

Abstract
We have recently described a system for the generation of dendritic cells (DC) and Langerhans cells (LC) from defined CD34+ precursors purified from peripheral blood of healthy adult volunteers (1). This study has now been extended by the characterization of two distinct subpopulations of CD34+ cells in normal human peripheral blood as defined by the expression of the skin homing receptor cutaneous lymphocyte-associated antigen (CLA). CD34+/CLA+ cells from normal peripheral blood were found to be CD71LOW/CD11a+/CD11b+/CD49d+/ CD45RA+ whereas CD34+/CLA− cells displayed the CD71+/CD11aLOW/CD11bLOW/CD49d(+)/ CD45RALOW phenotype. To determine the differentiation pathways of these two cell populations, CD34+ cells were sorted into CLA+ and CLA− fractions, stimulated with GM-CSF and TNF-α in vitro, and then were cultured for 10 to 18 d. Similar to unfractionated CD34+ cells, the progeny of both cell populations contained sizable numbers (12–22%) of dendritically shaped, CD1a+/HLA-DR+++ cells. In addition to differences in their motility, the two dendritic cell populations generated differed from each other by the expression of LC-specific structures. Only the precursors expressing the skin homing receptor were found to differentiate into LC as evidenced by the presence of Birbeck granules. In contrast, CLA− precursor cells generated a CD1a+ DC population devoid of Birbeck granule–containing LC. Provided that comparable mechanisms as found in this study are also operative in vivo, we postulate that the topographic organization of the DC system is already determined, at least in part, at the progenitor level.
Dendritic cells (DC) are bone marrow–derived leukocytes with potent immunostimulatory properties. They occur in small numbers in most non-lymphoid tissues and are well equipped to take up various types of antigen. After an antigenic challenge, these cells undergo phenotypic changes that allow them to leave their residence, to migrate to the regional lymphoid organs, and, upon arrival in the T-dependent zones, to activate resting T cells (2). This is best exemplified by the antigen-induced metamorphosis of epidermal Langerhans cells (LC; CD45+/CD1a+/CD32+/ E-cadherin+/Birbeck granule+/ATPase+/MHC class I+ and II+) into interdigitating reticulum cells (CD45+/CD1a−/ CD32−/E-cadherin−/Birbeck granule−/ATPase−/MHC class I++ and II+++) (3, 4). In contrast to the substantial information about the factors governing DC trafficking from the periphery to the lymphoid organs, relatively little is known about the reverse process, e.g., about the mechanisms underlying the immigration of circulating LC precursors in their ultimate tissue of residence, i.e., the skin and epidermis.
Together with the findings that the HECA-452–defined E-selectin ligand cutaneous lymphocyte-associated antigen (CLA) functions as a skin homing molecule for certain memory T cells (5), that LC within the skin are CLA+ (6), and that intravenously injected LC specifically home to the skin (7), our recent observation that approximately half of the CD34+ hematopoietic progenitor cells circulating in the peripheral blood of healthy adults (PBPC) react with the mAb HECA-452 (1) led us to the hypothesis that CLA might also be involved in the migration and/or the maturation of LC precursors. Here, we provide evidence that circulating CD34+/CLA+ but not CD34+/CLA− PBPC can differentiate into LC in vitro.
Materials and Methods
Purification of CD34+ PBPC from Healthy Volunteers.
CD34 + PBPC were isolated essentially as described (1). Following this procedure, 2 × 106 CD34+ PBPC with a purity of >95% and a viability of >96% can be reproducibly procured from one leukapheresis product (1.5–3 × 109 PBMC) (1).
Three-Color Flow Cytometry of PBPC.
Freshly isolated PBPC were adjusted to 5 × 105 cells/ml in PBS containing 1% FCS and 0.1% NaN3 and quenched with normal sheep serum (10% vol/ vol, 30 min on ice) to reduce nonspecific reactivity. Cells were reacted for 30 min on ice with a panel of mouse mAbs against human leukocyte differentiation antigens including CD45RA (clone 2H4; Coulter, Hialeah, FL), CD71 and HLA-DR (clones LO1.1 and L243; Becton Dickinson Immunocytometry Systems, BD-IS, San Jose, CA), CD11a, CD11b, CD18, CD49d, CD49e, CD29 (clones 25.3, Bear 1, BL5, HP2.1, SAM1, K20; all Immunotech, Marseille, France) as well as two different anti-CD1a mAb, which also served as isotype controls (clone OKT6, mouse IgG1; Ortho, Raritan, NJ, and B17.20.9, mouse IgG2a; Immunotech). With washings after each incubation, cells were consecutively exposed to biotinylated F(ab′)2 fragments of a polyclonal sheep anti–mouse Ig (10 min, on ice; Amersham Inc., Arlington Heights, IL), normal mouse serum (10% vol/vol, 20 min, on ice) and, finally, streptavidin-PerCP (BD-IS) simultaneously with mouse anti–human CD34-PE (8G12-PE; BD-IS) and HECA-452-FITC (5) (kindly provided by Dr. L. Picker, University of Texas Southwestern Medical Center, Dallas, TX) or the respective isotype control mAbs (30 min, on ice). For data analysis, CLA+/CD34+ and CLA−/ CD34+ events were gated and reactivity with the third mAb was measured as mean fluorescence intensity using a FACScan® flow cytometer (BD-IS) equipped with Lysys™ II (BD-IS) software.
Suspension Culture of PBPC Subpopulations.
To separate CLA+/ CD34+ from CLA−/CD34+ cells, 24-well cell culture plates (Costar, Cambridge, MA) were coated with purified HECA-452 mAb (10 μg/ml, overnight, 4°C) and then extensively washed with PBS. Freshly isolated CD34+ PBPC were incubated in HECA-452– coated wells for 60 min at 37°C (105 cells/well). Thereafter, plates were rinsed 3× with 1 ml HBSS to collect the nonadherent cell population.
Both the adherent and nonadherent PBPC populations were cultured in RPMI 1640 containing 2 mM l-glutamine, 0.1 mM nonessential amino acids, 100 U/ml penicillin, and 100 μg/ml streptomycin (all GIBCO BRL, Gaithersburg, MD; complete medium) supplemented with 20% heat-inactivated FCS, 200 U/ml recombinant human GM-CSF (kindly provided by Sandoz AG, Basle, Switzerland), and 50 U/ml TNF-α (Genzyme, Cambridge, MA). Alternatively, aliquots of unfractionated freshly isolated PBPC were cultured at 5–7 × 104 cells/ml in the same medium in 24-well culture plates, which were either coated with the HECA-452 mAb as described above or mock (HBSS)-coated. Cultures were maintained for at least 3 wk (37°C, humidified atmosphere, 5% CO2) by replacing one-fourth of the medium with fresh cytokine-supplemented complete medium every 4–5 d.
During the daily routine inspection of the cultures, we observed differences in the motility of cells derived from the two PBPC populations and used time lapse microphotography (Axiovert 135 equipped with a Contax 167MT camera; Zeiss, Oberkochen, Germany) to document these differences.
Phenotypic Analysis of PBPC-derived Cells by Immunocytochemistry, Flow Cytometry, and Electron Microscopy.
Cells derived from unfractionated PBPC or the HECA-452–sorted subpopulations were harvested after defined periods of culture with GM-CSF/ TNF-α (10, 14, 16, 18 d) and subjected to immunohistochemistry, flow cytometry, and immuno-electron microscopy.
For immunohistochemistry, cells were placed onto adhesion slides (Bio-Rad, Richmond, CA), allowed to attach for 10 min at room temperature (RT), fixed with methanol (15 min, −20°C), and, after quenching with normal goat serum (1% vol/vol in PBS, with 1% H2O2 to exhaust endogenous peroxidase; 30 min, 4°C), coincubated overnight at 4°C in a humidified chamber with the mAb Lag (mouse IgG1, directed against LC-associated Birbeck granules; kindly provided by Dr. S. Imamura [8]) and antiCD1a (clone B17.20.9, mouse IgG2a; Immunotech) or with the appropriate isotype controls. The specimens were then reacted simultaneously with goat anti–mouse IgG1 and biotinylated goat anti–mouse IgG2a (30 min, RT; both Southern Biotechnology Associates Inc., Birmingham, AL). IgG1-binding was visualized in red using mouse alkaline phosphatase–anti-alkaline phosphatase (APAAP, 30 min, RT; Dakopatts A/S, Glostrup, Denmark) with Fast Red (Sigma) as the developing reagent followed by visualization of IgG2a reactivity in brown with StreptABComplex/HRP (30 min, RT; Dakopatts) and diaminobenzidine (Sigma) according to previously published procedures (1, 6).
For additional flow cytometric analyses, PBPC-derived cells were labeled with anti-human CD1a-PE (clone SFCI19Thy1A8, T6RD1; Coulter), and counterstained with either mouse anti– human CD14-FITC (clone MEM 18; An der Grub, Kaumberg, Austria), mouse anti–human HLA-DR (clone L243, BD-IS) or mouse anti–human CD80 (clone BB1B7, BD-IS) plus biotinylated F(ab′)2 sheep anti–mouse Ig (Amersham) and then subjected to streptavidin–PerCP (BD-IS).
For immunoelectron microscopic analysis, PBPC-derived cells were reacted with mouse anti–human CD1a (BL6; Immunotech) or mouse IgG1 isotype control mAbs in the presence of normal goat serum (30 min, 4°C; British BioCell Laboratories, Cardiff, UK). After two washes at 4°C, they were incubated with goldlabeled (10 nm) goat anti–mouse Ab (30 min, 4°C; BioCell), fixed for 60 min at 4°C in 3% glutaraldehyde (Electron Microscopy Sciences, Euromedex, Strasbourg, France) in 0.1 M sodium cacodylate buffer containing 2% sucrose (pH 7.3; both Merck, Darmstadt, Germany) and for 45 min at 4°C in 1.5% glutaraldehyde. Cells were then incubated in 1% tannic acid (Merck) in 0.05 M sodium cacodylate buffer (45 min, RT), postfixed in 2% osmium tetroxide (Merck) in 0.1 M sodium cacodylate buffer (1 h, RT), dehydrated in a graded series of ethanol (50, 70, 80, 95, 100%), incubated overnight in Epon (Electron Microscopy Sciences)-absolute alcohol (1:1 vol/vol), and finally embedded in Epon. Ultrathin sections, stained with lead citrate (Leica, Bron, France) and uranyl acetate (Merck), were examined under a Philips CM 120 BioTwin electron microscope (120 kV).
Allogeneic Mixed Leukocyte Reaction.
Allogeneic mixed leukocyte reactions (MLR) were performed to test for the presence of potent accessory cells within the populations of PBPC-derived cells. Various numbers of mitomycin C (50 μg/ml, 30 min, 37°C; Sigma Chem. Co., St. Louis, MO)–treated PBPC-derived cells and, for control purposes, plastic-adherent monocytes from the same individual were cocultured with 105 purified allogeneic T cells in complete medium supplemented with 10% plasma of PBPC donor origin in 96-well round-bottomed culture plates (Costar). MLRs were maintained for 4, 5, or 6 d (37°C, humidified atmosphere, 5% CO2) before [3H]thymidine (1 μCi/well; Amersham) was added for additional 16 h. Incorporation of the radionucleotide was measured with a β-scintillation spectroscope (Packard Instruments, Meriden, CT). Results are expressed as mean cpm SD of triplicate cultures. Proliferation of 105 T cells or mitomycin C–treated stimulators alone resulted in an incorporation rate of <500 cpm.
Results and Discussion
Distinct Phenotype of CLA+ and CLA− PBPC.
In a first series of experiments, we used three-color flow cytometry to determine the immunophenotype of HECA-452–reactive and HECA-nonreactive CD34+ PBPC. We found that both populations were CD1a−, expressed equal levels of HLA-DR and common CD45 molecules, but exhibited pronounced quantitative and qualitative differences in their distribution of various leukocyte differentiation antigens. Quantitatively, CLA+/CD34+ cells showed a three- to fivefold higher expression of the β2-integrins LFA-1 (CD11a/CD18) and Mac-1/C3bi (CD11b/CD18), and a 1.5-fold higher expression of the β1-integrins VLA-4 (CD49d/CD29) and VLA-5 (CD49e/CD29) as compared with CLA−/CD34+ cells (Fig. 1). It will be interesting to determine whether the higher level of LFA-1/VLA-4 expression on CLA+/ CD34+ PBPC also allows for a more efficient binding to VCAM-1+/E-selectin+ endothelia (5, 9).
Figure 1.

Distinct phenotype of CLA+ and CLA− PBPC in normal peripheral blood. Using three-color flow cytometry, freshly isolated PBPC from healthy volunteers were analyzed for the distribution of the antigens shown on the y-axis. Reactivity was measured for gated CLA+/ CD34+ (black bars) and CLA-/CD34+ (hatched bars) PBPC. Results of one experiment are displayed as mean fluorescence intensity (MFI). Two additional experiments with cells from different donors yielded similar results. The lack of anti-CD1a-reactivity was used as a negative control.
Qualitatively, the major phenotypic differences between the two progenitor cell populations are the almost selective anti-CD45RA reactivity of CLA+ PBPC and the eightfold higher transferrin receptor (CD71) expression on CLA−/ CD45RA− PBPC (Fig. 1). Since CD45RALOW/−/CD34+ cells in bone marrow, cord blood, and growth factor–mobilized blood represent the more immature progenitor cell subset when compared with CD45RA+/CD34+ cells (10), CLA+/CD45RA+ PBPC probably constitute an already committed cell population of only limited proliferative potential (see below). Their low expression of transferrin receptors supports this assumption since the anti-CD71 reactivity of a given cell usually correlates with its state of proliferation (11).
Langerhans Cells Originate from CD34+ PBPC Bearing the Skin Homing Receptor CLA.
To determine whether both CLA+ and CLA− PBPC can differentiate into LC, we established a subtractive culture system that is based on the panning of one subpopulation of PBPC (i.e., CLA+ cells) directly in the culture well and on the transfer and subsequent culture of the nonadherent subtracted (i.e., CLA−) cells. This procedure allows for excellent cell recovery in both the panned and nonpanned (<0.1% contamination with CLA+ cells) fractions. When the two PBPC subsets were cultured for 2 wk with GM-CSF/TNF-α, the total number of nucleated cells recovered from the CLA− fraction was three to five times higher than that collected from the CLA+ subset. These differences in the multiplication rate reflect the phenotypic and maturational differences between the precursor cells (see above).
To test the hypothesis that CLA expression is involved in the generation of LC, immunohistochemistry was used to search for the presence of CD1a+ and Lag-reactive cells (8, 12). After 10 d of stimulation with GM-CSF/TNF-α, CD1a+ dendritically shaped cells were easily detectable in cultures from either CLA+ or CLA− precursors. The absolute numbers of CD1a+ cells were always higher in the cell fraction originating from CLA− precursors because these cells showed an approximately three- to fivefold higher proliferation compared to the CLA+ fraction (data not shown).
With regard to the progeny of CLA+/CD34+ PBPC, the percentage of CD1a+ cells ranged from 12% (day 10) to 22.5% (day 18) and that of Lag+ cells from 5% (day 10) to 20% (day 18; Table 1, Fig. 2 A). Double labeling on day 18 revealed that Lag reactivity of a given cell was restricted to the CD1a+ population (data not shown). Whereas on day 10 the percentage of Lag+ among CD1a+ cells ranged between 35–40%, ∼90% of CD1a+ cells gave positive Lag immunostaining on day 18 of culture. The majority of CD1a+/Lag+ cells tended to form clusters (Fig. 2 A) and were dendritic in shape. Occasionally, we observed isolated cells with a distinctive dendritic morphology devoid of Lag reactivity (data not shown).
Table 1.
Phenotype of HPC-derived DC
| % Antibody-reactive cells generated* | ||||||||
|---|---|---|---|---|---|---|---|---|
| CD1a+ total | Lag+ total | Lag+ within CD1a+ | Lag+ within CD1a− | |||||
| CD34+/CLA+ | ||||||||
| precursor | 22.5 | 20.1 | 89.3 | 0 | ||||
| CD34+/CLA− | ||||||||
| precursor | 11.7 | 0 | 0 | 0 | ||||
Sorted CLA+ and CLA-depleted CD34+ PBPC were cultured for 18 d in the presence of GM-CSF plus TNF-α. Lag reactivity indicating the presence of Birbeck granules was detected exclusively within CD1a+ cells derived from CD34+/CLA+ precursor cells.
Mean percentage of antibody-reactive cells calculated from three independent immunostaining experiments.
Figure 2.
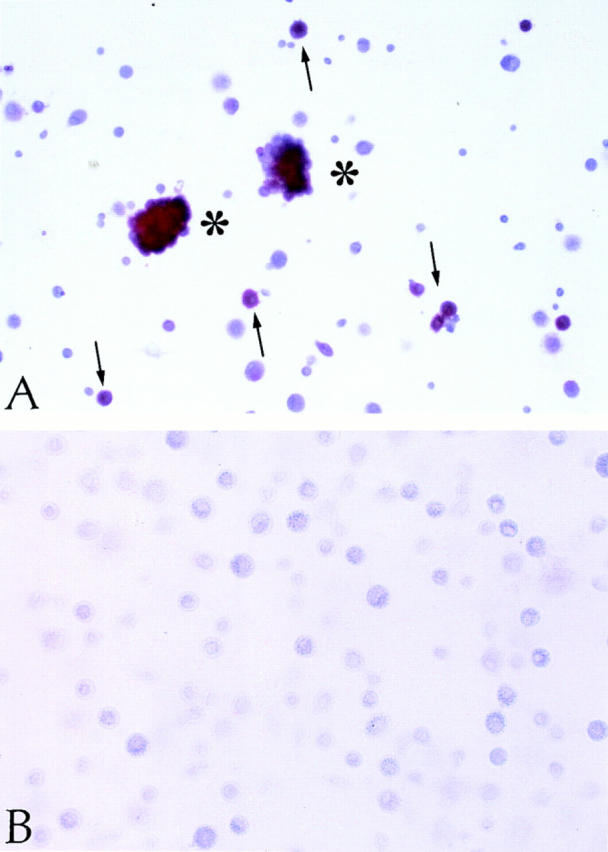
Immunohistochemical analysis of CLA+ and CLA− PBPC progeny. CLA+ and CLA− PBPC were cultured for two weeks in the presence of GM-CSF/TNF-α and then subjected to Lag immunolabeling using the APAAP procedure. Clustered (asterisks) as well as single (arrows) Lag+ cells (red) were detectable within the CLA+/CD34+ progeny (A). No Lag reactivity was found within the progeny of CLA− PBPC (B). Original magnification, ×150.
In sharp contrast to these findings, cells derived from CLA−/CD34+ PBPC, while containing various numbers of CD1a+ dendritic cells (Table 1), proved to be completely Lag− at all time points investigated (Fig. 2 B, Table 1). These data were confirmed at the ultrastructural level. Although Birbeck granules were easily detected in one third of CD1a+ LC (5/15 cells investigated) derived from CLA+ progenitors (Fig. 3), the search for these organelles in the CD1a+ progeny of CLA− cells yielded negative results (data not shown). Concerning other phenotypic features of the CD1a+ progeny of CLA+ and CLA− progenitors, we found that, on day 18 of culture, CD1a+ cells derived from either CLA+ or CLA− progenitors uniformly expressed MHC class II and CD80 (data not shown). CD14 was detected on a minor subpopulation of CLA+ PBPC–derived (15%) and CLA− PBPC–derived (6%) CD1a+ cells.
Figure 3.
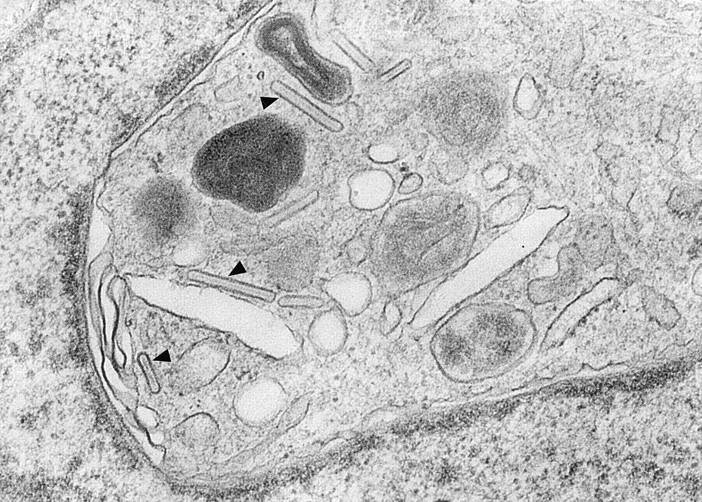
Detection of Birbeck granules in CD1a+ cells derived from CLA+ progenitors. Representative ultramorphology of a dendritically shaped CD1a+ LC that was generated from CLA+ PBPC during 16 d of culture in the presence of GM-CSF and TNF-α. Numerous trilaminar Birbeck granules (arrowheads) can be easily seen in the perinuclear area. Original magnification, ×83,500.
Other differences emerged when we investigated the functional properties of the two populations. Although cells derived from both CLA+ and CLA− progenitors induced vigorous proliferation of allogeneic T cells at all time points tested and, in this capacity, were 10–50 times more potent than syngeneic monocytes (Fig. 4), they clearly differed from each other in their migratory properties. During the routine daily inspection of the PBPC progeny, we noticed that cultures derived from either CLA+/CD34+ or unfractionated CD34+ cells, but not from CLA-depleted CD34+ cells, contain a population of dendritically shaped cells which, in contrast to their symbionts, moved continuously in the culture well. Microphotography revealed that these cells, per minute, cover distances which were multiples of their own diameter (Fig. 5). Since LC, upon receipt of an antigenic (danger? [13]) signal, leave their epidermal residence and migrate to the regional lymphoid organs (2), the migrating DC observed in our cultures (assuming that they are CD1a+ LC) might reflect this in vivo situation and may therefore serve as a paradigm of any peripheral non-lymphoid DC in its transition to a terminally differentiated stimulator cell of primary immune responses.
Figure 4.

MLR-stimulatory capacity of cells derived from CLA+ and CLA− PBPC. 3.3 × 103 cells generated in a 2-wk culture in GM-CSF/ TNF–containing medium from CLA+ (circles) and CLA− (diamonds) PBPC and, for comparison, monocytes from the same donor (squares) were tested for their capacity to stimulate 105 allogeneic T cells. Cells were pulsed with [3H]thymidine for 16 h after 4, 5, or 6 d of coculture. The [3H]thymidine incorporation rate is expressed as mean cpm values ± SD of triplicate cultures. 105 mitomycin C–treated APC or purified T cells gave <500 cpm. One representative experiment out of three performed is shown.
Figure 5.


A subpopulation of cells derived from CLA+/CD34+ PBPC exhibits a high motility in culture. Time-lapse microphotography was performed with cells derived from CLA+ PBPC after 3 wk of culture in the presence of GM-CSF/TNF-α. Pictures of cells migrating within the culture well were taken every minute. One (arrowhead) of the two dendritically shaped cells shown (A, time point zero) covers a distance being multiples of its own diameter within 3 min (B). Original magnification, ×150.
We were concerned that Birbeck granule formation in the progeny of CLA+ PBPC, instead of being an intrinsic property of this cellular subset, may have resulted from a HECA-452–induced cross-linking of CLA epitopes. In fact, the occurrence of a similar event has been described for murine dendritic epidermal T cells upon stimulation with a mitogenic anti–Thy-1 mAb (14). We consider this possibility highly unlikely for several reasons: (a) we (1) and others (15) have shown that unfractionated CD34+ cells can differentiate into Birbeck granule+ LC without engaging the CLA molecule; (b) comparable absolute numbers of CD1a+/ Lag+ LC were generated from unfractionated or CLAsorted starting cell populations containing equivalent numbers of CLA+ cells; (c) freshly isolated, unfractionated PBPC, when cultured in the presence or absence of the mAb HECA-452, exhibited similar proliferation rates and Lag frequencies upon stimulation with GM-CSF/TNF-α (data not shown). The latter observation implies that the differences in the multiplication rate between CLA− and CLA+/ CD34+ PBPC are not due to an antiproliferative signal resulting from the HECA-452–induced occupancy of CLA epitopes but rather reflect differences in the maturation state of the two cell populations.
At the present time, it is not known whether the CLA− PBPC can develop/mature into CLA+ ones and, if so, which factor(s) is (are) needed for this to occur. Also, it remains to be determined whether the expression on PBPC of HECA-452–reactive CLA and of other molecules necessary for adhesion to and transmigration through microvascular endothelial cells (e.g., CD49d/CD29) allows for the homing of progenitors to the skin. The mutually nonexclusive possibility exists that CLA expression merely reflects a distinct maturational stage of the progenitor cell that renders it susceptible to stimuli favoring LC development. In this context, recent attention has focused on TGF-β1 whose presence is apparently needed for the development of LC in vitro (16) and in vivo (17). We are currently investigating whether the CLA+ cell population differs from its CLA− counterpart in the responsiveness to TGF-β1 and, conversely, whether factors promoting the development of lymphoid DC (e.g., relB) (18) are selectively/predominantly expressed in CLA− PBPC and their progeny. Should this be the case, we would conclude that the topographic organization of the DC system is already determined at the progenitor level.
Acknowledgments
The authors are grateful to H. Bausinger, Dr. A. Bohbot, and Dr. D. Spehner for their help with electron microscopy. We wish to thank Dr. S. Imamura (Kyoto, Japan) for providing us with the mAb Lag.
Footnotes
This study was supported, in part, by grant S06702-MED from the Austrian Science Foundation (Vienna, Austria) and by grant FORTS 96 from the Agence Française du Sang (Paris, France).
Address reprint requests to Georg Stingl, Division of Immunology, Allergy and Infectious Diseases, Department of Dermatology, University of Vienna Medical School, Waehringer Guertel 18-20, A-1090 Vienna, Austria. Dr. D. Strunk's present address is Division of Hematology, Department of Internal Medicine, Karl Franzens University, Graz, Austria.
A preliminary report on this work was presented at the 4th International Symposium on Dendritic Cells in Fundamental and Clinical Immunology; Lido, Venice, October 5–10, 1996.
References
- 1.Strunk D, Rappersberger K, Egger C, Strobl H, Krömer E, Elbe A, Maurer D, Stingl G. Generation of human dendritic cells/Langerhans cells from circulating CD34+hematopoietic progenitor cells. Blood. 1996;87:1292–1302. [PubMed] [Google Scholar]
- 2.Steinman RM. The dendritic cell system and its role in immunogenicity. Annu Rev Immunol. 1991;9:271–296. doi: 10.1146/annurev.iy.09.040191.001415. [DOI] [PubMed] [Google Scholar]
- 3.Schuler G, Steinman RM. Murine epidermal Langerhans cells mature into potent immunostimulatory dendritic cells in vitro. J Exp Med. 1985;161:526–546. doi: 10.1084/jem.161.3.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aiba S, Katz SI. Phenotypic and functional characteristics of in vivo-activated Langerhans cells. J Immunol. 1990;145:2791–2796. [PubMed] [Google Scholar]
- 5.Picker LJ, Michie SA, Rott LS, Butcher EC. A unique phenotype of skin-associated lymphocytes in humans. Am J Pathol. 1990;136:1053–1068. [PMC free article] [PubMed] [Google Scholar]
- 6.Koszik F, Strunk D, Simonitsch I, Picker LJ, Stingl G, Payer E. Expression of monoclonal antibody HECA-452-defined E-selectin ligands on Langerhans cells in normal and diseased skin. J Invest Dermatol. 1994;102:773–780. doi: 10.1111/1523-1747.ep12377706. [DOI] [PubMed] [Google Scholar]
- 7.Cruz PD, Jr, Tigelaar RE, Bergstresser PR. Langerhans cells that migrate to skin after intravenous infusion regulate the induction of contact hypersensitivity. J Immunol. 1990;144:2486–2492. [PubMed] [Google Scholar]
- 8.Kashihara M, Ueda M, Horiguchi Y, Furukawa F, Hanaoka M, Imamura S. A monoclonal antibody specifically reactive to human Langerhans cells. J Invest Dermatol. 1986;87:602–607. doi: 10.1111/1523-1747.ep12455849. [DOI] [PubMed] [Google Scholar]
- 9.Berlin C, Bargatze RF, Campbell JJ, von Adrian UH, Szabo MC, Hasslen SR, Nelson RD, Berg EL, Erlandsen SL, Butcher EC. α4 integrins mediate lymphocyte attachment and rolling under physiologic flow. Cell. 1995;80:413–422. doi: 10.1016/0092-8674(95)90491-3. [DOI] [PubMed] [Google Scholar]
- 10.Lansdorp PM, Sutherland HJ, Eaves CJ. Selective expression of CD45 isoforms on functional subpopulations of CD34+hematopoietic cells from human bone marrow. J Exp Med. 1990;172:363–366. doi: 10.1084/jem.172.1.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neckers LM, Cossman J. Transferrin receptor induction in mitogen-stimulated human T lymphocytes is required for DNA synthesis and cell division and is regulated by interleukin 2. Proc Natl Acad Sci USA. 1983;80:3494–3498. doi: 10.1073/pnas.80.11.3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Romani N, Gruner S, Brang D, Kämpgen E, Lenz A, Trockenbacher B, Konwalinka G, Fritsch PO, Steinman RM, Schuler G. Proliferating dendritic cell progenitors in human blood. J Exp Med. 1994;180:83–93. doi: 10.1084/jem.180.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015. [DOI] [PubMed] [Google Scholar]
- 14.Romani, N., G. Stingl, E. Tschachler, K.C. Gunter, E.M. Shevach, and G. Schuler. 1985. Induction of Birbeck granule-like structures in murine T lymphocytes and dendritic Thy-1-positive epidermal cells by a T cell-activating antiThy-1 monoclonal antibody. J. Invest. Dermatol. 84(Abstr.):327.
- 15.Caux C, Dezutter-Dambuyant C, Schmitt D, Banchereau J. GM-CSF and TNF-α cooperate in the generation of dendritic Langerhans cells. Nature (Lond) 1992;360:258–261. doi: 10.1038/360258a0. [DOI] [PubMed] [Google Scholar]
- 16.Strobl H, Riedl E, Scheinecker C, Bello-Fernandez C, Pickl WF, Rappersberger K, Majdic O, Knapp W. TGF-β1 promotes in vitro development of dendritic cells from CD34+hemopoietic progenitors. J Immunol. 1996;157:1499–1507. [PubMed] [Google Scholar]
- 17.Borkowski TA, Letterio JJ, Farr AG, Udey MC. A role for endogenous transforming growth factor β1 in Langerhans cell biology: the skin of transforming growth factor β1 null mice is devoid of epidermal Langerhans cells. J Exp Med. 1996;184:2417–2422. doi: 10.1084/jem.184.6.2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burkly L, Hession C, Ogata L, Reilly C, Marconi LA, Olson D, Tizard R, Cate R, Lo D. Expression of relBis required for the development of thymic medulla and dendritic cells. Nature (Lond) 1995;373:531–535. doi: 10.1038/373531a0. [DOI] [PubMed] [Google Scholar]