [PubMed] [Google Scholar]
- Alexander, S.S., G. Colonna, and H. Edelhoch. 1979. The structure and stability of human plasma cold-insoluble globulin. J. Biol. Chem. 254:1501–1505. [PubMed] [Google Scholar]
- Bultmann, H., A.J. Santas, and D.M. Pesciotta Peters. 1998. Fibronectin fibrillogenesis involves the heparin II binding domain of fibronectin. J. Biol. Chem. 273:2601–2609. [DOI] [PubMed] [Google Scholar]
- Chen, H., and D.F. Mosher. 1996. Formation of sodium dodecyl sulfate-stable fibronectin multimers. J. Biol. Chem. 271:9084–9089. [DOI] [PubMed] [Google Scholar]
- Chernousov, M.A., A.I. Faerman, M.G. Frid, O.Y. Printseva, and V.E. Koteliansky. 1987. Monoclonal antibody to fibronectin which inhibits extracellular matrix assembly. FEBS Lett. 217:124–128. [DOI] [PubMed] [Google Scholar]
- Chernousov, M.A., F.J. Fogerty, V.E. Koteliansky, and D.F. Mosher. 1991. Role of the I-9 and III-1 modules of fibronectin in the formation of an extracellular fibronectin matrix. J. Biol. Chem. 266:10851–10858. [PubMed] [Google Scholar]
- Erickson, H.P. 1994. Reversible unfolding of fibronectin type III and immunoglobulin domains provides the structural basis for stretch and elasticity of titin and fibronectin. Proc. Natl. Acad. Sci. USA. 91:10114–10118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson, H.P., and N.A. Carrell. 1983. Fibronection in extended and compact conformations. Electron microscopy and sedimentation analysis. J. Biol. Chem. 258:14539–14544. [PubMed] [Google Scholar]
- Hocking, D.C., J. Sottile, and P.J. McKeown-Longo. 1994. Fibronectin's III-1 module contains a conformation-dependent binding site for the amino-terminal region of fibronectin. J. Biol. Chem. 269:19183–19191. [PubMed] [Google Scholar]
- Hocking, D.C., R.K. Smith, and P.J. McKeown-Longo. 1996. A novel role for the integrin-binding III-10 module in fibronectin matrix assembly. J. Cell Biol. 133:431–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes, R.O. 1990. Fibronectins. Springer-Verlag, New York. 544 pp.
- Ingham, K.C., S.A. Brew, S. Huff, and S.V. Litvinovich. 1997. Cryptic self-association sites in type III modules of fibronectin. J. Biol. Chem. 272:1718–1724. [DOI] [PubMed] [Google Scholar]
- Johnson, K.J., H. Sage, G. Briscoe, and H.P. Erickson. 1999. The compact conformation of fibronectin is determined by intramolecular ionic interactions. J. Biol. Chem. 274:15473–15479. [DOI] [PubMed] [Google Scholar]
- Krammer, A., H. Lu, B. Isralewitz, K. Schulten, and V. Vogel. 1999. Forced unfolding of the fibronectin type III module reveals a tensile molecular recognition switch. Proc. Natl. Acad. Sci. USA. 96:1351–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald, J.A., B.J. Quade, T.J. Broekelman, R. LaChance, K. Forsman, E. Hasegawa, and S. Akiyama. 1987. Fibronectin's cell-adhesive domain and an amino-terminal matrix assembly domain participate in its assembly into fibroblast pericellular matrix. J. Biol. Chem. 262:2957–2967. [PubMed] [Google Scholar]
- McKeown-Longo, P.J., and D.F. Mosher. 1983. Binding of plasma fibronectin to cell layers of human skin fibroblasts. J. Cell Biol. 97:466–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeown-Longo, P.J., and D.F. Mosher. 1985. Interaction of the 70,000-mol. wt. amino terminal fragment of fibronectin with matrix-assembly receptor of fibroblasts. J. Cell Biol. 100:364–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morla, A., and E. Ruoslahti. 1992. A fibronectin self-assembly site involved in fibronectin matrix assembly: reconstruction in a synthetic peptide. J. Cell Biol. 118:421–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morla, A., Z. Zhang, and E. Ruoslahti. 1994. Superfibronectin is a functionally distinct form of fibronectin. Nature. 367:193–196. [DOI] [PubMed] [Google Scholar]
- Mosher, D.F. 1989. Fibronectin. Academic Press, New York. 474 pp.
- Moyano, J.V., B. Carnemolla, C. Dominguez-Jimenez, M. Carcia-Gila, J.P. Albar, P. Sanchez-Aparicio, A. Leprini, G. Querze, L. Zardi, and A. Garcia-Pardo. 1997. Fibronectin type III5 repeat contains a novel cell adhesion sequence, KLDAPT, which binds activated α4β1 and α4β7 integrins. J. Biol. Chem. 272:24832–24836. [DOI] [PubMed] [Google Scholar]
- Rocco, M., M. Carson, R. Hantgan, J. McDonagh, and J. Hermans. 1983. Dependence of the shape of the plasma fibronectin molecule on solvent composition. J. Biol. Chem. 258:14545–14549. [PubMed] [Google Scholar]
- Schwarzbauer, J.E. 1991. Identification of the fibronectin sequences required for assembly of a fibrillar matrix. J. Cell Biol. 113:1463–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzbauer, J.E., and J.L. Sechler. 1999. Fibronectin fibrillogenesis: a paradigm for extracellular matrix assembly. Curr. Opin. Cell Biol. 11:622–627. [DOI] [PubMed] [Google Scholar]
- Sechler, J.L., S.A. Corbett, and J.E. Schwarzbauer. 1997. Modulatory roles for integrin activation and the synergy site of fibronectin during matrix assembly. Mol. Biol. Cell. 8:2563–2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sechler, J.L., A.M. Cumiskey, D.M. Gazzola, and J.E. Schwarzbauer. 2000. A novel RGD-independent fibronectin assembly pathway initiated by α4β1 integrin binding to the alternatively spliced V region. J. Cell Sci. 113:1491–1498. [DOI] [PubMed] [Google Scholar]
- Sechler, J.L., Y. Takada, and J.E. Schwarzbauer. 1996. Altered rate of fibronectin matrix assembly by deletion of the first type III repeats. J. Cell Biol. 134:573–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sottile, J., J. Schwarzbauer, J. Selegue and D.F. Mosher. 1991. Five type I modules of fibronectin form a functional unit that binds to fibroblasts and Staphylococcus aureus. J. Biol. Chem. 266:12840–12843. [PubMed] [Google Scholar]
- Sottile, J., and D.F. Mosher. 1997. N-terminal type I modules required for fibronectin binding to fibroblasts and to fibronectin's III1 module. Biochem. J. 323:51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugarova, T.P., C. Zamaroon, Y. Veklich, R.D. Bowditch, M.H. Ginsberg, J.W. Weisel, and E.F. Plow. 1995. Conformational transitions in the cell binding domain of fibronectin. Biochemistry. 34:4457–4466. [DOI] [PubMed] [Google Scholar]
- Wennerberg, K., L. Lohikangas, D. Gullberg, M. Pfaff, S. Johansson, and R. Fassler. 1996. β1 integrin-dependent and -independent polymerization of fibronectin. J. Cell Biol. 132:227–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, E.C., P.A. Janmey, J.D. Ferry, and D.F. Mosher. 1982. Conformational states of fibronectin. Effects of pH, ionic strength, and collagen-binding. J. Biol. Chem. 257:14973–14978. [PubMed] [Google Scholar]
- Wu, C., V. Kevins, T.E. O'Toole, J.A. McDonald, and M.H. Ginsberg. 1995. Integrin activation and cytoskeletal interaction are critical steps in the assembly of a fibronectin matrix. Cell. 83:715–724. [DOI] [PubMed] [Google Scholar]
- Wu, C., P.E. Hughes, M.H. Ginsberg, and J.A. McDonald. 1996. Identification of a new biological function for the integrin αvβ3: initiation of fibronectin matrix assembly. Cell Adhes. Commun. 4:149–158. [DOI] [PubMed] [Google Scholar]