

Abstract
Hepatocyte growth factor (HGF) is a potent hepatocyte mitogen that exerts opposing effects depending on cell density. Glutathione (GSH) is the main non-protein thiol in mammalian cells that modulates growth and apoptosis. We previously showed that GSH level is inversely related to cell density of hepatocytes and is positively related to growth. Our current work examined whether HGF can modulate GSH synthesis in a cell density dependent manner and how GSH in turn influence HGF’s effects. We found HGF treatment of H4IIE cells increased cell GSH levels only under subconfluent density. The increase in cell GSH under low density was due to increased transcription of GSH synthetic enzymes. This correlated with increased protein levels and nuclear binding activities of c-Jun, c-Fos, p65, p50, Nrf1 and Nrf2 to the promoter region of these genes. HGF acts as a mitogen in H4IIE cells under low cell density and protects against tumor necrosis factor α (TNFα)-induced apoptosis by limiting JNK activation. However, HGF is pro-apoptotic under high cell density and exacerbates TNFα-induced apoptosis by potentiating JNK activation. The increase in cell GSH under low cell density allows HGF to exert its full mitogenic effect but is not necessary for its anti-apoptotic effect.
Keywords: Hepatocyte growth factor, glutathione, glutamate-cysteine ligase, glutathione synthetase, tumor necrosis factor α, cell growth, apoptosis, JNK, AP-1, NFκB
INTRODUCTION
Glutathione (GSH) is the main non-protein thiol in mammalian cells that participates in many critical cellular functions including antioxidant defense and cell growth [1]. GSH synthesis occurs in the cytosol of all mammalian cells via two enzymatic steps: the formation of γ-glutamylcysteine from glutamate and cysteine catalyzed by glutamate-cysteine ligase (GCL), and formation of GSH from γ-glutamylcysteine and glycine catalyzed by GSH synthetase (GSS) [1]. GCL, the rate-limiting enzyme, is made up of a catalytic (GCLC) and a modifier (GCLM) subunit [2]. Coordinated up-regulation of GCL and GSS further enhances the GSH synthetic capacity [3]. We showed previously that GSH levels and GCLC transcription increased when cultured hepatocytes are plated under low cell density [4, 5] and during rat liver regeneration [6]. If the increase in GSH was blocked, regeneration was impaired [7]. A positive correlation between GSH level and cell growth was also observed in vitro in HepG2 cells [7].
Hepatocyte growth factor (HGF) is a potent mitogen for a variety of cells through activation of its receptor c-Met [8–10]. HGF acts as a hepatotropic factor in vivo after partial hepatectomy and was initially isolated as a potent mitogen for hepatocytes in primary culture [11, 12]. However, HGF’s mitogenic effect in primary cultures of rat hepatocytes depends on the cell density of the culture. It induced growth at low cell density whereas it induced albumin synthesis at high cell density [13]. The role of HGF in liver cancer has been highly controversial [14–17] and its effect in liver cancer cells often contradictory [18–23]. Recently, we reported that the effect of HGF in liver cancer cell line HepG2 depends on the cell density [24]. HGF acts as a mitogen at low cell density but causes cell cycle arrest at high cell density. This can help to reconcile conflicting reports in the literature regarding the effect of HGF in vitro and may explain why HGF only acts as a mitogen in vivo when there is loss of cell-cell contact.
Given our previous observation that cell GSH is increased under low cell density and is positively correlated with hepatocyte growth, the aims of our current work were to examine whether HGF can modulate GSH synthesis in a cell density dependent manner and how GSH in turn influence HGF’s effects. Our current work was carried out using rat hepatoma H4IIE cells to facilitate transfection studies with rat GSH synthetic enzyme promoter constructs. We found that HGF has growth and death modulatory effects, depending on the cell density and that GSH plays an important role in HGF’s mitogenic but not its anti-apoptotic effects.
MATERIALS AND METHODS
Materials
Fetal bovine serum was obtained from Atlas Biologicals (Fort Colins, CO). HGF and buthionine sulfoximine (BSO) were obtained from Sigma (St. Louis, MS). Recombinant rat TNF -α was obtained from Biosource (Camarillo, CA). [32P]dCTP and γ-32P ATP (3000 Ci/mmol) were purchased from PerkinElmer Life Sciences (DuPont, Boston, MA). Methyl-[3H]thymidine (25 Ci/mmol) was purchased from GE Healthcare (Piscataway, NJ). All other reagents were of analytical grade and were obtained from commercial sources.
Cell Culture and HGF treatment
H4IIE cells were obtained from the Cell Culture Core of the USC Liver Disease Research Center and grown according to instructions provided by the American Type Culture Collection (Rockville, MD) in Earle’s minimal essential medium supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine and 1% penicillin-streptomycin mixture. Cells were plated at different cell densities in 10% FBS for 24 hours and then switched to 0.1% FBS overnight. Cell density was determined at this time (2.5 to 6.2x104 cells/cm2) and medium was then changed to omit serum in the presence or absence of HGF (10 ng/ml) for 30 minutes to 48 hour for various assays described below.
RNA Isolation and Gene Expression Analysis
Total RNA was isolated by the TRIzol reagent (Invitrogen, Carlsbad, CA) from H4IIE cells. RNA concentration was determined spectrophotometrically before use and the integrity was checked by electrophoresis with subsequent ethidium bromide staining. Electrophoresis of RNA, gel blotting, and Northern hybridization analysis were performed on total RNA using standard procedures as described [25]. Specific GCLC, GCLM, GSS and β-actin cDNA probes were labeled with [32P]dCTP using a DECAprime II kit (Ambion, Austin, TX). Autoradiography and densitometry (Gel Documentation System, Scientific Technologies, Carlsbad, CA and NIH Image 1.63 software program) were used to quantitate relative RNA. Results of Northern blot analysis were normalized to β-actin.
Western Blot Analysis
H4IIE cells plated at different densities and treated with HGF (10 ng/ml) for varying duration were subjected to Western blot analysis as we described [24]. Equal amounts of protein (15 μg/well) were resolved in 12.5% SDS-polyacrylamide gels. Proteins were electrophoretically transferred to nitrocellulose membranes, blocked 1h with blocking buffer (Rockland, Gilbersville, PA), washed with tris-buffered saline/0.1% Tween 20, and incubated 1.5 hr with primary antibodies in tris-buffered saline/0.1% Tween 20. Blots were washed in tris-buffered saline/0.1% Tween 20 and incubated 1 hr with the secondary antibody in tris-buffered saline/0.1% Tween 20. Membranes were probed with anti-GCLC, GCLM (Novus Biologicals, Littleton, CO), GSS, c-Jun, JunD, c-Fos, FosB, p50, p65, RelB, c-Rel, Nrf1, Nrf2, phospho-JNK, and total JNK1 (Santa Cruz Biotechnology Inc., Santa Cruz, CA) antibodies. To ensure equal loading, membranes were stripped and re-probed with anti-actin antibodies (Santa Cruz Biotechnology, Santa Cruz, CA). A horseradish peroxidase-conjugated secondary antibody was used. Blots were developed by enhanced chemoluminescence.
Effect of Cell Density, HGF and TNFα on GSH Synthetic Enzymes’ Promoter Activity
H4IIE cells were plated under high cell density (HD, 6.2x104 cells/cm2) or low cell density (LD, 2.5x104 cells/cm2) and transfected with recombinant rat GCLC (−597/+2-GCLC-LUC), GCLM (−902/+3-GCLM-LUC) or GSS promoter construct (−561/+2-GSS-LUC) and treated with 15 ng/ml TNFα or 10 ng/ml HGF during the last 4 hours of the transfection. At this short duration, there was no detectable apoptosis or toxicity. We had previously shown that these promoter constructs contain maximal promoter activity that could be induced by TNFα [25]. Luciferase activities driven by these promoter luciferase gene constructs were measured as we described previously [25].
Electrophoretic Mobility Shift Assay (EMSA) and Supershift Assay
EMSAs and supershift assays were done as described [26]. Fifteen to 30 μg of nuclear protein from H4IIE cells plated at HD or LD and treated with HGF (10 ng/ml) for 30 minutes to 24 hours were preincubated with 2μg of poly(dI-dC) in a buffer containing 10 mM HEPES (pH 7.6), 50 mM KCl, 0.1 mM EDTA, 1 mM dithiothreitol, 5 mM MgCl2 and 10% glycerol for 10 minutes on ice. 32P-end labeled double-stranded DNA fragments used in the EMSAs and supershift assays contained AP-1 (−358 to −336) or NFκB (−380 to −358) elements of rat GCLC [25, 27], antioxidant response element (ARE) of rat GCLM (−300 to −279) [25], and AP-1/Nrf2 site of rat GSS (−194 to −171) [28]. Mixtures were incubated for 20 minutes on ice, loaded on a 4% nondenaturing polyacrylamide gel and subjected to electrophoresis in 50 mM Tris, 45 mM borate, and 0.5 mM EDTA (pH 8.0). Gels were dried and subjected to autoradiography. Further confirmation of the identity of the binding proteins was done by antibody supershift assays for c-Jun, JunB, JunD, c-Fos, FosB, p50, p65, Nrf1 and Nrf2 (Santa Cruz Biotechnology Inc., Santa Cruz) as we described [25, 26].
Measurement of Mitogenic Response
H4IIE cells were plated at high cell density (HD, 6.2x104 cells/cm2) or low cell density (LD, 2.5x104 cells/cm2) and treated with 10 ng/ml HGF for up to 48 hours. DNA synthesis was measured by [3H]thymidine incorporation into DNA (1 μCi per well) during the last 4 hours of HGF treatment as we described [24].
Measurement of GSH Levels
GSH levels were measured in H4IIE cells plated under high or low cell density by the recycling method of Tietze as we described [6].
Measurement of Apoptosis
Apoptosis was measured by DNA fragmentation and Hoeschst staining as we described [29].
JNK Activation
JNK activation was documented by measuring levels of phosphorylated JNK normalized to total JNK using Western blot analysis as described above.
Statistical analysis
Data are given as mean±s.e.m. and statistical analysis was performed using ANOVA followed by Fisher’s test for multiple comparisons. For changes in mRNA and protein levels, ratios of various genes to β-actin, or various proteins to actin densitometric values were compared by ANOVA. Significance was defined by p<0.05.
RESULTS
Effect of Cell Density and HGF on GSH Levels in H4IIE Cells
We had previously shown that there is an inverse relationship between plating cell density of primary rat hepatocytes and cell GSH levels [4]. The same is also true for rat hepatoma cell line H4IIE and HGF treatment increased GSH levels only under low cell density (Fig. 1).
Fig. 1.

Effect of HGF on cellular GSH levels. H4IIE cells were plated under low (2.5x104 cells/cm2) or high (6.2x104 cells/cm2) cell densities and treated with HGF (10 ng/ml) for 24 hours. At the end of treatment, low density cells were about 50% confluent and high density cells were fully confluent. Cell GSH levels were determined as described in Methods. *p<0.01 vs. untreated control, †p<0.05 vs. low density control from 4 to 10 experiments.
Effect of Cell Density and HGF on mRNA and Protein Levels of GCL and GSS
To see if the increase in cell GSH levels is due to increased expression of GSH synthetic enzymes, we examined mRNA and protein levels of GCLC, GCLM and GSS in H4IIE cells plated under high density (HD, 6.2x104 cells/cm2) or low density (LD, 2.5x104 cells/cm2). Figure 2A shows that HGF treatment under LD increased the mRNA levels of GCLC, GCLM and GSS in a time-dependent manner, reaching maximum effect at 4 hours after treatment. However, HGF treatment under HD had no effect on the mRNA levels of any of these genes (Fig. 2B). The increase in mRNA level translated to increased steady state levels of the proteins as well under LD (Fig. 3A). Note that the baseline protein levels of GCLC and GSS, but not GCLM, were higher under LD as compared to HD (see first two lanes of Fig. 3A, both GCLC and GSS baseline protein levels under LD were about 200% of HD). HGF treatment under LD increased the steady state levels of all three proteins, reaching a maximum about 4 to 8 hours after treatment (Fig. 3B, GCLC peaked at 4 hours at 423% of control, GCLM peaked at 8 hours at 450% of control, GSS peaked at 8 hours at 350% of control, values represent mean of two independent experiments). Again, HGF had no effect under HD (Fig. 3B).
Fig. 2.

HGF’s effect on the mRNA levels of GSH synthetic enzymes depends on the cell density. RNA (15 μg/lane) samples from H4IIE cells plated at low (part A, 2.5x104 cells/cm2) or high (part B, 6.2x104 cells/cm2) densities and treated with HGF (10 ng/ml) for 0 to 16 hours as described in Methods were analyzed by Northern blot analysis with different cDNA probes. The same membranes were rehybridized with a 32P-labeled β-actin cDNA probe. Representative Northern blots are shown. Numbers below each blot refer to densitometric changes expressed as % of 0 hour.
Fig. 3.
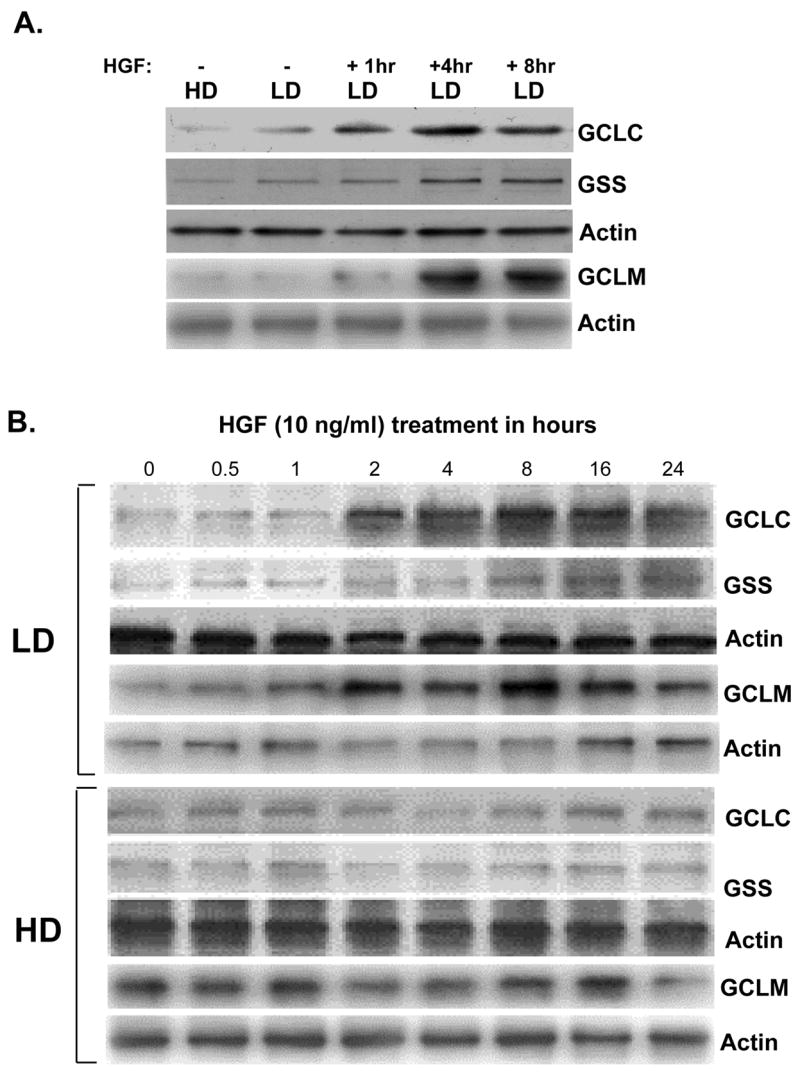
Effects of cell density and HGF on protein levels of GSH synthetic enzymes. H4IIE cells were plated at low density (LD) or high density (HD) as above and treated with HGF (10 ng/ml) for 0 to 24 hours and subjected to Western blot analysis for GCLC, GCLM, and GSS protein levels as described in Methods. Membranes were stripped and re-probed with actin for housekeeping control. Part A shows the effect of density alone and HGF treatment under low density (LD). Part B shows HGF treatment resulted in higher steady state GCLC, GCLM, and GSS protein levels only under LD.
Effects of Cell Density, HGF, and TNFα on Promoter Activities of GCLC, GCLM and GSS
We next examined whether increased expression of GSH synthetic enzymes lies at the level of gene transcription by measuring recombinant promoter activities of these genes. Figure 4 shows that HGF treatment for 4 hours induced the promoter activity of rat GCLC, GCLM, and GSS only under LD. TNFα also exerted a stimulatory effect on the same genes in a cell density dependent manner. Note that the basal promoter activity of GCLC and GSS was significantly higher under LD as compared to HD, but the basal promoter activity of GCLM was independent of cell density.
Fig. 4.

Effects of cell density and HGF on promoter activities of GSH synthetic enzymes. H4IIE cells were transfected with recombinant rat GCLC (A), GCLM (B), and GSS (C) promoter constructs and subsequently treated with HGF (10 ng/ml) or TNFα (15 ng/ml) for 4 hours. Promoter activities were measured as described in Methods. Results represent mean±s.e.m. from 3 to 4 experiments. *p<0.05 vs. HD control, †p<0.05 vs. LD control.
Effect of HGF on Binding Activity of Key Transcription Factors to GCLC, GCLM and GSS
HGF treatment in LD cells exerted a time-dependent increase in nuclear binding of c-Jun, JunB, FosB, c-Fos, p65 and p50 to the rat GCLC promoter fragments that contain functional AP-1 and NFκB sites [25–27] (Fig. 5). Similarly, HGF treatment in LD cells exerted a time-dependent increase in nuclear binding of c-Jun, c-Fos, Nrf1 and Nrf2 to the rat GCLM promoter fragment that contains a critical ARE site [25] (Fig. 6A), and a time-dependent increase in nuclear binding of Nrf2, c-Jun, c-Fos, JunB, JunD and FosB to the rat GSS promoter fragment that contains an AP-1/Nrf2 site [28] (Fig. 7A). HGF had no effect on nuclear binding activity to any of these promoter fragments under high cell density (Figs. 5, 6B and 7B).
Fig. 5.

Effect of HGF on AP-1 and NFκB binding to the rat GCLC promoter depends on cell density. H4IIE cells were plated under low or high cell density and treated with HGF (10 ng/ml) for 0 to 24 hours. EMSA with supershift was performed as described in Methods. Part A shows the time-dependent increase in nuclear binding to GCLC promoter fragment containing functional AP-1 site occurs only under low density (LD). Supershift revealed identity of the bound proteins to include c-Jun, JunB, FosB, and c-Fos. Part B shows higher nuclear binding to NFκB site under LD as compared to HD that is further induced by HGF treatment (10 ng/ml) for 8 hours. HGF treatment had no effect on NFκB nuclear binding under HD. Supershift analysis confirmed presence of both p65 and p50. Representative EMSAs with supershift analyses are shown.
Fig. 6.

Effect of HGF on nuclear binding to the rat GCLM ARE element depends on cell density. H4IIE cells were plated under low or high cell density and treated with HGF (10 ng/ml) for 0 to 24 hours. EMSA with supershift was performed as described in Methods. Part A shows the time-dependent increase in nuclear binding to GCLM promoter fragment containing a functional ARE site under low cell density. Supershift revealed identity of the bound proteins to include c-Jun, c-Fos, Nrf1 and Nrf2. Part B shows lack of effect of HGF on nuclear binding activity to the same promoter fragment when cells were plated under high density. Representative EMSAs with supershift analyses are shown.
Fig. 7.

Effect of HGF on nuclear binding to the rat GSS promoter fragment depends on cell density. H4IIE cells were plated under low or high cell density and treated with HGF (10 ng/ml) for 0 to 24 hours. EMSA with supershift was performed as described in Methods. Part A shows the time-dependent increase in nuclear binding to GSS promoter fragment containing an AP-1/Nrf2 site under low cell density. Supershift revealed identity of the bound proteins to include Nrf2, c-Jun, c-Fos, JunB, JunD and FosB. Part B shows lack of effect of HGF on nuclear binding activity to the same promoter fragment when cells were plated under high density. Representative EMSAs with supershift analyses are shown.
Effect of HGF on Expression of Transcription Factors
Increased nuclear binding activity can be due to increased level of the transcription factor or increased nuclear binding activity without a change in the level. Figure 8 shows that HGF treatment of LD cells led to a time-dependent increase in NFκB family members (p65, p50, RelB, c-Rel), AP-1 family members (c-Fos, FosB, c-Jun, JunD), Nrf1 and Nrf2. HGF treatment of HD cells had minimal to no effect on any of these proteins except for Nrf1.
Fig. 8.

HGF treatment increases the protein levels of key transcription factors under low cell density. H4IIE cells were plated under low or high cell densities as described above and treated with HGF (10 ng/ml) for up to 16 hours. Western blot analyses were performed for p65, p50, RelB, c-Rel, c-Fos, FosB, c-Jun, JunD, Nrf1 and Nrf2 as described in Methods. Actin served as housekeeping control. Numbers below the blots represent densitometric measurements expressed as % of control (0 hour). Representative Western blots are shown.
Effects of HGF and GSH on Mitogenic Response
Similar to our observations in HepG2 cells [24], HGF is only mitogenic in H4IIE cells plated under LD (Fig. 9A). To see if the HGF-mediated increase in GSH is important for the mitogenic response, we blocked the increase in GSH using BSO, an irreversible inhibitor of GCL [7]. We first established the optimal dose of BSO that resulted in maximal GSH depletion after 24 hours without causing any toxicity. This was achieved with 200 μM BSO (GSH was 39% of control). Lowering baseline GSH itself had no significant effect on [3H]thymidine incorporation but blocking the increase in GSH by HGF under LD condition significantly inhibited HGF’s mitogenic response (Fig. 9B, see Fig. 11B for cell GSH levels).
Fig. 9.

GSH increase contributes to HGF’s mitogenic effect in H4IIE cells plated under low cell density. Part A: H4IIE cells were plated at LD or HD at the time of HGF (10 ng/ml) treatment for up to 48 hours. DNA synthesis was measured by 3H-thymidine incorporation into DNA over the last 4 hours of the treatment or before the start of HGF treatment (time 0). *p<0.005 versus 0 hour, †p<0.05 versus 0 hour from 4 independent determinations. Part B: Increase in GSH under LD contributes to HGF’s mitogenic effect. H4IIE cells were plated under LD and treated with HGF (10 ng/ml), BSO (200 μM) or both BSO and HGF for 24 hours. DNA synthesis was measured as above. *p<0.005 vs. LD control, **p<0.05 vs. both HGF and BSO from 3 to 5 experiments.
Fig. 11.

Role of GSH in HGF’s anti-apoptotic effect under LD. H4IIE cells were plated under LD and treated with HGF (10 ng/ml), TNFα (15 ng/ml), BSO (200 μM) alone or together in various combinations for 24 hours. At the end of the treatment, cells were processed for measurements of apoptosis or GSH levels as described in Methods. Results are expressed as mean±s.e.m. from 5 to 9 experiments. Part A: Effects on apoptosis is shown; *p<0.005 vs. control, †p<0.01 vs. TNFα, **p<0.01 vs. BSO or TNFα, ††p<0.05 vs. BSO, HGF+TNFα, or BSO+TNFα. Part B: Effects on cell GSH is shown; *p<0.01 vs. control, †p<0.01 vs. HGF, ††p<0.05 vs. TNFα, **p<0.01 vs. control or HGF+TNFα.
Effect of HGF on Apoptosis is Cell Density-Dependent
TNFα induced apoptosis in H4IIE cells, regardless of the cell density, although the magnitude is higher under high cell density (Fig. 10). However, HGF’s effect on apoptosis depends on the cell density. Under LD, HGF protected against TNFα-induced apoptosis; under HD, HGF induced apoptosis by itself and potentiated TNFα-induced apoptosis (Fig. 10). To see if the HGF-mediated increase in GSH is important in the anti-apoptotic response under LD, we used BSO to block the increase in GSH. Figure 11 shows that BSO treatment, which reduced the cell GSH level by 61% (Fig. 11B), greatly potentiated TNFα-induced apoptosis (Fig. 11A). However, despite blocking the increase in GSH by BSO, HGF was still protective against TNFα-induced apoptosis (compare BSO+TNFα to BSO+HGF+TNFα). These results suggest lowering GSH exacerbated TNFα-induced apoptosis but HGF’s anti-apoptotic effect is GSH-independent.
Fig. 10.

HGF modulates apoptosis in a cell density-dependent manner. H4IIE cells were plated under low or high cell density and treated with HGF (10 ng/ml), TNFα (15 ng/ml) separately or together for 24 hours. Part A DNA fragmentation occurred after TNFα treatment in both densities but after HGF treatment only under HD. Furthermore, combination of HGF with TNFα resulted in less DNA fragmentation as compared to TNFα alone under LD, but more DNA fragmentation as compared to TNFα alone under HD. Part B shows quantitative apoptosis measurements by Hoeschst staining as described in Methods. *p<0.01 vs. respective controls, †p<0.05 vs. TNFα from 3 experiments.
Molecular Mechanism of Cell Density’s Influence on HGF’s Apoptotic Effects
HGF has been reported to either activate JNK [30–32] or inhibit JNK [33] and to be either pro-apoptotic [20, 31, 32] or anti-apoptotic [33, 34]. TNFα is known to induce apoptosis via sustained JNK activation [35, 36]. GSH depletion is known to sensitize hepatocytes to TNFα-induced apoptosis in part via sustained JNK activation [35]; whereas in retinal epithelial cells, HGF protected against cell death induced by GSH depletion by in part raising mitochondrial GSH [34]. These complex interactions and contradictory reports prompted us to examine the effects of HGF, GSH and TNFα on JNK activation under different cell densities.
HGF’s effect on JNK activation is dependent on cell density. Under high cell density, HGF activated both JNK forms; while under low cell density, HGF exerted minimal effect on JNK (Fig. 12A). TNFα activated JNK regardless of cell density, although the magnitude was higher under high cell density. BSO treatment potentiated TNFα-induced JNK activation. Interestingly, HGF lowered TNFα-mediated JNK activation under low cell density, but it potentiated TNFα-induced JNK activation under high cell density (Fig. 12B).
Fig. 12.

Modulation of JNK activation by HGF is cell density dependent. Part A: H4IIE cells were plated under HD or LD and treated with HGF (10 ng/ml) for up to 24 hours. Western blot analysis of pJNK1/2 and total JNK1 was performed as described in Methods. Part B: H4IIE cells were plated under HD or LD and treated with HGF (10 ng/ml), TNFα (15 ng/ml), BSO (200 μM) alone or together for 24 hours. Western blot analysis of pJNK1/2 and total JNK1 was performed as described in Methods. Densitometric changes from three experiments are shown in the table. Representative Western blots are shown.
DISCUSSION
We recently showed that the effect of HGF on growth depends on the plating cell density in HepG2 cells [24]. Under low cell density HGF acts as a mitogen and induces the expression of genes associated with growth; whereas under high confluent cell density, HGF induces p21 and p27 and causes cell cycle arrest. This observation may explain why HGF only acts as a mitogen in vivo when there is loss of cell-cell contact [37]. It also helps to reconcile many contradictory reports in the literature regarding the effect of HGF on growth both in vitro and in vivo [14–23, 31, 38]. In the current work we examined the influence of HGF on GSH status because GSH is another parameter known to influence hepatocyte growth. Thus, GSH level increases during rapid liver growth [6] and GSH status directly correlates with the growth of liver cancer cells [7]. Others have also shown that increased GSH promotes growth of metastatic melanoma cells in the liver [39]. In cultured rat hepatocytes, GSH level is inversely correlated with plating cell density [4]. Thus, it is of interest to know whether HGF influences GSH status in liver cancer cells and whether this depends on the cell density. Furthermore, it is also of interest to know whether GSH in turn modulates the effects of HGF. In this regards, we examined how GSH status influenced HGF’s effect on both growth and apoptosis.
Similar to what we reported in primary cultures of rat hepatocytes [4], cell GSH levels are also inversely correlated to plating cell density in rat hepatoma cells H4IIE. HGF treatment increased cell GSH levels only in subconfluent low density cells. HGF has been reported to increase GSH level and GCL activity in rat hepatocytes [40]. However, the molecular mechanism for the increase in GCL activity was not investigated in that work. We found that the increase in cell GSH levels can be explained by increased expression of genes involved in GSH biosynthesis, namely GCL (both GCLC and GCLM subunits) and GSS. The mechanism lies at the transcriptional level as HGF induced the recombinant promoter activity of all three genes. Cell density by itself also influenced the promoter activity of GCLC and GSS but not GCLM. This is reflected at the protein level as well. Interestingly, cell density also exerted a strong influence on TNFα-mediated increase in promoter activity, being inductive only at low cell density. These findings stress the importance of cell density as a variable that will influence experimental outcome.
To further investigate the molecular mechanisms of the increase in GCL and GSS gene expression, we examined the nuclear binding activities of key transcription factors that we have shown to regulate the expression of these genes. For the rat genes, both AP-1 and NFκB can trans-activate GCLC at functional AP-1 and NFκB sites, respectively [25–28]; whereas Nrf2 and c-Jun trans-activate a functional ARE site in the rat GCLM promoter [25], and multiple functional AP-1 sites exist in the rat GSS [28]. Furthermore, Nrf1 and Nrf2 can positively influence rat GCLC and GSS expression despite lack of ARE sites because these transcription factors can positively influence the expression of c-Jun, c-Fos, p50 and p65 [26]. Consistently, HGF treatment of low density cells led to increased AP-1 and NFκB binding to the rat GCLC promoter fragments containing the functional AP-1 and NFκB sites, c-Jun and Nrf2 to the rat GCLM fragment containing the functional ARE site, and Nrf2, c-Jun and c-Fos binding to the rat GSS promoter containing an AP-1/Nrf2 site. We further showed that this increased nuclear binding is largely due to increased expression of these transcription factors. Collectively, these results show that HGF treatment of low density cells resulted in increased expression of AP-1, NFκB and Nrf2, with subsequent increase in gene transcriptional rates of GCL and GSS and increased steady state cell GSH levels.
Similar to HepG2 cells, HGF treatment of H4IIE cells led to increased DNA synthesis only when cells were plated under low density. Since GSH synthesis is induced under low cell density by HGF and GSH level positively correlates with growth, we next asked the question whether the increase in GSH is important for the mitogenic effect of HGF. We found that when the increase in GSH was blocked, the mitogenic activity of HGF is significantly inhibited but not entirely eliminated. Thus, increased GSH contributes to increased DNA synthesis. GSH is known to modulate DNA synthesis by maintaining reduced glutaredoxin or thioredoxin, which are required for the activity of ribonucleotide reductase, the rate-limiting enzyme in DNA synthesis [41]. Alternatively, an increase in the cellular GSH content may change the thiolredox status of the cell which is proportional to [GSH]2/[GSSG] [42]. This may affect the expression or activity of factors important for cell cycle progression.
GSH also modulates TNFα-induced apoptosis [35]. This prompted us to examine whether HGF can modulate TNFα-induced apoptosis in a cell density-dependent manner. We found that HGF by itself had no toxicity under low cell density and protected against TNFα-induced apoptosis. However, the scenario is quite different under high cell density. HGF induced apoptosis by itself and potentiated TNFα-induced apoptosis. To see if the HGF’s protection against TNFα-induced apoptosis under low cell density is due to increased GSH, we blocked the increase in GSH with BSO. Lowering GSH itself exacerbated TNFα-induced apoptosis, which is consistent with previous report [35]. However, HGF was still protective even though GSH synthesis was blocked. These results suggest that while GSH does modulate TNFα-induced apoptosis, HGF’s protective effect is independent of GSH.
An apparent discrepancy is the finding that despite the ability of TNFα to increase the promoter activity of GSH synthetic enzymes to even higher levels than HGF (Fig. 4), cell GSH levels were lower in TNFα treated cells (Fig. 11B). Two reasons explain this discrepancy. First is that the promoter analysis was done with only 4 hours of treatment with TNFα, whereas GSH levels were measured after 24 hours. TNFα induced apoptosis in H4IIE cells when treated for 24 hours irregardless of cell density (but no toxicity at 4 hours). Second, toxic dose of TNFα is well known to induce oxidative stress, which will consume GSH as GSH is oxidized to GSSG and GSSG is pumped out of the cell. Thus, cellular GSH level is determined by the balance between biosynthesis and consumption (such as oxidative stress) [1]. Indeed, Morales et al showed TNFα’s effect on cell GSH levels depended on the dose. At lower doses, cell GSH levels were increased but at higher doses, cell GSH levels were lower. This was due to increased oxidative stress [43].
TNFα is known to induce hepatocyte apoptosis via sustained JNK activation, which is prevented by NFκB [36, 44–46]. JNK exists in two isoforms, JNK1 and JNK2, in most cell types. Jnk1 and jnk2 null mice are viable but double knockout is embryonically lethal, suggesting JNK1 and JNK2 have redundant functions [46]. With respect to c-Jun, JNK1 and JNK2 have been shown to play distinct roles, with JNK1 mediating the majority of c-Jun phosphorylation whereas JNK2 lacks this kinase activity and actually can decrease c-Jun stability when it binds to c-Jun [36, 47]. There are conflicting reports on the role of the two JNK forms in mediating the pro-apoptotic effects of TNFα, with JNK1 being responsible in fibroblasts [45] but JNK2 being responsible in galactosamine/lipopolysaccharide-induced liver injury in vivo [46]. HGF’s effect on JNK is highly controversial, as both activation [30–32] and inhibition [33] have been reported. We next investigated the influence of cell density and GSH on this pathway. Interestingly, we found that at high density, HGF treatment by itself led to JNK activation of both JNK forms. TNFα also activated JNK and together the level of JNK activation was further enhanced. At low cell density, HGF by itself had little influence on JNK activation, but it reduced TNFα-mediated JNK activation. Since NFκB is known to keep JNK activation in check, it is likely that the increase in NFκB by HGF under low cell density contributes to the reduction in JNK activation by TNFα. Lowering cell GSH by BSO enhanced JNK activation by TNFα. This is consistent with previous report [35]. Taken together, these results show that cell density has a strong influence on HGF-activation of JNK. Under high cell density, HGF is pro-apoptotic and further exacerbates TNFα-induced apoptosis because of higher and more sustained JNK activation. The opposite occurs under low cell density. How cell density influence HGF’s effect on JNK activation is unknown and will require further investigation. However, these results can explain the contradictory reports in the literature regarding HGF’s effect on JNK activation and apoptosis.
Question can be raised about the relevance of our findings in vivo. Loss of cell-cell contact is a critical step in cancer metastasis, including liver cancer [48]. Others have shown that increased GSH content in B16 melanoma cells protect them from hepatic sinusoidal cell-mediated oxidative damage and promote survival and metastasis [39]. We showed that HGF and its receptor c-Met are both up-regulated in HCC [24]. Our current data would suggest that HGF acts to promote further growth and protect liver cancer cells from apoptotic insult when they lose cell-cell contact. This way HGF can further facilitate the invasive and metastatic potential of liver cancer cells.
In summary, HGF induces the expression of GSH synthetic enzymes and raises cellular GSH levels when H4IIE cells are plated under low cell density. This is a result of HGF’s ability to increase the expression of key transcription factors known to trans-activate these genes. The increase in GSH is important for the mitogenic response of HGF under low cell density. HGF also modulates apoptosis in a cell density-dependent manner, being pro-apoptotic under high cell density but anti-apoptotic under low cell density. The mechanism of anti-apoptosis is GSH-independent, and is due to the influence of cell density on HGF-mediated changes in the JNK signaling pathway.
Acknowledgments
This work was supported by NIH grant DK45334 (S. C. Lu). H4IIE cells were provided by the Cell Culture Core and confocal microscope was provided by the Cell Biology Core of the USC Research Center for Liver Diseases (DK48522).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lu SC. Regulation of hepatic glutathione synthesis: Current concept and controversies. FASEB J. 1999;13:1169–1183. [PubMed] [Google Scholar]
- 2.Huang C, Chang L, Anderson ME, Meister A. Catalytic and regulatory properties of the heavy subunit of rat kidney γ-glutamylcysteine synthetase. J Biol Chem. 1993;268:19675–19680. [PubMed] [Google Scholar]
- 3.Huang ZZ, Yang HP, Chen CJ, Zheng ZH, Lu SC. Inducers of γ-glutamylcysteine synthetase and their effects on glutathione synthetase expression. Biochim Biophys Acta. 2000;1493:48–55. doi: 10.1016/s0167-4781(00)00156-1. [DOI] [PubMed] [Google Scholar]
- 4.Lu SC, Ge J. Loss of suppression of GSH synthesis under low cell density in primary cultures of rat hepatocytes. Am J Physiol. 1992;263:C1181–1189. doi: 10.1152/ajpcell.1992.263.6.C1181. [DOI] [PubMed] [Google Scholar]
- 5.Cai J, Sun WM, Lu SC. Hormonal and cell density regulation of hepatic γ-glutamylcysteine synthetase gene expression. Mol Pharmacol. 1995;48:212–218. [PubMed] [Google Scholar]
- 6.Huang ZZ, Li H, Cai J, Kuhlenkamp J, Kaplowitz N, Lu SC. Changes in glutathione homeostasis during liver regeneration in the rat. Hepatology. 1998;27:147–153. doi: 10.1002/hep.510270123. [DOI] [PubMed] [Google Scholar]
- 7.Huang ZZ, Chen CJ, Zeng ZH, Yang HP, Oh J, Chen LX, Lu SC. Mechanism and significance of increased glutathione level in human hepatocellular carcinoma and liver regeneration. FASEB J. 2001;15:19–21. doi: 10.1096/fj.00-0445fje. [DOI] [PubMed] [Google Scholar]
- 8.Zarnegar R, Michalopoulos GK. The many faces of hepatocyte growth factor: From hepatopoiesis to hematopoiesis. J Cell Biol. 1995;129:1177–1180. doi: 10.1083/jcb.129.5.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bottaro DP, Rubin JS, Faletto DL, Chan AM, Kmiecik TE, Vande Woude GF, Aaronson SA. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science. 1991;251:802–804. doi: 10.1126/science.1846706. [DOI] [PubMed] [Google Scholar]
- 10.Naldini L, Vigna E, Narsimhan RP, Gaudino G, Zarnegar R, Michalopoulos GK, Comoglio PM. Hepatocyte growth factor (HGF) stimulates the tyrosine kinase activity of the receptor encoded by the proto-oncogene c-MET. Oncogene. 1991;6:501–504. [PubMed] [Google Scholar]
- 11.Borowiak M, Garratt AN, Wustefeld T, Strehle M, Trautwein C, Birchmeier C. Met provides essential signals for liver regeneration. Proc Natl Acad Sci U S A. 2004;101:10608–10613. doi: 10.1073/pnas.0403412101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nakamura T, Nishizawa T, Hagiya M, Seki T, Shimonishi M, Sugimura A, Tashiro K, Shimizu S. Molecular cloning and expression of human hepatocyte growth factor. Nature. 1989;342:440–443. doi: 10.1038/342440a0. [DOI] [PubMed] [Google Scholar]
- 13.Takehara T, Matsumoto K, Nakamura T. Cell density-dependent regulation of albumin synthesis and DNA synthesis in rat hepatocytes by hepatocyte growth factor. J Biochem. 1992;112:330–334. doi: 10.1093/oxfordjournals.jbchem.a123900. [DOI] [PubMed] [Google Scholar]
- 14.Liu ML, Mars WM, Michalopoulos GK. Hepatocyte growth factor inhibits cell proliferation in vivo of rat hepatocellular carcinomas induced by diethylnitrosamine. Carcinogenesis. 1995;16:841–843. doi: 10.1093/carcin/16.4.841. [DOI] [PubMed] [Google Scholar]
- 15.Yaono M, Hasegawa R, Mizoguchi Y, Futakuchi M, Nakamura T, Ito N, Shirai T. Hepatocyte growth factor enhancement of preneoplastic hepatic foci development in rats treated with diethylnitrosamine and N-ethyl-N-hydroxyethylnitrosamine. Jap J Cancer Res. 1995;86:718–723. doi: 10.1111/j.1349-7006.1995.tb02459.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Santoni-Rugiu E, Preisegger KH, Kiss A, Audolfsson T, Shiota G, Schmidt EV, Thorgeirsson SS. Inhibition of neoplastic development in the liver by hepatocyte growth factor in a transgenic mouse model. Proc Natl Acad Sci, USA. 1996;93:9577–9582. doi: 10.1073/pnas.93.18.9577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sakata H, Takayama H, Sharp R, Rubin JS, Merlino G, LaRochelle WJ. Hepatocyte growth factor/scatter factor overexpression induces growth, abnormal development, and tumor formation in transgenic mouse livers. Cell Growth Differ. 1996;7:1513–1523. [PubMed] [Google Scholar]
- 18.Lee HS, Huang AM, Huang GT, Yang PM, Chen PJ, Sheu JC, Lai MY, Lee SC, Chou CK, Chen DS. Hepatocyte growth factor stimulates the growth and activates mitogen-activated protein kinase in human hepatoma cells. J Biomed Sci. 1998;5:180–184. doi: 10.1007/BF02253467. [DOI] [PubMed] [Google Scholar]
- 19.Jiang Y, Xu W, Lu J, He F, Yang X. Invasiveness of hepatocellular carcinoma cell lines: contribution of hepatocytes growth factor, c-met, and transcription factor Ets-1. Biochem Biophys Res Comm. 2001;286:1123–1130. doi: 10.1006/bbrc.2001.5521. [DOI] [PubMed] [Google Scholar]
- 20.Matteucci E, Castoldi R, Desiderio MA. Hepatocyte growth factor induces pro-apoptotic genes in HepG2 hepatoma but not in B16-F1 melanoma cells. J Cell Physiol. 2001;186:387–396. doi: 10.1002/1097-4652(2000)9999:9999<000::AID-JCP1033>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 21.Tsukada Y, Tanaka T, Miyazawa K, Kitamura N. Involvement of down-regulation of Cdk2 activity in hepatocyte growth factor-induced cell cycle arrest at G1 in the human hepatocellular carcinoma cell line HepG2. J Biochem. 2004;136:701–709. doi: 10.1093/jb/mvh177. [DOI] [PubMed] [Google Scholar]
- 22.Wang SY, Chen B, Zhan YQ, Xu WX, Li CY, Yang RF, Zheng H, Yue PB, Larsen SH, Sun HB, Yang X. SU5416 is a potent inhibitor of hepatocyte growth factor receptor (c-Met) and blocks HGF-induced invasiveness of human HepG2 hepatoma cells. J Hepatol. 2004;41:267–273. doi: 10.1016/j.jhep.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 23.Han J, Tsukada Y, Hara E, Kitamura N, Tanaka T. Hepatocyte growth factor induces redistribution of the p21CIP1 and p27KIP1 through ERK-dependent p16INK4a up-regulation, leading to cell cycle arrest at G1 in HepG2 hepatoma cells. J Biol Chem. 2005;280:31548–31556. doi: 10.1074/jbc.M503431200. [DOI] [PubMed] [Google Scholar]
- 24.Yang HP, Magilnick N, Noureddin M, Mato JM, Lu SC. Effect of hepatocyte growth factor on methionine adenosyltransferase genes and growth is cell density-dependent in HepG2 cells. J Cell Physiol. 2007;210:766–773. doi: 10.1002/jcp.20891. [DOI] [PubMed] [Google Scholar]
- 25.Yang HP, Magilnick N, Ou XP, Lu SC. Tumor necrosis alpha induces coordinated activation of rat GSH synthetic enzymes via NFκB and AP-1. Biochem J. 2005;391:399–408. doi: 10.1042/BJ20050795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang HP, Magilnick N, Lee C, Kalmaz D, Ou XP, Chan JY, Lu SC. Nrf1 and Nrf2 regulate rat glutamate-cysteine ligase catalytic subunit transcription indirectly via AP-1 and NFκB. Mol Cell Biol. 2005;25:5933–5946. doi: 10.1128/MCB.25.14.5933-5946.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang HP, Wang JH, Huang ZZ, Ou XP, Lu SC. Cloning and characterization of the 5′-flanking region of the rat glutamate-cysteine ligase catalytic subunit. Biochem J. 2001;357:447–455. doi: 10.1042/0264-6021:3570447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang HP, Zeng Y, Lee TD, Yang Y, Ou XP, Chen LX, Haque M, Rippe R, Lu SC. Role of AP-1 in the co-ordinate induction of rat glutamate-cysteine ligase and glutathione synthetase by tert-butylhydroquinone. J Biol Chem. 2002;277:35232–35239. doi: 10.1074/jbc.M203812200. [DOI] [PubMed] [Google Scholar]
- 29.Yang HP, Sadda MR, Yu V, Zeng Y, Lee TD, Ou XP, Chen LX, Lu SC. Induction of human methionine adenosyltransferase 2A expression by tumor necrosis factor a: role of NFκB and AP-1. J Biol Chem. 2003;278:50887–50896. doi: 10.1074/jbc.M307600200. [DOI] [PubMed] [Google Scholar]
- 30.Rodrigues GA, Park M, Schlessinger J. Activation of the JNK pathway is essential for transformation by the Met oncogene. EMBO J. 1997;16:2634–2645. doi: 10.1093/emboj/16.10.2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Conner EA, Teramoto T, Wirth PJ, Kiss A, Garfield S, Thorgeirsson SS. HGF-mediated apoptosis via p53/bax-independent pathway activating JNK1. Carcinogenesis. 1999;20:583–590. doi: 10.1093/carcin/20.4.583. [DOI] [PubMed] [Google Scholar]
- 32.Kim WH, Matsumoto K, Bessho K, Nakamura T. Growth inhibition and apoptosis in liver myofibroblasts promoted by hepatocyte growth factor leads to resolution from liver cirrhosis. Am J Pathol. 2005;166:1017–1028. doi: 10.1016/S0002-9440(10)62323-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reveneau S, Paumelle R, Deheuninck J, Leroy C, Van De Launoit Y, Fafeur V. Inhibition of JNK by HGF/SF prevents apoptosis induced by TNF-α. Ann NY Acad Sci. 2003;1010:100–103. doi: 10.1196/annals.1299.016. [DOI] [PubMed] [Google Scholar]
- 34.Jin M, Yaung J, Kannan R, He S, Ryan SJ, Hinton DR. Hepatocyte growth factor protects RPE cells from apoptosis induced by glutathione depletion. IOVS. 2005;46:4311–4319. doi: 10.1167/iovs.05-0353. [DOI] [PubMed] [Google Scholar]
- 35.Matsumaru K, Ji C, Kaplowitz N. Mechanisms for sensitization to TNF-induced apoptosis by acute glutathione depletion in murine hepatocytes. Hepatology. 2003;37:1425–1434. doi: 10.1053/jhep.2003.50230. [DOI] [PubMed] [Google Scholar]
- 36.Schwabe RF, Brenner DA. Mechanisms of liver injury. I. TNF-α-induced liver injury: role of IKK, JNK, and ROS pathways. Am J Physiol. 2006;290:G583–589. doi: 10.1152/ajpgi.00422.2005. [DOI] [PubMed] [Google Scholar]
- 37.Michalopoulos GK, DeFrances MC. Liver regeneration. Science. 1997;276:60–66. doi: 10.1126/science.276.5309.60. [DOI] [PubMed] [Google Scholar]
- 38.Bell A, Chen Q, DeFrances MC, Michalopoulos GK, Zarnegar R. The five amino acid-deleted isoform of hepatocytes growth factor promotes carcinogenesis in transgenic mice. Oncogene. 1999;18:887–895. doi: 10.1038/sj.onc.1202379. [DOI] [PubMed] [Google Scholar]
- 39.Carretero J, Obrador E, Anasagasti MJ, Martin JJ, Vidal-Vanaclocha F, Estrela JM. Growth-associated changes in glutathione content correlate with liver metastatic activity of B16 melanoma cells. Clin Exp Metastasis. 1999;17:567–574. doi: 10.1023/a:1006725226078. [DOI] [PubMed] [Google Scholar]
- 40.Tsuboi S. Elevation of glutathione level in rat hepatocytes by hepatocyte growth factor via induction of γ-glutamylcysteine synthetase. J Biochem. 1999;126:815–820. doi: 10.1093/oxfordjournals.jbchem.a022521. [DOI] [PubMed] [Google Scholar]
- 41.Holmgren A. Regulation of ribonucleotide reductase. Curr Top Cell Regul. 1981;19:47–76. doi: 10.1016/b978-0-12-152819-5.50019-1. [DOI] [PubMed] [Google Scholar]
- 42.Hutter DE, Till BG, Greene JJ. Redox state changes in density-dependent regulation of proliferation. Exp Cell Res. 1997;232:435–438. doi: 10.1006/excr.1997.3527. [DOI] [PubMed] [Google Scholar]
- 43.Morales A, García-Ruiz C, Miranda M, Marí M, Colell A, Ardite E, Fernández-Checa JJ. Tumor necrosis factor increases hepatocellular glutathione by transcriptional regulation of the heavy subunit chain of γ-glutamylcysteine synthetase. J Biol Chem. 1997;272:30371–30379. doi: 10.1074/jbc.272.48.30371. [DOI] [PubMed] [Google Scholar]
- 44.Papa S, Zazzeroni F, Pham CG, Bubici C, Franzoso G. Linking JNK signaling to NF-B: a key to survival. J Cell Sci. 2004;117:5197–5208. doi: 10.1242/jcs.01483. [DOI] [PubMed] [Google Scholar]
- 45.Liu J, Minemoto Y, Lin A. c-Jun N-Terminal Protein Kinase 1 (JNK1), but not JNK2, is essential for tumor necrosis factor alpha-induced c-Jun kinase activation and apoptosis. Mol Cell Biol. 2004;24:10844–10856. doi: 10.1128/MCB.24.24.10844-10856.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang Y, Singh R, Lefkowitch JH, Rigoli RM, Czaja MJ. Tumor necrosis factor-induced toxic liver injury results from JNK2-dependent activation of caspase-8 and the mitochondrial death pathway. J Biol Chem. 2006;281:15258–15267. doi: 10.1074/jbc.M512953200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sabapathy K, Hochedlinger K, Nam SY, Bauer A, Karin M, Wagner EF. Distinct roles for JNK1 and JNK2 in regulating JNK activity and c-Jun-dependent cell proliferation. Mol Cell. 2004;15:713–725. doi: 10.1016/j.molcel.2004.08.028. [DOI] [PubMed] [Google Scholar]
- 48.Nakanishi K, Sakamoto M, Yasuda J, Takamura M, Fujita N, Tsuruo T, Todo S, Hirohashi S. Critical involvement of the phosphatidylinositol 3-kinase/Akt pathway in anchorage-independent growth and hematogeneous intrahepatic metastasis of liver cancer. Cancer Res. 2002;62:2971–2975. [PubMed] [Google Scholar]