

Abstract
We have used interleukin-10 (IL-10) gene knockout mice (IL-10−/−) to examine the role of endogenous IL-10 in allergic lung responses to Aspergillus fumigatus Ag. In vitro restimulated lung cells from sensitized IL-10−/− mice produced exaggerated amounts of IL-4, IL-5, and interferon-γ (IFN-γ) compared with wild-type (WT) lung cells. In vivo, the significance of IL-10 in regulating responses to repeated A. fumigatus inhalation was strikingly revealed in IL-10−/− outbred mice that had a 50–60% mortality rate, while mortality was rare in similarly treated WT mice. Furthermore, IL-10−/− outbred mice exhibited exaggerated airway inflammation and heightened levels of IL-5 and IFN-γ in bronchoalveolar lavage (BAL) fluids. In contrast, the magnitude of the allergic lung response was similar in intranasally (i.n.) sensitized IL-10−/− and wild-type mice from a different strain (C57BL/6). Using a different route of priming (intraperitoneal) followed by one i.n. challenge we found that IL-10−/− C57BL/6 mice had heightened eosinophilic airway inflammation, BAL–IL-5 levels, and numbers of αβT cells in the lung tissues compared with WT mice. We conclude that IL-10 can suppress inflammatory Th2-like lung responses as well as Th1-like responses given the constraints of genetic background and route of priming.
A spergillus fumigatus is an opportunistic fungal pathogen for humans and animals causing lung hypersensitivities with and without life-threatening growth in the lungs or sinuses (reviewed in reference 1). In humans, lung hypersensitivity to A. fumigatus can occur in different forms (2). The two opposite extremes are asthma with increased serum IgE titers (2) and hypersensitivity pneumonitis with increased serum IgG and low IgE titers (3, 4). Clinically, asthma presents as recurrent bouts of dyspnoea due to bronchoconstriction, whereas hypersensitivity pneumonitis is characterized by bouts of dyspnoea accompanied by influenza-like symptoms (e.g., fever, fatigue). Immunologically, asthma has been associated with an exaggerated Th2 response promoting IgE synthesis, eosinophil infiltration, and activation of mast cells in the lungs (reviewed in reference 5). In contrast, hypersensitivity pneumonitis is characterized by neutrophil influx into the lungs at the acute phase and T cell and macrophage influx during the chronic phase of the disease (6). It is thought to be caused by excessive macrophage activation (6) due to an immune complex–mediated Arthus reaction (reviewed in reference 3) together with a CD4 T cell response probably mediated by Th1 cytokines (7). Most patients who are hypersensitive to A. fumigatus suffer from a disease called allergic bronchopulmonary aspergillosis (ABPA)1. The main features of this disease are activated Th2 cells (8) and asthma; however, IgG-mediated Arthus reactions (2) and autoimmune reactions (9) can also contribute to the pathogenesis.
Because IL-10 is constitutively produced by bronchial epithelial cells (10) and potentially inhibits cytokine production by cultured alveolar macrophages and lung dendritic cells (11–15), there has been considerable interest in the role of IL-10 in regulating pulmonary immune responses. IL-10 has been shown to suppress acute inflammation induced by the formation of antigen–antibody complexes in the lungs of mice (localized Arthus reaction) (16). However, little information is available concerning a role for IL-10 in regulating Th2-like responses leading to asthmatic lung hypersensitivity reactions. Analysis of bronchoalveolar lavage (BAL) fluids from asthmatic patients have produced puzzling results as they showed increased IL-10 mRNA expression by BAL cells (17) but diminished IL-10 protein in BAL fluids (18). Based on the results of numerous murine studies, it has been proposed that IL-10 enhances Th2 responses, albeit indirectly, by inhibiting an accompanying Th1 response (reviewed in reference 19). Although these studies evaluated antigen-induced responses in organs other than the lung, the general findings would suggest that IL-10 may actually contribute to the preferential generation of a Th2 response deemed responsible for allergic pulmonary reactions. On the other hand, a recent study showed that mice sytemically primed with OVA exhibited diminished lung eosinophilia upon rechallenge with aerosolized OVA if IL-10 was also administered (20). These latter studies show that IL-10 is at least capable of suppressing eosinophilic inflammation (Th2-like response) under certain in vivo conditions and thus may have some therapeutic value.
The present study has focused on the role of endogenous IL-10 in regulating allergic pulmonary reactions. Previous studies have shown that sensitization of BALB/c mice with A. fumigatus Ag normally induces a strong Th2-like response resulting in pulmonary eosinophilia and elevated serum IgE levels (21, 22). We have compared the responses of IL-10−/− and wild-type (WT) mice after repeated challenges with A. fumigatus Ag to identify altered reactions that may occur in the absence of IL-10 regulation (i.e., cytokine production, airway inflammation, airway hyperresponsiveness, and serum antibody titers). Furthermore, different routes of sensitization and strains of mice were used as these variables have been shown to influence the type and/or magnitude of an immune response elicited in other experimental systems.
Materials and Methods
Animals.
IL-10−/− outbred mice, generated on a mixed C57BL/6 × 129Sv F2 background (23), and outbred WT littermate mice were derived by cesarean section under specific pathogen-free conditions at Simonsen Laboratory (Gilroy, CA) and maintained in micro isolator cages in the animal facility at DNAX Research Institute (Palo Alto, CA) (24). Inbred C57BL/6 IL-10−/− mice were derived from outbred IL-10−/− mice by 12 backcrosses to C57BL/6 WT mice and interbreeding of heterozygous offspring. Heterozygous littermates and homozygous WT C57BL/6 mice purchased from The Jackson Laboratory (Bar Harbor, ME) were used as controls. All of the mice were maintained in the DNAX animal facility under identical conditions. IL-10−/−, WT, and sentinel mice were periodically examined by the Research Animal Diagnostic and Investigative Laboratory (University of Missouri, Columbia, MO). Bacterial cultures, parasitological examinations, serologic tests, and special histological stains were negative for known murine viral and bacterial pathogens. 6–9-wk-old mice were used in the experiments.
A. fumigatus Ag.
The A. fumigatus Ag preparation is a mixture of culture filtrate and mycelial extract prepared free of living organisms as described (21). This complex preparation has been particularly useful in detecting serum antibody levels and skin reactivity in human patients (25). Two batches of A. fumigatus Ag with equivalent activities (lot numbers 5323R1, 5325) were aliquotted at a concentration of 10 mg/ml and stored at −70°C for use in all the experiments described herein. The LPS content of the Ag extracts was less than 0.2 EU/100 μg of Ag when tested with the Limulus Amebocyte Lysate test (BioWhittaker, Walkersville, MD).
Sensitization with A. fumigatus Ag.
Wild-type and IL-10−/− mice were repeatedly primed either i.p. or intranasally (i.n.) followed by a final i.n. challenge (21). In all cases, 100 μg of Ag was used after dilution in PBS so as to deliver a total volume of 50 or 200 μl for i.n. or i.p. administration, respectively. All mice receiving an i.n. challenge were anaesthetized with isofluorane and held upright until all of the Ag administered onto their nostrils with a micropipette tip was inhaled.
Airway Inflammation.
Airway inflammation was analyzed using BAL. Lungs were lavaged three times with 1 ml of PBS. Approximately 2.4 ml of fluid was routinely recovered. Total cell numbers were determined using a hemacytometer. Differential cell counts were obtained from BAL cells spun onto slides with a cytocentrifuge and treated with Wright-Giemsa stain (Sigma Chem. Co., St. Louis, MO). 400 cells were enumerated. Absolute numbers of specific cell types present in BAL fluids were calculated from the recovered volume, total cell count, and the relative frequency of that particular cell type. BAL supernatants were collected by centrifugation and stored at −70°C until assayed for cytokine levels.
In Vitro Restimulation of Lung Cells.
Lung tissue, trimmed of bronchial lymph nodes and trachea, were minced through a sterile no. 100 steel mesh screen (Tylinter, Inc., Mentor, OH) into a petri dish containing 5 ml of HBSS and antibiotics. The lung cells were washed once with HBSS and resuspended at 8 × 106 cells/ ml in medium (RPMI-1640 supplemented with 10% FCS, 50 mM 2-ME, 100 U/ml penicillin, and 100 μg/ml streptomycin). 500 μl to 1 ml of lung cell suspension was pipetted into three sets of 24-well flat-bottomed plates (Falcon; Becton Dickinson Labware, Lincoln Park, NJ) containing an equal volume of medium or medium supplemented with A. fumigatus Ag (20 μg/ml). One set of plates had been previously coated with 12 μg/ml hamster anti–mouse CD3 mAb (clone 32C11). In the case of cultures containing wildtype lung cells, additional wells were supplemented with a neutralizing anti–IL-10 mAb (clone 2A5) at 10 μg/ml. Cultures were incubated at 37°C in a humidified atmosphere with 5% CO2. Supernatants were harvested 72 h later and stored at −70°C until assayed.
Measurement of Cytokine Levels in Culture Supernatants and BAL Fluids and Determination of Serum Antibody Levels.
Cytokine levels in BAL fluids and cell culture supernatants were determined by two-site sandwich ELISA assays using the following Ab pairs provided by Dr. J. Abrams and Dr. R.L. Coffman (DNAX): for IFN-γ, XMG1.2 and R504 polyclonal rabbit Ab; for IL-4, 11B11 and biotinylated BVD24G2.3; and for IL-5, TRFK5 and biotinylated TRFK4. Before cytokine analysis, BAL supernatants were concentrated six- to tenfold using Centricon 10 microconcentrators (Amicon, Beverly, MA) with a 10,000 MW cut off. The results were then corrected for the degree of concentration.
Serum IgE titers were determined by two-site sandwich ELISA assay using the mAb EM99 and NIP-conjugated rabbit anti– mouse IgE (provided by Dr. R.L. Coffman). IgG1 and IgG2a levels were determined using ELISA kits (Southern Biotechnology, Birmingham, AL) as per manufacturer's specifications.
Flow Cytometric Analysis of Intracellular Cytokine Production by Lung Cells.
The assay was performed according to Openshaw et al. (26) with slight modifications. In brief, cells recovered from murine lungs as described above were treated with 0.9% ammonium chloride to lyse erythrocytes, washed once with HBSS, resuspended in medium, and cultured at 4 × 106 cells/ml in 6-well flat-bottomed culture plates in 5-ml volumes. Cells from the HDK1 Th1 cell line (produces IFN-γ but not IL-4), or from the CDC25 Th2 cell line (produces IL-4 but not IFN-γ) were included as positive controls. All cell cultures were stimulated with PMA (50 ng/ml) and ionomycin (500 ng/ml) for 2–2.5 h in the presence of Fas–Ig fusion protein (5 μg/ml) (27) to prevent activation induced cell death. Brefeldin A (10 μg/ml) was then added to the cultures. After 2 h of incubation, DNAse I (10 μg/ml, Boehringer Mannheim, Mannheim, Germany) was added for a few min. The cells were removed, washed with PBS, fixed with formaldehyde (20–25 min), washed with PBS, and then stored in FACS® buffer (PBS, 1% BSA, 1 mM sodium azide) at 1–2 × 106 cells/ml at 4°C. The staining procedure was carried out at room temperature within 24 h. The presence of intracellular IL-4 and IFN-γ was detected with PE-conjugated 11B11 and FITC-conjugated AN18 mAb (provided by Dr. A. O'Garra; DNAX); PE- and FITC-conjugated rat IgG1 isotype-matched Ab (PharMingen, San Diego, CA) were used as negative controls. The cells were treated for 10 min with permeabilization buffer (0.5% saponin in FACS® buffer) containing 25 μg/ml Fc blocking solution (PharMingen), spun down, and then incubated with the anti-cytokine or control Ab for 30 min. The cells were washed with permeabilization buffer for two times and with FACS® buffer once before staining with Tri-Color™-conjugated anti-mouse αβTCR mAb (Caltec, San Francisco), or hamster Ig control Ab (Caltec). Samples were examined using a FACScan® flow cytometer (Beckton Dickinson, San Jose, CA). The data were analyzed using CellQuest software (Becton Dickinson).
Airway Responsiveness.
Airway responsiveness to i.v. acetylcholine (ACh) challenge was measured as previously described (28). In brief, mice were anesthetized with Nembutal sodium (Abbott Laboratories, North Chicago, IL) administered i.p. at 0.04 mg/g body weight. The tracheas were surgically exposed, cannulated with a blunt-ended, 20-gauge angiocatheter, and connected to a rodent ventilator (Harvard Apparatus, South Natick, MA). Mice were ventilated with 100% oxygen at a rate of 150 breaths per min and a tidal volume of 9 μl/g. Following paralysis with pancuronium bromide (4 μg/g; Gensia Laboratories, Irvine, CA), i.v. access was established using a 27-gauge needle placed into a tail vein and mice were placed into a rodent plethysmograph capable of determining tidal volume, airflow, dynamic compliance, and transthoracic resistance continuously. Airway responses were expressed as the amount of ACh required to double baseline transthoracic resistance (PC200).
Microscopic Analysis.
Lungs from control and sensitized mice were inflated with one ml of 10% (vol/vol) neutral buffered formaldehyde. The inflated lungs were then placed into 10% (vol/ vol) neutral buffered formaldehyde. Tissues were routinely processed, sectioned at 6 μm in a dorsal plane through the center of the lung, and stained with hematoxylin and eosin (Sigma Chem. Co.). Sections were analyzed by two independent examiners who were blinded to treatment information.
Statistical Analysis.
Experimental groups were compared to each other or to control groups using the nonparametric, unpaired, two-tailed Mann-Whitney U test. For airway hyperresponsiveness, statistical analysis was performed using analysis of variance (ANOVA) and the Bonferroni T test, on data expressed as the logarithm of PC200. The statistical comparison of mortality seen in different experimental groups was performed using Fisher's Exact Test.
Result
Experimental Protocol.
WT and IL-10−/− mice on the C57BL/6 background were repeatedly primed i.p. (four times) or i.n. (four times) and then given a final i.n. challenge (Fig. 1). Additionally, WT and IL-10−/− mice on a mixed C57BL/6 × 129Sv background (outbred) were repeatedly exposed to A. fumigatus Ag delivered only by the i.n. route (Fig. 1) for a limited number of times (2 to 3) due to the high mortality rate of the IL-10−/− mutants (see below). In all experimental groups, the final i.n. challenge was followed by a comparative analysis of cytokine production, airway inflammation, antibody responses, airway physiology, and histopathology.
Figure 1.

Sensitization protocols. Three different experimental protocols were used involving two different strains of mice and two different routes of priming. The same dose of A. fumigatus Ag without adjuvant was delivered either i.n. or i.p. at the times indicated.
Lung T cells from Sensitized IL-10− /− Mice Have Increased Potential to Secrete Th2 and Th1 Cytokines In Vitro.
Lung cells from sensitized IL-10−/− and WT mice and from unsensitized control mice were restimulated in vitro with immobilized anti-CD3 mAb. Irrespective of the mouse strain or the route of priming, lung T cells from sensitized IL-10−/− mice consistently produced higher levels of IL-4 and IL-5 than lung T cells from similarly treated WT mice (Fig. 2). Furthermore, IL-10−/− cells also produced large quantities of IFN-γ, while WT cells produced low to undetectable levels of IFN-γ (Fig. 2). It seems unlikely that the decreased levels of IL-4, IL-5, and IFN-γ produced by WT T cells when compared with IL-10−/− T cells was due to the suppressive effects of endogenously produced IL-10 in the WT cultures, as the addition of neutralizing anti–IL-10 mAb did not lead to increased production of these cytokines (Fig. 2).
Figure 2.

Cytokine levels made by in vitro restimulated lung cells from WT and IL-10−/− mice. Lung cell suspensions were prepared from outbred mice 4–5 d after the second sensitization and from C57BL/6 mice 4–5 d after the final i.n. challenge. Lung cell suspensions were restimulated with immobilized anti-CD3 in vitro. Closed dots represent data from single sensitized WT mice; open dots represent data from WT lung cell suspensions that were cultured in the presence of anti–IL-10 mAb. Squares represent data from single sensitized IL-10−/− mice. Dashed lines represent upper 95% confidence limits of cytokine levels secreted by restimulated lung cells from unsensitized control mice. As data from unsensitized WT and IL-10−/− mice were not statistically different they were combined for the calculation of upper 95% confidence levels.
Given that in vitro restimulation of IL-10−/− lung T cells resulted in the enhanced production of Th2 (IL-4 and IL-5) as well as Th1 (IFN-γ) cytokines, we questioned whether the absence of IL-10–mediated regulation led to the generation of polarized Th1 and Th2 cells and/or of Th0 cells capable of producing both types of cytokines. Therefore, αβT cells from the lungs of nonsensitized controls and sensitized IL-10−/− and WT mice were analyzed for their intracellular cytokines by flow cytometry (26). T cells from both WT and IL-10−/− mice displayed a similar pattern of cytokine-producing cells irrespective of their strain or route of priming. Representative data (Fig. 3) show that sensitization with A. fumigatus Ag invariably induced the generation of highly polarized IL-4–producing T cells in WT mice as they represented a very small proportion of the cytokine-producing cells present in the lungs of unsensitized control mice. Because the same results were also obtained with IL-10−/− cells, the generation of polarized Th2 cells was not dependent on the regulatory activities of IL-10. Our data also show that exposure to A. fumigatus Ag did not lead to an equivalent increase in Th0 cells or in polarized Th1-like cells in the lungs of either IL-10−/− or WT mice. These cells were present in approximately the same proportion as they were in samples from unsensitized controls. This latter finding was somewhat surprising in the case of IL-10−/− mice because the experiments described above demonstrated that their lung T cells were capable of producing high levels of IFN-γ after restimulation with anti-CD3 mAb.
Figure 3.

Flow cytometric analysis of lung αβT cells from outbred and C57BL/6 mice for intracellular IL-4 and IFN-γ. Lung cells were prepared from outbred mice 4–5 d after the second sensitization and from C57BL/6 mice 4–5 d after the final i.n. challenge. Lung cells were briefly restimulated in vitro with PMA and ionomycin as described in Materials and Methods. The cells were then triple-stained with FITC-labeled anti–IFN-γ mAb, PE-labeled anti–IL-4 mAb, and Tri-Color–labeled antiTCRαβ mAb. A separate cell sample was stained with FITC-, PE-, and Tri-Color–labeled isotype control antibodies. The gates for positive labeling were set so that less than 0.5% of the cells were stained with the isotype control antibodies. Lymphoid cells were gated using forward and side scatter. Within this gate, a subgate was created for cells labeled by the anti-αβTCR mAb. The αβTCR-positive cells were then examined for the presence of intracellular cytokines. Four to five mice were studied in each experimental group. Each dot plot represents a typical result. The median percentage of cells present in each of the four quadrants is represented in the corresponding cross section.
The absolute numbers of IL-4–producing and/or IFNγ–producing cells recovered from the lungs of sensitized C57BL/6 mice are presented in Fig. 4. We detected higher numbers of cytokine-producing T cells in the lungs of IL-10−/− mice primed i.p. when compared with similarly treated WT mice. This particular comparison suggested that increased T cell numbers in the lungs of IL-10−/− mice may account for the higher levels of IL-4, IL-5, and IFN-γ detected in their lung cultures when restimulated in vitro with anti-CD3 mAb (see Fig. 2). However, the number of cytokine-producing T cells recovered from IL-10−/− mice primed i.n. were roughly equivalent to the number recovered from WT mice primed i.n. (Fig. 4), although mutant cells consistently produced more IL-4, IL-5, and IFN-γ after anti-CD3-stimulation (see Fig. 2). Therefore, it appeared that sensitization with A. fumigatus Ag in the absence of IL-10 regulation, generated cytokine producing T cells in larger numbers and/or T cells that had acquired the inherent potential to produce larger quantities of cytokines.
Figure 4.

Absolute numbers of αβT cells capable of making cytokines in lungs of C57BL/6 mice. Lung cell suspensions were prepared from C57BL/6 mice as described in the legend to Fig. 3. The absolute number of lung cells recovered was determined before the brief restimulation with PMA and ionomycin in vitro. The cells were then stained and analyzed as described in the legend to Fig. 3. As numbers of lung αβT cells capable of making cytokines did not differ between unsensitized IL-10−/− (two separate pools of two lungs each) and WT (three separate pools of two lungs each) control mice, data were combined for comparison to sensitized groups. Data are reported as means ± SD of 4–5 mice per sensitized group.
Attempts to use A. fumigatus Ag to restimulate in vitro cytokine production by lung cells from primed WT or IL-10−/− mice proved uninformative. A. fumigatus Ag-induced IL-5 production was low, IL-4 production was barely at the detection limit, and IFN-γ production was undetectable (data not shown). Therefore, we focused on experiments designed to measure A. fumigatus Ag-induced cytokine production in vivo and to correlate these findings with numbers and types of inflammatory cells infiltrating the airways.
Outbred IL-10− /− Mice Showed Increased Cytokine Production, Had Increased Airway Inflammation, and Experienced Morbidity and Early Mortality Following Intranasal Sensitization.
Morbidity was evident after the second i.n. exposure to A. fumigatus Ag. Compared with unsensitized controls, sensitized IL-10−/− and WT mice lost weight. However, weight loss was more pronounced in IL-10−/− mice (87.4 ± 6.3% of the initial wt; n = 19) than in WT mice (93.2 ± 3.0%, n = 17) (P = 0.0014). Furthermore, IL-10−/− mice became dehydrated and hypothermic. Approximately 2/3 of the IL-10−/− mice died within 2–3 d after the third sensitization, whereas mortality was rare in similarly treated WT mice (Fig. 5). Because of the high mortality rate of sensitized IL-10−/− mice, in vivo cytokine production and airway inflammation was assessed after the second sensitization. The BAL fluids from IL-10−/− and WT mice contained equivalent levels of IL-4 (Fig. 6). However, the same fluids from IL-10−/− mice contained much higher levels of IL-5 and IFN-γ (Fig. 6). The cellular composition of BAL fluids recovered from IL-10−/− and WT mice was also different as increased numbers of neutrophils were present in BAL fluids of IL-10−/− mice (Fig. 6). In the BAL fluids of IL-10−/− mice, neutrophils (59 ± 8%) predominated, while eosinophils (11 ± 5%) were less frequent. In contrast, neutrophils (19 ± 3%), and eosinophils (25 ± 10%) were present in approximately equal ratios in the BAL fluids of WT mice. These results fit well with the fact that IL-10−/− mice appeared to suffer from an uncontrolled acute inflammatory response as evidenced by their high morbidity and mortality rates with increasing exposure to A. fumigatus. Although eosinophils in the BAL samples from both groups were already increased well above the levels exhibited by unsensitized controls, the numbers detected in the IL-10−/− samples were not as high as those in the WT samples. This finding was surprising given that IL-5 production was higher in IL-10−/− than in WT mice. One possible explanation for this outcome is that the lung eosinophilia in IL-10−/− mice was suppressed because of their simultaneous production of IFN-γ, which is known to inhibit the influx of eosinophils during an inflammatory response (29–31).
Figure 5.

Mortality induced by sensitization with A. fumigatus Ag in outbred mice. Survival curves are shown for IL-10−/− (n = 18) and WT (n = 18) outbred mice sensitized with A. fumigatus Ag i.n.
Figure 6.

Cytokine levels and numbers of neutrophils and eosinophils in BAL fluids of outbred mice. BAL was performed 18–24 h after the second i.n. sensitization. Dots and squares represent the data from single sensitized WT or IL-10−/− mice, respectively. Dashed lines represent upper 95% confidence limits of data obtained from unsensitized control mice. As data from unsensitized WT (n = 3) or IL-10−/− (n = 5) mice were not statistically different, data from both groups were combined to calculate upper 95% confidence limits.
IL-10− /− and WT Mice on the C57BL/6 Background Showed Similar Levels of Cytokine Production and Airway Inflammation Following Intranasal Sensitization.
When C57BL/6 IL-10−/− and WT mice repeatedly inhaled A. fumigatus Ag (five times), significant levels of IL-5 and low levels of IL-4 were detected in their BAL fluids as compared with unsensitized controls (Fig. 7). However, there were no differences between sensitized IL-10−/− and WT mice as equivalent amounts of IL-4 and IL-5 were detected and IFN-γ levels were negligible. Moreover, the total number of cells present in their BAL fluids were similar, including eosinophils (Fig. 7). In both groups, eosinophils (63 ± 11%) predominated and neutrophils (0.3 ± 0.4%) were rare. Furthermore, when we examined crude lung cell suspensions, IL-10−/− and WT mice had similar numbers of αβT cells capable of making IL-4, or IFN-γ (see Fig. 4) and of αβT cells that made neither cytokine (data not shown).
Figure 7.

Cytokine levels and numbers of eosinophils in BAL fluids from C57BL/6 mice sensitized i.n. BAL was performed 18–24 h after the final i.n. challenge. Dots and squares represent the data from single sensitized WT or IL-10−/− mice, respectively. Dashed lines represent upper 95% confidence limits of data obtained from unsensitized control mice. As data from unsensitized WT (n = 7) or IL-10−/− (n = 9) mice were not statistically different, data from both groups were combined to calculate upper 95% confidence limits.
C57BL/6 IL-10− /− Mice Primed Intraperitoneally Showed Increased IL-5 Production and Airway Inflammation Following Intranasal Challenge.
A striking difference in the response of C57BL/6 IL-10−/− as compared with WT mice was observed when A. fumigatus Ag-priming was given i.p. (four times) followed by a single i.n. challenge. IL-5 levels in BAL fluids of IL-10−/− mice were significantly increased. Both groups produced significant but similar levels of IL-4, and IFN-γ was extremely low or undetectable (Fig. 8). This cytokine profile was consistent with the significantly increased numbers of eosinophils in the BAL fluids of IL-10−/− mice compared with similarly treated WT mice (Fig. 8). Similar ratios of cell types with predominance of eosinophils (82.6 ± 5.9%) were present in BAL fluids from WT and IL-10−/− mice primed i.p., suggesting qualitatively similar but quantitatively increased airway inflammation in IL-10−/− mice. Furthermore, numbers of αβT cells capable of producing IL-4 and/or IFN-γ (see Fig. 4) as well as numbers of αβT cells that made neither of these two cytokines (data not shown) were significantly increased in lung tissues of IL-10−/− mice over levels seen in similarly treated WT mice.
Figure 8.

Cytokine levels and number of eosinophils in BAL fluids of C57BL/6 mice primed i.p. and challenged i.n. BAL was performed 18– 24 h after the i.n. challenge. Dots and squares represent the data from single sensitized WT or IL-10−/− mice, respectively. Dashed lines represent upper 95% confidence limits of data obtained from unsensitized control mice. As data from unsensitized WT (n = 7) or IL-10−/− (n = 9) mice were not statistically different, data from both groups were combined to calculate upper 95% confidence limits.
Serum Antibody Titers and Airway Hyperresponsiveness Elicited by A. fumigatus Ag Are Not Altered in IL-10− /− Mice.
Because of the sensitization-induced mortality in IL-10−/− outbred mice, serum antibody titers and airway responsiveness were evaluated in C57BL/6 mice only. Both IL-10−/− and WT mice exhibited increased total serum IgE and IgG1 titers compared with unsensitized controls (Table 1). Independent of the route of sensitization, IgE and IgG1 serum titers in IL-10−/− mice were not statistically different from those of WT mice. Neither IL-10−/− nor WT mice showed increased IgG2a titers as compared with unsensitized controls (data not shown). The antibody response (IgE and IgG1) was always higher in mice primed by the i.n. route as opposed to the i.p. route.
Table 1.
Antibody Titers and Airway Responsiveness in Wild-type and IL-10−/− C57BL/6 Mice
| Sensization | Wild type | IL-10−/− | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Control | i.n. | i.p. and i.n. | Control | i.n. | i.p. and i.n. | |||||||
| μg/ml | ||||||||||||
| Serum IgE titers | ||||||||||||
| Median | 0.40 | 22.90* | 3.60* | 1.10 | 12.60* | 2.30* | ||||||
| Range | 0.2–2.1 | 3.8–80.7 | 1.3–7.9 | 0.3–2.7 | 1.9–52.7 | 1.3–4.0 | ||||||
| n | 16 | 15 | 11 | 16 | 11 | 11 | ||||||
| mg/ml | ||||||||||||
| Serum IgG1 titers | ||||||||||||
| Median | 0.08 | 0.97* | 0.25* | 0.11 | 1.05* | 0.33* | ||||||
| Range | 0.02–0.18 | 0.56–1.62 | 0.08–0.40 | 0.02–0.43 | 0.56–1.62 | 0.17–0.87 | ||||||
| n | 11 | 14 | 14 | 12 | 15 | 15 | ||||||
| μg ACh/g bodyweight | ||||||||||||
| PC200 | ||||||||||||
| Mean | 2.24 | 0.22* | 0.31* | 1.13 | 0.30* | 0.47* | ||||||
| Range | 0.89–4.17 | 0.04–0.41 | 0.11–0.56 | 0.51–2.40 | 0.13–0.59 | 0.14–1.02 | ||||||
| n | 6 | 12 | 11 | 6 | 12 | 12 | ||||||
Antibody titers and airway responsiveness in WT and IL-10−/− C57BL/6 mice. Total serum antibody titers and airway responsiveness were examined 4–6 d after the final i.n. challenge of C57BL/6 mice primed i.n. or i.p. Airway responsiveness is expressed as PC200, the amount of ACh required to double baseline resistance. Antibody titers and PC200 values from unsensitized WT and IL-10−/− mice were not statistically different. Baseline airway resistance was similar in sensitized and unsensitized mice (data not shown). For the statistical comparison between PC200 values from experimental groups and controls, data from unsensitized IL-10−/− and WT mice were combined.
Data are significantly different from unsensitized control mice (P <0.05).
Sensitization via either route elicited marked airway hyperresponsiveness (decreases in PC200) in both IL-10−/− and WT mice as compared with unsensitized controls (Table 1). However, PC200 values were less variable in mice primed by the i.n. route rather than by the i.p. route (Table 1). PC200 values measured in i.n. or i.p. primed IL-10−/− mice and similarly treated WT mice were statistically indistinguishable (Table 1).
Lung Lesions in Sensitized WT and IL-10− /− Mice.
Lungs from unsensitized mice (Fig. 9 A) served as controls. Microscopic changes in all groups of sensitized mice (Fig. 9, B–F), were qualitatively similar, but differed in severity. Changes tended to be multifocal in less severely affected mice, but coalesced and became more diffuse in severely affected animals. Lung changes were generally characterized by inflammatory cell infiltrates adjacent to airways (Fig. 9, open arrows) and blood vessels (closed arrows), and by filling of alveoli with inflammatory cells and by thickening of alveolar septa (arrowheads). The epithelium lining airways in affected regions of lung was often hyperplastic and contained numerous goblet cells. Inflammatory infiltrates adjacent to airways and blood vessels consisted primarily of eosinophils, accompanied by smaller numbers of lymphocytes, macrophages, plasma cells, and neutrophils. Inflammatory cells within alveoli consisted of macrophages, lymphocytes, eosinophils, neutrophils, and multinucleated giant cells. Alveolar septa in affected areas of lung were often thickened by similar inflammatory cells, with the exception of multinucleated giant cells, which were only seen within alveoli.
Figure 9.
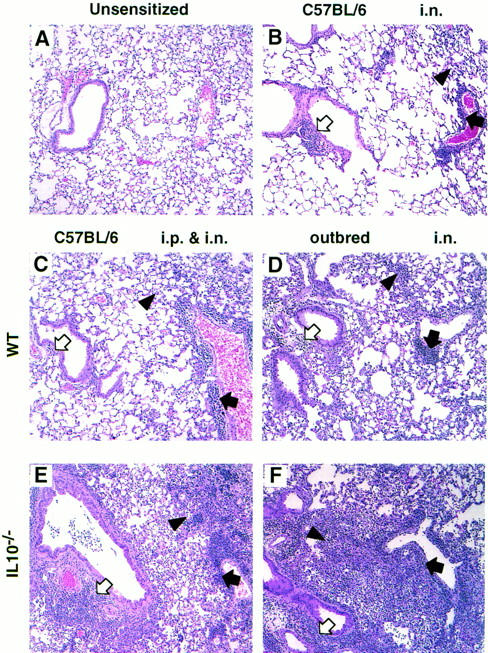
Lung lesions induced by sensitization with A. fumigatus Ag in outbred and C57BL/6 mice. Sections were prepared from formaldehyde-fixed lungs and stained with hematoxylin and eosin. Representative sections of a lung from each group of mice are shown (magnification, × 80). Lungs were removed 4 d after the second i.n. sensitization of outbred IL-10−/− (n = 9) and WT (n = 8) mice. Lungs were removed 4–7 d after the final i.n. challenge of C57BL/6 mice that had been primed i.n. or i.p. (n = 6–9). Open arrows point to peribronchial inflammation, closed arrows point to perivascular inflammation, and arrowheads point to alveolar lesions. (A) As lung sections from unsensitized WT and IL-10−/− mice were similar regardless of the mouse strain, one representative section is shown. (B) As lesions did not differ between WT or IL-10−/− C57BL/6 mice sensitized i.n., only one representative section is shown. (C and E) Lungs from C57BL/6 mice primed i.p. and challenged i.n. (C) WT, (E) IL-10−/−. (D and F) Lungs from outbred mice sensitized i.n. (D) WT, (F) IL-10−/−.
Comparisons among the sensitized groups showed that alveolar lesions that developed in WT mice were least severe in C57BL/6 mice (Fig. 9, B and C) and most severe in outbred mice (Fig. 9 D). When comparisons were made between IL-10−/− mice and their WT counterparts, C57BL/6 IL-10−/− and WT mice sensitized i.n. developed minimal to mild multifocal lesions that were very similar (refer to representative section shown in Fig. 9 B). No differences could be detected between these groups morphologically. In contrast, C57BL/6 IL-10−/− mice primed i.p. and challenged i.n. had more severe lesions than similarly treated C57BL/6 WT mice (Fig 9, C and E), especially with respect to the inflammatory infiltrates around airways and blood vessels. In addition, changes in the C57BL/6 IL-10−/− mice were often more diffuse than in the similarly treated C57BL/6 WT mice. Both WT and IL-10−/− outbred mice sensitized i.n. developed the most severe alveolar changes of all the experimental groups, and the changes in the IL-10−/− mice were more severe and diffuse than those seen in the WT outbred mice (Fig. 9, D and F).
Discussion
We have examined the response to inhaled A. fumigatus Ag in mice deficient in IL-10 production. Previous studies have demonstrated that A. fumigatus Ag elicit a dominant Th2-like response in the lungs of WT mice (21, 22). There were two main hypotheses regarding the outcome of our experiments. Sensitization of IL-10−/− mice would lead to an increased macrophage and Th1 response (reviewed in reference 19) or to increased Th1 and Th2 responses as observed in human studies (reviewed in references 32, 33). Our results showed clearly that the presence of IL-10 is not necessary for the development of polarized IL-4–producing Th2 cells as this population was consistently generated regardless of the route of sensitization or the mouse strain used. Furthermore, compared with WT mice, lung T cells from sensitized IL-10−/− mice consistently secreted more Th2 (IL-4 and IL-5) and Th1 (IFN-γ) cytokines when restimulated in vitro. Therefore, our findings are more in agreement with those of human studies, suggesting that IL-10 negatively regulates both Th1 and Th2 responses to Ag most probably by inhibiting antigen-presenting cells (11, 12, 32–36).
Although our in vitro studies have shown that sensitizing IL-10−/− mice with A. fumigatus Ag led to the generation of T cells capable of producing high levels of Th1 and Th2 cytokines, it was not clear how this potential would be manifested in vivo where other regulatory factors could come into play. Would increased Th1 and Th2 cytokine production lead to more inflammation in the lungs? Alternatively, would IFN-γ production antagonize potentially increased IL-4 and IL-5 levels and protect mice from increased eosinophilic inflammation of the lungs, IgE production, and airway hyperresponsiveness (29–31)? We found that each of the three different experimental protocols used provided us with a different answer that appeared to be profoundly influenced by the genetic strain and the route of Ag-exposure.
The disease in outbred IL-10−/− mice sensitized i.n. appears to represent a mixture between ABPA and Aspergillus-induced hypersensitivity pneumonitis. Although the increased BAL–neutrophilia, BAL–IFN-γ levels, morbidity, and alveolar lesions in the sensitized IL-10−/− outbred mice could be interpreted as typical for hypersensitivity pneumonitis, the large numbers of eosinophils in the BAL fluid of these mice and increased BAL–IL-5 levels do not fit with the classification of hypersensitivity pneumonitis (2, 3). Although lung lesions in sensitized IL-10−/− outbred mice were more extensive than those elicited in WT mice, they were of similar quality. Therefore, we suspect that mortality in IL-10−/− mice induced by repeated inhalation of A. fumigatus Ag was caused by the uncontrolled production of proinflammatory cytokines (e.g., IL-5, IFN-γ), which led to an acute shock-like state. This outcome was also seen in IL-10−/− mice challenged with LPS (24) or infected with Toxoplasma gondii (37).
In striking contrast with the outbred strain, C57BL/6 mice sensitized i.n. with A. fumigatus Ag developed mild lung lesions. These changes, together with the pronounced airway hyperresponsiveness and high IgE serum titers, are typical for murine experimental asthma (28, 38). Surprisingly, airway inflammation (BAL eosinophilia) that developed in the IL-10−/− mice was indistinguishable from that of WT mice. Moreover, the BAL fluids of the IL-10−/− and WT mice contained equivalent amounts of IL-4 and IL-5 and no IFN-γ. Therefore, it appears that IL-10−/− mice developed a dominant Th2 lung response of the same magnitude as that of WT mice. Despite the fact that IL-10−/− mice, repeatedly exposed to A. fumigatus Ag through inhalation, developed T cells with the capacity to secrete large amounts of Th1 and Th2 cytokines when polyclonally activated in vitro, their in vivo response appeared to be just as tightly regulated as that of WT mice.
Thus far, our data have illustrated the profound part that genetic factors can play in lung disease induced by A. fumigatus Ag. Even though the same Ag preparation was administered i.n. to both outbred and C57BL/6 mice, the outbred mice developed a complex disease resembling a mixture of asthma and hypersensitivity pneumonitis, whereas C57BL/6 mice developed primarily an asthma-like disease. This adds to the body of evidence that genetic factors may modify lung responses to A. fumigatus Ag. A previous study has shown that inhalation of A. fumigatus Ag by mice from two different mouse strains produced lung lesions of different severity (39). Genetic susceptibility factors in the case of human ABPA have been suggested by the familial occurrence of the disease (40, 41) and by an increased frequency of mutations in the cystic fibrosis transmembrane conductance regulator gene in ABPA patients (42). In addition, our studies showed that the inability to produce IL-10 leads to marked dysregulation and heightened disease on the outbred but not the C57BL/6 background. A logical conclusion would be that C57BL/6 mice have the inherent ability to compensate for the absence of IL-10 by using other negative regulatory mechanisms that could be provided by alveolar macrophages (43) or by TGF-β (44).
The specific mechanism evoked in C57BL/6 mice to compensate for the absence of IL-10 regulation is unknown. However, it is insufficient to prevent exaggerated eosinophilia and IL-5 production in the airways and heightened infiltration of the lungs with αβT cells, when the route of priming with A.fumigatus Ag is i.p. rather than i.n. This priming protocol appeared to elicit strong Th2 responses in both IL-10−/− and WT mice as inflammatory lung lesions were of intermediate severity typical of experimental ABPA (21) and BAL–IL-4 levels were high, whereas IFN-γ was undetectable. In this particular model of ABPA, the absence of IL-10 results in dysregulated IL-5 production, a factor that appears to play a pivotal role in generating allergic eosinophilic lung inflammation (38). In support of the view that IL-10 can suppress IL-5–dependent eosinophilic inflammation, IL-10 administered at the time of the Ag recall has been shown to diminish numbers of eosinophils in the peritoneal cavity of primed BALB/c mice (45).
It has been reported previously that antibody responses in IL-10−/− mice are not different from WT mice following Ag stimulation (23). Our results confirm this observation as serum IgE and IgG1 titers elicited in C57BL/6 IL-10−/− and WT mice were roughly equivalent. Our results show additionally that endogenous IL-10 neither promotes nor reduces airway hyperresponsiveness. Sensitization elicited similar airway hyperresponsiveness in WT and IL-10−/− mice. The equivalent antibody and airway responses in WT and IL-10−/− mice might reflect equivalent in vivo IL-4 production, or elicitation of compensatory mechanisms in IL-10−/− mice. Throughout our experiments, BAL–IL-4 levels were similar in sensitized IL-10−/− and WT mice, supporting the former hypothesis.
In conclusion, our in vitro studies show that endogenous IL-10 limits the potential to make large Th2 and Th1 responses elicited by A. fumigatus Ag in the lungs. In vivo, endogenous IL-10 suppresses lung cytokine secretion and airway inflammation induced by inhaled A. fumigatus Ag. Therefore, IL-10 may be effective in ameliorating inflammation in human allergic lung diseases. However, the success of IL-10 therapy for allergic lung diseases may depend on as yet undefined genetic factors and the route of exposure.
Acknowledgments
The authors thank Drs. R. Kühn, W. Müller, and K. Rajewsky (Institute for Genetics, University of Cologne, Cologne, Germany) for providing outbred IL-10−/− mice. We gratefully acknowledge the generous contributions of antibodies by Drs. R.L. Coffman, A. O'Garra, and J. Abrams (DNAX), and of Fas–Ig fusion protein by Dr. Miyuki Azuma (Department of Immunology, Jutendo University, School of Medicine, Tokyo, Japan), Dr. N.A. Hosken, and S. Zurawski (DNAX). We also thank the staff of the Histology Laboratory at the Schering-Plough Research Institute.
Footnotes
DNAX Research Institute of Molecular and Cellular Biology is supported by Schering-Plough Corporation. This work was partially supported by a US Veterans Affairs Medical research grant (V.P. Kurup) and by grant HL 03344 from the National Institutes of Health (D.B. Corry).
1 Abbreviations used in this paper: ABPA, allergic bronchopulmonary aspergillosis; ACh, Acetylcholine; BAL, bronchoalveolar lavage; IL-10−/− mice, IL-10 gene knockout mice; i.n., intranasal; PC200, provocative challenge dose 200; WT mice, wild-type mice.
References
- 1.Kauffman HF, Tomee JFC, Van der Werf TS, de Monchy JGR, Koeter GK. Review of fungus-induced asthmatic reactions. Am J Respir Crit Care Med. 1995;151:2109–2116. doi: 10.1164/ajrccm.151.6.7767565. [DOI] [PubMed] [Google Scholar]
- 2.Kurup VP, Kumar A. Immunodiagnosis of aspergillosis. Clin Microbiol Rev. 1991;4:439–456. doi: 10.1128/cmr.4.4.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fink JN. Hypersensitivity pneumonitis. Clin Chest Med. 1992;13:303–309. [PubMed] [Google Scholar]
- 4.Trompelt J, Becker W-M, Schlaak M. Analysis of IgG subclass and IgE response in allergic disease caused by Aspergillus fumigatusby immunoblotting techniques. Int Arch Allergy Immunol. 1994;104:390–398. doi: 10.1159/000236697. [DOI] [PubMed] [Google Scholar]
- 5.Drazen JM, Arm JP, Austen KF. Sorting out the cytokines of asthma. J Exp Med. 1996;183:1–6. doi: 10.1084/jem.183.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Denis M. Proinflammatory cytokines in hypersensitivity pneumonitis. Am J Respir Crit Care Med. 1995;151:164–169. doi: 10.1164/ajrccm.151.1.7812548. [DOI] [PubMed] [Google Scholar]
- 7.Schuyler, M., K. Gott, G. Shopp, and L. Crooks. 1993. CD3+, CD4+, CD8-, Ia- T cells adoptively transfer murine experimental hypersensitivity pneumonitis. Chest 103(Suppl.): 143S–145S. [DOI] [PubMed]
- 8.Knutsen AP, Mueller KR, Levine AD, Chauhan B, Hutcheson PS, Slavin RG. Asp fICD4+ Th2-like T-cell lines in allergic bronchopulmonary aspergillosis. J Allergy Clin Immunol. 1994;94:215–221. doi: 10.1016/0091-6749(94)90043-4. [DOI] [PubMed] [Google Scholar]
- 9.Crameri R, Faith A, Hemmann S, Jaussi R, Ismail C, Menz G, Blaser K. Humoral and cell-mediated autoimmunity in allergy to Aspergillus fumigatus. . J Exp Med. 1996;184:265–270. doi: 10.1084/jem.184.1.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bonfield TL, Konstan MW, Burfeind P, Panuska JR, Hilliard JB, Berger M. Normal bronchial epithelial cells constitutively produce the anti-inflammatory cytokine interleukin-10, which is downregulated in cystic fibrosis. Am J Respir Cell Mol Biol. 1995;13:257–261. doi: 10.1165/ajrcmb.13.3.7544594. [DOI] [PubMed] [Google Scholar]
- 11.Nicod, L.P., F. el Habre, J.M. Dayer, and N. Boehringer. 1995. Interleukin-10 decreases tumor necrosis factor alpha and beta in alloreactions induced by human lung dendritic cells and macrophages. Am. J. Respir. Cell. Mol. Biol. 13:83–90. [DOI] [PubMed]
- 12.Wilkes DS, Neimeier M, Mathur PN, Soliman DM, Twigg HLR, Bowen LK, Heidler KM. Effect of human lung allograft alveolar macrophages on IgG production: immunoregulatory role of interleukin-10, transforming growth factor-beta, and interleukin-6. Am J Respir Cell Mol Biol. 1995;13:621–628. doi: 10.1165/ajrcmb.13.5.7576699. [DOI] [PubMed] [Google Scholar]
- 13.Armstrong L, Jordan N, Millar A. Interleukin 10 (IL-10) regulation of tumour necrosis factor α (TNF-α) from human alveolar macrophages and peripheral blood monocytes. Thorax. 1996;51:143–149. doi: 10.1136/thx.51.2.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thomassen MJ, Divis LT, Fisher CJ. Regulation of human alveolar macrophage inflammatory cytokine production by interleukin-10. Clin Immunol Immunopathol. 1996;80:321–324. doi: 10.1006/clin.1996.0130. [DOI] [PubMed] [Google Scholar]
- 15.Zissel G, Schlaak J, Schlaak M, Müller-Quernheim J. Regulation of cytokine release by alveolar macrophages treated with interleukin-4, interleukin-10, or transforming growth factor β. Eur Cytokine Netw. 1996;7:59–66. [PubMed] [Google Scholar]
- 16.Shanley TP, Schmal H, Friedl HP, Jones ML, Ward PA. Regulatory effects of intrinsic IL-10 in IgG immune complex–induced lung injury. J Immunol. 1995;154:3454–3460. [PubMed] [Google Scholar]
- 17.Robinson DS, Tsicopoulos A, Meng Q, Durham S, Kay AB, Hamid Q. Increased interleukin-10 messenger RNA expression in atopic allergy and asthma. Am J Respir Cell Mol Biol. 1996;14:113–117. doi: 10.1165/ajrcmb.14.2.8630259. [DOI] [PubMed] [Google Scholar]
- 18.Borish L, Aarons A, Rumbyrt J, Cvietusa P, Negri J, Wenzel S. Interleukin-10 regulation in normal subjects and patients with asthma. J Allergy Clin Immunol. 1996;97:1288–1296. doi: 10.1016/s0091-6749(96)70197-5. [DOI] [PubMed] [Google Scholar]
- 19.Abbas AK, Murphy KM, Sher A. Functional diversity of helper T lymphocytes. Nature (Lond) 1996;383:787–793. doi: 10.1038/383787a0. [DOI] [PubMed] [Google Scholar]
- 20.Zuany-Amorim C, Haile S, Leduc D, Dumarey C, Huerre M, Vargaftig BB, Pretolani M. Interleukin10 inhibits antigen-induced cellular recruitment into the airways of sensitized mice. J Clin Invest. 1995;95:2644–2651. doi: 10.1172/JCI117966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kurup VP, Mauze S, Choi H, Seymour BWP, Coffman RL. A murine model of allergic bronchopulmonary Aspergillosis with elevated eosinophils and IgE. J Immunol. 1992;148:3783–3788. [PubMed] [Google Scholar]
- 22.Wang JM, Denis M, Fournier M, Laviolette M. Experimental allergic bronchopulmonary aspergillosis in the mouse: immunological and histological features. Scand J Immunol. 1994;39:19–26. doi: 10.1111/j.1365-3083.1994.tb03334.x. [DOI] [PubMed] [Google Scholar]
- 23.Kühn R, Löhler J, Rennick D, Rajewsky K, Müller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–274. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- 24.Berg DJ, Kühn R, Rajewsky K, Müller W, Menon S, Davidson N, Grünig G, Rennick D. Interleukin-10 is a central regulator of the response to LPS in murine models of endotoxic shock and the Shwartzman reaction but not endotoxin tolerance. J Clin Invest. 1995;96:2339–2347. doi: 10.1172/JCI118290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kurup VP, Ramasamy M, Greenberger PA, Fink JN. Isolation and characterization of a relevant Aspergillus fumigatus antigen with IgG- and IgE-binding activity. Int Arch Allergy Appl Immunol. 1988;86:176–182. doi: 10.1159/000234568. [DOI] [PubMed] [Google Scholar]
- 26.Openshaw P, Murphy E, Hosken NA, Maino V, Davis K, Murphy K, O'Garra A. Heterogeneity of intracellular cytokine synthesis at the single-cell level in polarized T helper 1 and T helper 2 populations. J Exp Med. 1995;182:1357–1367. doi: 10.1084/jem.182.5.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hanabuchi S, Koyanagi M, Kawasaki A, Shinohara N, Matsuzawa A, Nishimura Y, Kobayashi Y, Yonehara S, Yagita H, Okumura K. Fas and its ligand in a general mechanism of T-cell–mediated cytotoxicity. Proc Natl Acad Sci USA. 1994;91:4930–4934. doi: 10.1073/pnas.91.11.4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Corry DB, Folkesson HG, Warnock ML, Erle DJ, Matthay MA, Wiener KJ, Locksley RM. Interleukin 4, but not interleukin 5 or eosinophils, is required in a murine model of acute airway hyperreactivity. J Exp Med. 1996;183:109–117. doi: 10.1084/jem.183.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Iwamoto I, Nakajima H, Endo H, Yoshida S. Interferon γ regulates antigen-induced eosinophil recruitment into the mouse airways by inhibiting the infiltration of CD4+ T cells. J Exp Med. 1993;177:573–576. doi: 10.1084/jem.177.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lack G, Renz H, Saloga J, Bradley KL, Loader J, Leung DYM, Larsen G, Gelfand EW. Nebulized but not parenteral IFN-γ decreases IgE production and normalizes airways function in a murine model of allergen sensitization. J Immunol. 1994;152:2546–2554. [PubMed] [Google Scholar]
- 31.Li X-M, Chopra RK, Chou T-Y, Schofield BH, Wills-Karp M, Huang S-K. Mucosal IFN-γ transfer inhibits pulmonary allergic responses in mice. J Immunol. 1996;157:3216–3219. [PubMed] [Google Scholar]
- 32.Yssel, H., and R. De Waal Malefyt. 1995. IL-10 and human T cells. In Interleukin-10. J.E. de Vries and R. de Waal Malefyt, editors. Springer-Verlag, Heidelberg, Germany. 19–27.
- 33.de Waal Malefyt, R., C.G. Figdor, and J.E. de Vries. 1995. Regulation of human monocyte functions by interleukin 10. In Interleukin 10. J.E. de Vries and R. de Waal Malefyt, editors. Springer-Verlag, Heidelberg, Germany. 37–52.
- 34.Del Prete G, De Carli M, Almerigogna F, Guidizi MG, Biagiotti R, Romagnani S. Human IL-10 is produced by both type 1 helper (Th1) and type 2 helper (Th2) T cell clones and inhibits their antigen-specific proliferation and cytokine production. J Immunol. 1993;150:353–360. [PubMed] [Google Scholar]
- 35.Punnonen J, de Waal R, Malefyt, van Vlasselaer P, Gauchat J-F, de Vries JE. IL-10 and viral IL-10 prevent IL-4-induced IgE synthesis by inhibiting the accessory cell function of monocytes. J Immunol. 1993;151:1280–1289. [PubMed] [Google Scholar]
- 36.Takanaski S, Nonaka R, Xing Z, O'Byrne P, Dolovich J, Jordana M. Interleukin 10 inhibits lipopolysaccharide-induced survival and cytokine production by human peripheral blood eosinophils. J Exp Med. 1994;180:711–715. doi: 10.1084/jem.180.2.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gazzinelli RT, Wysocka M, Hieny S, Scharton-Kersten T, Cheever A, Kühn R, Müller W, Trinchieri G, Sher A. In the absence of endogenous IL-10, mice acutely infected with Toxoplasma gondiisuccumb to a lethal immune response dependent on CD4+ T cells and accompanied by overproduction of IL-12, IFN-γ, and TNF-α. J Immunol. 1996;157:798–805. [PubMed] [Google Scholar]
- 38.Foster PS, Hogan SP, Ramsay AJ, Matthaei KI, Young IG. Interleukin 5 deficiency abolishes eosinophilia, airways hyperreactivity, and lung damage in a mouse asthma model. J Exp Med. 1996;183:195–201. doi: 10.1084/jem.183.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kurup VP, Choi H, Resnick A, Kalbfleisch J, Fink JN. Immunopathological response of C57BL/6 and C3H/HeN mice to Aspergillus fumigatus antigens. Int Arch Allergy Immunol. 1990;91:145–154. doi: 10.1159/000235106. [DOI] [PubMed] [Google Scholar]
- 40.Graves TS, Fink JN, Patterson R, Kurup VP, Scanlon GT. A familial occurrence of allergic bronchopulmonary aspergillosis. Ann Intern Med. 1979;91:378–382. doi: 10.7326/0003-4819-91-3-378. [DOI] [PubMed] [Google Scholar]
- 41.Shah A, Khan ZU, Chaturvedi S, Malik GB, Randhawa HS. Concomitant allergic Aspergillus sinusitis and allergic bronchopulmonary aspergillosis associated with familial occurrence of allergic bronchopulmonary aspergillosis. Ann Allergy. 1990;64:507–512. [PubMed] [Google Scholar]
- 42.Weiner Miller, P., A. Hamosh, M. Macek, P.A. Greenberger, J. MacLean, S.M. Walden, R.G. Slavin, and G.R. Cutting. Cystic fibrosis transmembrane conductance regulator (CFRTR) gene mutations in allergic bronchopulmonary aspergillosis. Am J Hum Genet. 1996;59:45–51. [PMC free article] [PubMed] [Google Scholar]
- 43.Bilyk N, Holt PG. Inhibition of the immunosuppressive activity of resident pulmonary alveolar macrophages by granulocyte/macrophage colony-stimulating factor. J Exp Med. 1993;177:1773–1777. doi: 10.1084/jem.177.6.1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, Allen R, Sidman C, Proetzel G, Calvin D, et al. Targeted disruption of the mouse transforming growth factor–beta 1 gene results in multifocal inflammatory disease. Nature (Lond) 1992;359:693–699. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zuany-Amorim C, Creminon C, Nevers MC, Nahori MA, Vargraftig BB, Pretolani M. Modulation by IL-10 of antigen-induced IL-5 generation, and CD4+ T lymphocyte and eosinophil infiltration into the mouse peritoneal cavity. J Immunol. 1996;157:377–384. [PubMed] [Google Scholar]