

Nucleic acids can adopt a variety of well-defined conformations. DNA exists predominantly as a double-stranded right-handed helix but can also adopt alternative structures such as bulges, hairpins, and four-stranded quadruplexes. Such DNA secondary structures have been identified in genomic DNA and many of these are associated with regulatory mechanisms. DNA secondary structures in regulatory regions are potentially attractive targets for chemical intervention by agents that bind and either block or enhance the binding of transcription factors. Thus far, the most successful approaches for the development of artificial transcriptional repressors have included both small molecules and proteins that recognize RNA or DNA structures. These transcriptional antagonists include oligonucleotides (or oligonucleotide mimics) that recognize the major groove of double-stranded DNA through triple-helix[1] or synthetic pyrrole/imidazole-based polyamides that recognize the minor groove of the DNA double helix.[2] Recently, a small molecule was reported to suppress expression of the oncogene c-myc by targeting a proximal four-stranded G-quadruplex DNA secondary structure element.[3]
It is desirable to develop new approaches and new molecules that recognize particular nucleic acid structure(s) with both high affinity and specificity. Dynamic covalent chemistry (DCC) is a reversible exchange process that allows noncovalent interactions to template covalent bond formation.[4] Since its conception, there have been very few examples that have used biologically relevant nucleic acids as a template.[5] We recently reported the use of dynamic covalent chemistry for the generation of G-quadruplex ligands from a small library of nine different species.[6] Herein, we demonstrate the general application of DCC as both a qualitative and quantitative method for the selection and discrimination of ligands that bind distinct nucleic acid structures (quadruplex and duplex DNA).
Distamycin-like polyamides containing N-methylpyrrole and N-methylimidazole amino acids can bind to the minor groove of double-stranded DNA with an affinity similar to naturally occurring DNA-binding proteins in a sequence-specific manner.[7] During the past decade, extensive work, particularly by Dervan and co-workers, has demonstrated that covalently linking polyamides that contain three pyrrole or imidazole units along a γ-aminobutyric acid turn can lead to a six-heterocycle hairpin that recognizes five base pairs with increased affinity and specificity.[8] More recently, it was shown that distamycin can recognize the G-quadruplex motif.[9, 10] Initial studies by NMR spectroscopy and mass spectrometry suggested that distamycin interacts as a dimer with two opposing grooves of a quadruplex with an overall stoichiometry of 4:1.[9] However, recent NMR spectroscopic studies proposed a structurally distinct model in which two distamycin molecules are stacked on the terminal planar G quartet of a quadruplex.[10] Herein, we describe the selection of dimers of pyrrole-based polyamides that bind duplex and/or quadruplex DNA by using an optimized dynamic combinatorial approach. Based on previous studies by ourselves and others, thiol-disulfide chemistry was chosen to mediate the reversible exchange process because it is water-compatible, relatively fast, and pH-controlled.[6, 11]
Polyamides P1, P2, and P3, which respectively contain one, two, and three N-methylpyrrole heterocycles, were designed to closely mimic the chemical structure of distamycin. The formyl group present at the terminus of distamycin was replaced with a β-thiocarbonyl group that provides the aliphatic thiol, and the amidine function at the other terminus of distamycin was replaced by an amino group. Previous studies on distamycin and distamycin analogues suggest that such molecules have the potential to bind quadruplex and duplex DNA as dimers.[7] We set out to optimize the number of heteroaromatic units for binding to each of the two DNA templates. The molecular targets for this study were: 1) a four-stranded intramolecular G quadruplex formed by human telomeric DNA, which is a target under study for anticancer agents,[12] and 2) an A/T-rich duplex DNA sequence identified in the promoter region of the oncogene c-kit,[13] which could serve as a site for intervention of transcriptional regulation.
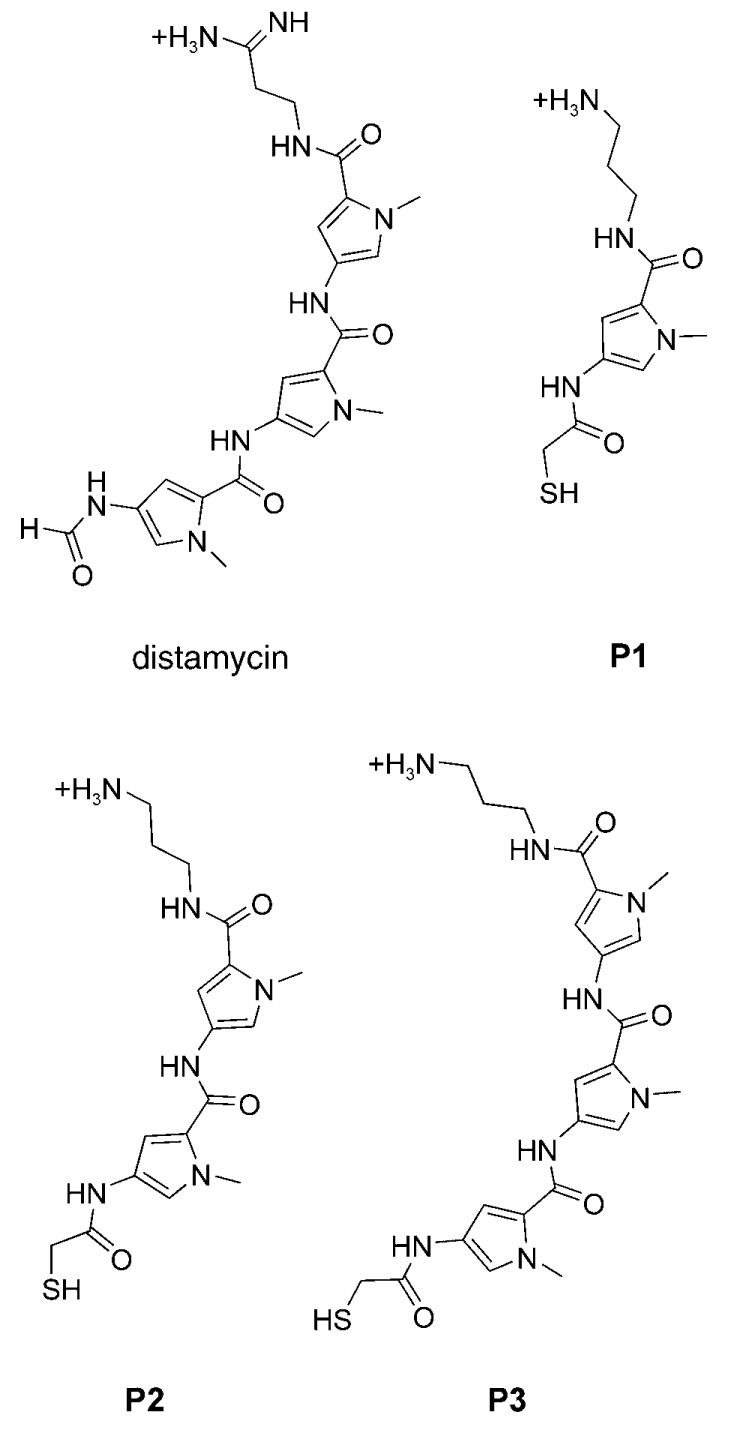
The Fmoc-protected N-methylpyrrole monomers were synthesized as previously reported[14] and assembled on a 1,3-diaminopropane-derivatized trityl resin (Merck Biosciences, loading 1.19 mmol g−1) by using standard solid-phase Fmoc chemistry (Fmoc = 9-fluoromethoxycarbonyl). Three distamycin analogues containing either one, two, or three N-methylpyrrole units were end-derivatized with S-trityl-β-thioacetic acid. Treatment with a solution of TFA/TIS/H2O (95:2.5:2.5) afforded the desired molecules, which were subsequently purified by HPLC (TFA = trifluoroacetic acid, TIS = triisopropylsilane). The nucleic acid targets used were the intramolecular human telomeric quadruplex formed from the folded deoxyoligonucleotide 5′-biotin-(GTTAGG)5,[15] and an 11-mer duplex DNA formed from 5′-biotin-(CTTTTATTTTG) hybridized with its complementary sequence (also 5′-biotinylated). The experimental design principles were essentially the same as those we reported previously.[6] Briefly, an equimolar mixture of monomers P1, P2, and P3 were allowed to react at pH 7.4 in an exchange buffer containing an excess of both oxidized (G–G) and reduced (G) glutathione. Once equilibrium was reached, the exchange process was stopped by lowering the pH to 2, and the biotinylated target, with associated ligands, was isolated by immobilization onto magnetic streptavidin beads. The DNA was then heat-denatured to release any bound ligands; HPLC was used to identify and quantify each species represented in the dynamic combinatorial library (DCL). Comparison of the DCL composition with that obtained in a parallel control experiment that lacked nucleic acid target enabled the identification of the species that were amplified by the presence of template.
A major limitation of the DCC concept is the possibility for library components to strongly self-associate or to associate with another building block in the absence of template.[11] This effect could mask the template-induced amplification of species that are already present in large quantities in the absence of template.[16] To overcome this intrinsic limitation, we reasoned that a large excess of a competing thiol such as glutathione could be used to drive the thermodynamic equilibrium toward the formation of glutathione conjugates in the absence of target, thus decreasing the presence of stable disulfide adducts of library components. This was expected to sensitize the system toward the detection of amplified species that are otherwise significantly pre-formed in the absence of template.
We initially used reduced glutathione (G, 1.5 mm) and oxidized disulfide-bridged glutathione (G–G, 0.375 mm) in Tris-HCl buffer (300 mm, pH 7.4), KCl (150 mm), and 100 μm of each monomer to mediate thiol–disulfide exchange. Under such conditions, monomer P3, in particular, existed predominantly as the homodisulfide P3–P3 (representing 73% of the total amount of thiol P3) at equilibrium (Figure 1 a, blue trace). In such conditions, further amplification of P3–P3 induced by the addition of DNA target would have been very difficult to observe. Exchange experiments were then carried out with a 10-fold increase in the glutathione concentration (G, 15mm; G–G, 3.75 mm) as well as a corresponding 20-fold increase in concentration (G, 30 mm; G–G, 7.5 mm). The HPLC traces representing the composition of the DCL mixtures at equilibrium are shown in Figure 1 a (red and green traces). Elevated glutathione concentrations caused the amounts of P3 and P2 glutathione conjugates and free thiols to increase significantly at the expense of both homodisulfides and of heterodisulfide P3–P2. For example, whereas the P3–P3 homodisulfide represented 75% of thiol P3 at the lowest concentration of glutathione (G, 1.5 mm; G–G, 0.375 mm) it represented only 15% when the amount of glutathione was increased 20-fold.
Figure 1.
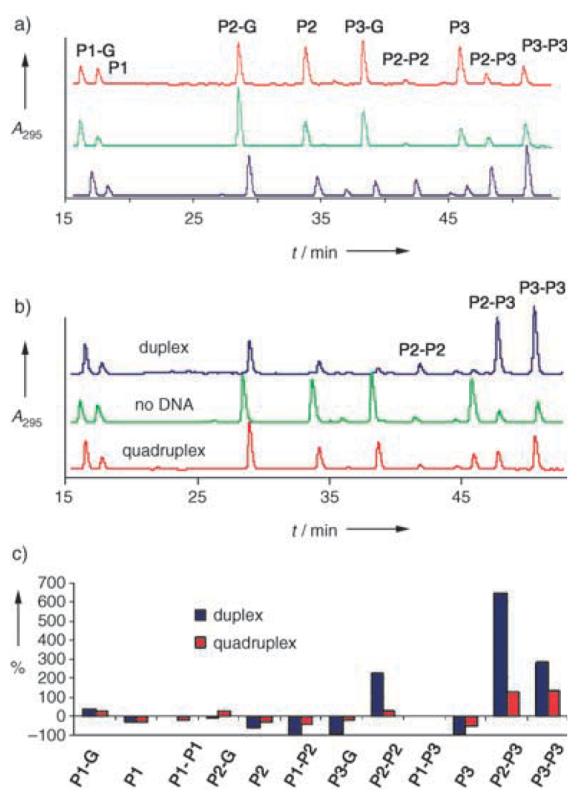
a) HPLC traces showing the component composition at equilibrium after 18 h for exchange experiments starting from P1, P2, and P3 as thiols (100 μm each) in Tris buffer (300 mm Tris-HCl pH 7.4, 150 mm KCl) containing different concentrations of glutathione (G). From top to bottom: G 30 mm + G–G 7.5 mm, G 15 mm + G–G 3.75 mm, and G 1.5 mm + G–G 0.375 mm; b) HPLC traces showing the component composition at equilibrium after 18 h for exchange experiments starting from P1, P2, and P3 as thiols (100 μm each) in Tris buffer (300 mm Tris-HCl pH 7.4, 150 mm KCl) containing G (30 mm) + G–G (7.5 mm) and either in the absence of template or in the presence of quadruplex DNA (100 μm, bottom) or duplex DNA (100 μm, top). c) Histogram showing the percent change in each species of the equilibrium mixture upon introduction of either quadruplex (red) or duplex DNA (blue).
Among the conditions tested, the highest glutathione concentrations were optimal for the detection of DNA-template-induced amplification of species containing either P2 or P3. The concept of dynamic combinatorial chemistry strictly requires the system under study to establish equilibrium under the conditions of the selection experiments. To confirm true thermodynamic equilibrium for the disulfide exchange, the experiment was repeated starting from P1, P2, and P3–P3, instead of P1, P2, and P3 thiol. The same distribution of thiols and disulfides was obtained after 18 h despite the change from initial conditions, which confirms that the system was reversible and under thermodynamic control. Parallel experiments were then carried out with conditions identical to those described above, but in the presence of 100 μm biotinylated quadruplex or duplex DNA. Equilibrium was reached within 24 h and the same distribution of species was observed starting with either all three monomers as thiols or P1 and P2 as thiols and P3 in disulfide form. This confirmed a true equilibration of the system in the presence of template. HPLC traces showing the distribution of every species at equilibrium in the presence of either quadruplex or double-stranded DNA are shown in Figure 1 b.
Compared with the experiment carried out in the absence of template, a moderate amplification of disulfides P2–P3 and P3–P3 (130% and 140%, respectively) was observed upon the addition of quadruplex DNA. The quantities of all other DCL species were either unchanged or significantly decreased. Interestingly, substantially greater amplification effects were observed with double-stranded DNA as a template. In the presence of duplex DNA (Figure 1 c) the amounts of disulfides P2–P3, P3–P3 and P2–P2 at equilibrium increased significantly, corresponding to amplifications of 660, 295, and 220%, respectively. These results suggest that homo- and heterodisulfides of P2 and P3 are amplified, albeit to varying extents, in the presence of quadruplex and duplex DNA templates. Duplex DNA elicits a much stronger amplification of these selected species than does quadruplex DNA (for example, the increase in P2–P3 is 660% with duplex DNA and 130% for quadruplex DNA), which also suggests that the disulfides of P2 and P3 show a much greater affinity for duplexes than for quadruplexes.
Pyrrole polyamides have already been shown to bind A/T-rich duplexes as antiparallel dimers with a very high affinity (Kd values in the nm range). Our results suggest such polyamides bind quadruplex DNA with much lower affinity, in accordance with a comparative NMR spectroscopic study, which showed that both distamycin and netropsin possess a higher affinity for duplex DNA than for quadruplex DNA.[17] To confirm the relative binding of selected species (amplified or not) to both DNA targets, thermal melting studies were carried out under conditions similar to those of the DCC experiments (100 μm DNA, 300 mm Tris-HCl, 150 mm KCl) in the absence or presence of 100 μm selected disulfide ligand (Figure 2). The melting temperature of the duplex alone was found to be 43.5°C. No stabilization effect was observed upon incubation of DNA with disulfide P1–P1, which was not amplified. However, addition of either P2–P2 or P2–P3 to the double-stranded DNA produced an increase in the Tm value to 46.5°C (ΔTm = +3°C) and 48.5°C (ΔTm = +5°C), respectively. No significant shift in melting temperature was observed with quadruplex DNA and either P1–P1 or P2–P3 (data not shown), which may be a consequence of weaker (or less-stabilized) binding. This shows a correlation between the melting temperature values and the level of amplification observed in the DCC experiment, consistent with observations reported previously by Bugaut et al. for the dynamic combinatorial selection of nucleic acid modifications that stabilize DNA duplexes.[5c]
Figure 2.
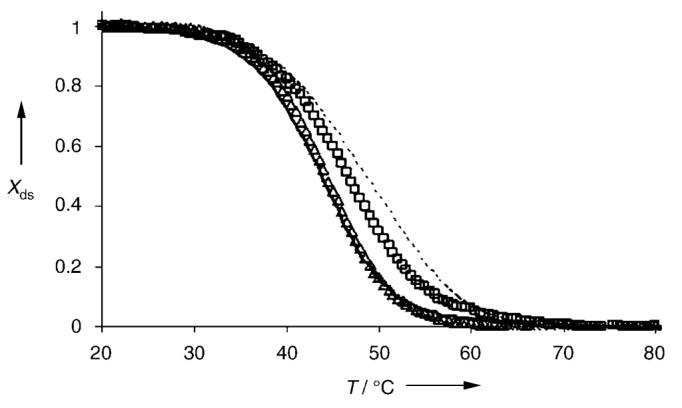
UV/Vis thermal denaturation curves obtained from 100 μm duplex DNA alone (——) and in the presence of either P1–P1 (△), P2–P2 (□), or P2–P3 (·····) at a concentration of 100 μm. Experiments were carried out in duplicate in buffer (300 mm Tris-HCl, pH 7.4 containing 150 mm KCl) in a cuvette with a 1-mm path length. Xds = fraction double-stranded DNA.
Recently, Poulin-Kerstien and Dervan showed that double-stranded DNA is capable of accelerating the irreversible formation of tandem hairpin dimers.[18] There is also a significant body of work on the use of DNA or RNA as a template for directing chemical reactions, primarily under irreversible conditions.[19] The study reported herein shows that DNA secondary structures (duplex or quadruplex) can also be used as templates that are capable of inducing, under reversible conditions, the formation of ligands that are thermodynamically stabilized through interactions with the DNA. A key feature of our studies is the demonstration that an increase in the concentration of buffering thiol (glutathione) is an effective means to sensitize the dynamic selection process to amplifications of pre-stabilized disulfide species. This may be particularly useful for more complex and larger libraries, in which there is an increased likelihood for mutual recognition between library members. Theoretical studies have demonstrated that the correlation between host–guest binding and amplification could vary from poor (with template in large excess) to good (with substoichiometric template).[20] The optimized dynamic combinatorial approach reported herein could facilitate the detection of only the strongest-binding ligands by preventing the formation of weaker-binding ligands with a large excess of competitor glutathione. This study also suggests that it is possible to compare the level of amplification of the same species obtained in experiments carried out in parallel but with different templates. This will be of particular interest for the discovery of DNA-binding ligands that bind selectively and with high-affinity.
Footnotes
This work was funded by the BBSRC and Cancer Research U.K. We thank Dr. A. Bugaut for proofreading the manuscript.
References
- 1.Vasquez KM, Glazer PM. Quart. Rev. Biophys. 2002;35:89. doi: 10.1017/s0033583502003773. [DOI] [PubMed] [Google Scholar]
- 2.Gottesfeld JM, Neely L, Trauger JW, Baird EE, Dervan PB. Nature. 1998;396:202. doi: 10.1038/387202a0. [DOI] [PubMed] [Google Scholar]
- 3.Siddiqui-Jain A, Grand CL, Bearss DJ, Hurley LH. Proc. Natl. Acad. Sci. USA. 2002;99:11593. doi: 10.1073/pnas.182256799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.For selected reviews, see: Lehn J-M. Chem. Eur. J. 1999;5:2455. Ramstrom O, Bunyapaiboonsri T, Lohmann S, Lehn J-M. Biochim. Biophys. Acta. 2002;1572:178. Rowan SJ, Cantrill SJ, Cousins GRL, Sanders JKM, Stoddart JF. Angew. Chem. 2002;114:938. doi: 10.1002/1521-3773(20020315)41:6<898::aid-anie898>3.0.co;2-e. Angew Chem. Int. Ed. 2002;41:898.
- 5.a) Klekota B, Miller BL. Tetrahedron. 1999;55:11687. [Google Scholar]; b) Karan C, Miller BL. J. Am. Chem. Soc. 2001;123:7455. doi: 10.1021/ja010325v. [DOI] [PubMed] [Google Scholar]; c) Bugaut A, Toulmé JJ, Rayner B. Angew. Chem. 2004;116:3206. doi: 10.1002/anie.200454041. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2004;43:3144. [Google Scholar]; d) Bugaut A, Bathany K, Schmitter J-M, Rayner B. Tetrahedron Lett. 2005;46:687. [Google Scholar]
- 6.Whitney AM, Ladame S, Balasubramanian S. Angew. Chem. 2004;116:1163. doi: 10.1002/anie.200353069. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2004;43:1143. [Google Scholar]
- 7.For a recent review, see: Dervan PB, Edelson BS. Curr. Opin. Struct. Biol. 2003;13:284. doi: 10.1016/s0959-440x(03)00081-2.
- 8.Baird ED, Dervan PB. J. Am. Chem. Soc. 1996;118:6141. [Google Scholar]
- 9.a) Randazzo A, Galeone A, Mayol L. Chem. Commun. 2001:1030. [Google Scholar]; b) David WM, Brodbelt J, Kerwin SM, Thomas PW. Anal. Chem. 2002;74:2029. doi: 10.1021/ac011283w. [DOI] [PubMed] [Google Scholar]
- 10.Cocco MJ, Hanakahi LA, Huber MD, Maizels N. Nucleic Acids Res. 2003;31:2944. doi: 10.1093/nar/gkg392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krishnan-Ghosh Y, Balasubramanian S. Angew. Chem. 2003;115:2221. doi: 10.1002/anie.200250551. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2003;42:2171. [Google Scholar]
- 12.Cuesta J, Read MA, Neidle S. Mini-Rev. Med. Chem. 2003;3:11. doi: 10.2174/1389557033405502. [DOI] [PubMed] [Google Scholar]
- 13.Yamamoto K, Tojo A, Aoki N, Shibuya M. Jpn. J. Cancer Res. 1993;84:1136. doi: 10.1111/j.1349-7006.1993.tb02813.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wurtz NR, Turner JM, Baird EE, Dervan PB. Org. Lett. 2001;3:1201. doi: 10.1021/ol0156796. [DOI] [PubMed] [Google Scholar]
- 15.The G quadruplex was folded under standard conditions. The single-stranded oligonucleotide, 5′-biotinylated (GTTAGG)5, was heated in buffer at 90°C and slowly cooled to room temperature overnight. Its fully folded structure was confirmed by circular dichroism spectroscopy and UV/Vis melting experiments.
- 16.Huc I, Nguyen R. Comb. Chem. High Throughput Screening. 2001;4:53. doi: 10.2174/1386207013331273. [DOI] [PubMed] [Google Scholar]
- 17.Randazzo A, Galeone A, Esposito V, Varra M, Mayol L. Nucleosides Nucleotides Nucleic Acids. 2002;21:535. doi: 10.1081/NCN-120015067. [DOI] [PubMed] [Google Scholar]
- 18.Poulin-Kerstien AT, Dervan PB. J. Am. Chem. Soc. 2003;125:15811. doi: 10.1021/ja030494a. [DOI] [PubMed] [Google Scholar]
- 19.a) Li X, Liu DR. Angew. Chem. 2004;116:4956. [Google Scholar]; Angew. Chem. Int. Ed. 2004;43:4848. [Google Scholar]; b) Gartner ZJ, Tse BN, Grubina R, Doyon JB, Snyder TM, Liu DR. Science. 2004;305:1601. doi: 10.1126/science.1102629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.a) Corbett PT, Otto S, Sanders JKM. Chem. Eur. J. 2004;10:3139. doi: 10.1002/chem.200400300. [DOI] [PubMed] [Google Scholar]; b) Corbett PT, Otto S, Sanders JKM. Org. Lett. 2004;6:1825. doi: 10.1021/ol049398k. [DOI] [PubMed] [Google Scholar]