

Abstract
The induction by IFN-γ of reactive nitrogen intermediates has been postulated as a major mechanism of host resistance to intracellular pathogens. To formally test this hypothesis in vivo, the course of Toxoplasma gondii infection was assessed in nitric oxide synthase (iNOS)−/− mice. As expected, macrophages from these animals displayed defective microbicidal activity against the parasite in vitro. Nevertheless, in contrast to IFN-γ−/− or IL-12 p40−/− animals, iNOSdeficient mice survived acute infection and controlled parasite growth at the site of inoculation. This early resistance was ablated by neutralization of IFN-γ or IL-12 in vivo and markedly diminished by depletion of neutrophils, demonstrating the existence of previously unappreciated NO independent mechanisms operating against the parasite during early infection. By 3-4 wk post infection, however, iNOS knockout mice did succumb to T. gondii. At that stage parasite expansion and pathology were evident in the central nervous system but not the periphery suggesting that the protective role of nitric oxide against this intracellular infection is tissue specific rather than systemic.
Reactive nitrogen intermediates (RNI)1, including nitric oxide (NO), have been identified as important effector molecules which restrict pathogen growth in infected hosts (1). Expression of distinct NO synthase enzymes results in synthesis of RNI by constitutive and/or inducible pathways in a number of cell types including endothelial cells, epithelial cells, fibroblasts, hepatocytes, muscle cells, neutrophils, and phagocytes (1, 2). Murine macrophages are one of the best characterized of these RNI sources. In the latter cells, NO synthesis is not constitutive but requires stimulation by lipopolysaccharide and/or cytokines which enhance transcription of the inducible nitric oxide synthase (iNOS) enzyme, leading to the conversion of l-arginine to l-citrulline and NO. Induction of the high output NO pathway in macrophages is preferentially driven by Type I immune responses in which production of IFN-γ is prominent. Interruption of the RNI pathway with enzyme antagonists inhibits the ability of macrophages to kill pathogens both in vitro and in vivo (3–16). Based on the above evidence, a paradigm of host resistance has emerged in which RNI, produced by cytokine activated macrophages, are important effectors of the Type 1 immune response against bacterial, fungal, helminth, and protozoan infectious agents (1).
A well studied inducer and target of the NO pathway is the intracellular protozoan Toxoplasma gondii (17). This opportunistic pathogen is able to infect and propagate in virtually all nucleated host cells (18). Nonetheless, in immunocompetent individuals infection is largely asymptomatic and is characterized by a brief acute stage in which rapidly replicating tachyzoites disseminate to peripheral host tissues (19). Fulminant infection is prevented by a potent innate immune response that is largely T cell independent and leads to the transformation of the parasite into a dormant bradyzoite form, which is confined primarily to the central nervous system (CNS). Parasite latency in the chronic stage of infection is maintained by an adaptive T cell response. IFN-γ has been shown to be crucial both for the early control of tachyzoite expansion and for preventing reactivation of dormant parasite stages. Thus, anti–IFN-γ mAb–treated, as well as IFN-γ−/− mice rapidly succumb to primary infection with normally avirulent parasite strains and this enhanced susceptibility is associated with uncontrolled tachyzoite replication in the periphery (20–22). Similarly, acute disease can be triggered in chronically infected animals by treatment with the same IFN-γ–neutralizing mAb (23, 24). Finally, exogenous administration of rIFN-γ increases resistance to acute infection while decreasing the incidence of encephalitis in the chronic stage (25, 26).
Several hypotheses have been proposed to explain the role of IFN-γ in host resistance to T. gondii. One mechanism that is readily demonstrable in vitro is the ability of the cytokine to activate macrophages to kill intracellular parasites (27, 28). The involvement of the RNI in this IFN-γ–mediated protection is based on the observation that l-NMMA, a competitive analog of l-arginine, simultaneously inhibits NO synthesis and intracellular tachyzoite killing by cytokine activated peritoneal and bone marrow– derived macrophages as well as microglial cells (9, 29, 30). A key function for NO in control of T. gondii infection is also supported by in vivo observations. Mice in which NO synthesis is impaired as a result of genetic disruptions of the IFN-γ or interferon regulatory factor-1 (IRF-1) genes succumb to acute infection within 14 d of parasite exposure (22, 31). Similarly, animals treated with the RNI inhibitor aminoguanidine also display enhanced susceptibility (11). However, in the latter case, the mice survive the acute stage but develop accelerated disease progression later in infection as assessed by the presence of increased parasite numbers and inflammatory infiltration in CNS tissue. The basis of this discrepancy is not clear but may relate to the impairment of effector functions unrelated to NO synthesis in the IFN-γ and IRF-1 knockout (ko) mice or to incomplete inhibition of NO in the drug-treated animals.
Recently mice with a targeted disruption of the NOS2 (iNOS) gene have been generated by homologous recombination technology (32). Macrophages from these mutant animals fail to express detectable iNOS mRNA, protein or enzyme activity and consequently are unable to produce significant levels of nitrite (NO2 −) or nitrate (NO3 −). iNOS ko animals have been shown to display increased susceptibility to infection with the gram-positive bacterium Listeria monocytogenes (32) as well as the intracellular protozoan, Leishmania major (33). In the present study, we have used iNOS ko mice to formally assess the requirement for RNI in host resistance to T. gondii. As predicted, macrophages from these mutant animals were defective in parasite killing in vitro. Surprisingly, however, control of acute infection in vivo was unaffected by the iNOS deficiency. Our data thus challenge the view that synthesis of RNI, an established correlate of in vitro killing, is a primary mechanism of innate resistance to T. gondii in vivo and argue instead that the major role of this effector function is to maintain control of established infections.
Materials and Methods
Experimental Animals.
Mice with a targeted disruption of the NOS2 gene (iNOS ko) were generously provided by Drs. J.D. MacMicking, C. Nathan (Cornell University Medical College, New York), and J.S. Mudgett (Merck Research Laboratories, Rahway, NJ). These mice were generated as previously described (32) with a gene replacement vector pINOS-RV1 that was designed to delete the 5′ end of the NOS2 gene (proximal 585 bases of the promoter and the exons 1–4). The iNOS ko animals used for our experiments were obtained from homozygous inbreeding in the F2 generation (129SvEv × C57BL/6). As shown in Fig. 1 (inset), initial experiments comparing C57BL/6 (Division of Cancer Treatment, National Cancer Institute, Frederick, MD) and C57BL/6 × 129/J F1 (The Jackson Laboratory, Bar Harbor, ME) mice revealed no difference in the outcome of Toxoplasma infection over the time period analyzed (3 mo). The latter finding is consistent with previously published data on the susceptibility of the parental strains, 129 and C57BL/6, to avirulent T. gondii (34, 35). Furthermore, in unrelated experiments other 129 × C57BL/6 hybrid strains (129 Sv Ev or 129/SvJ or 129/Ola) survived at least 3 mo after i.p. infection with 20 cysts of ME49 (data not shown). Due to their greater availability, C57BL/6 mice were used as controls in all subsequent experiments. As previously described (36), IL-12 p40−/− mice were generated by genetically disrupting exon 3 through a homologous recombination event between the wildtype gene sequence and a mutant allele carrying the PGK-1 neo gene. Mice carrying the IL-12 p40 mutant allele were backcrossed five times to the C57BL/6 genetic background followed by intercross of the heterozygotes in order to generate mice homozygous for the targeted mutation (IL-12 p40−/−). Breeding pairs of mice with a targeted disruption of the IFN-γ gene were originally provided by Dyana Dalton and T. Stewart (Genentech, San Bruno, CA) (37). The IFN-γ knockout mice used were at the seventh generation of back-crossing to the C57BL/6 strain. Animals were housed in specific pathogen-free conditions and both male and female mice were used for experiments at 5–12 wk of age.
Figure 1.

In contrast to IFN-γ ko and IL-12 p40−/− mice, iNOS ko animals survive acute infection with T. gondii (ME49). Mice were infected either by the i.p. (A) or p.o. (B) route of infection with 20 ME49 cysts. The data shown in A are pooled from three independent experiments and involve a minimum of 10 ko mice per group. The experiment presented in B involved five mice per group and is representative of three performed. As described in Materials and Methods, infected C57BL/6 × 129/J F1 and C57BL/6 animals displayed similar survival patterns (inset).
Parasites and Experimental Infection.
Tachyzoites of the virulent RH were maintained in vitro by infection of human foreskin fibroblasts and biweekly passage in DMEM (GIBCO BRL, Gaithersburg, MD) supplemented with 1% FCS (Hyclone Laboratories, Logan UT), penicillin (100 U/ml), and streptomycin (100 ug/ml). Cysts of the avirulent ME49 strain (initially provided by Dr. J. Remington, Palo Alto Research Foundation) were harvested from the brains of C57BL/6 mice which had been inoculated with ∼20 cysts intraperitoneally 1 mo prior. For experimental infections, mice received 20 ME49 cysts or PBS (Biowhitaker, Walkersville, MD) by either the intraperitoneal (i.p.) or peroral (p.o) route. Control inoculations with normal brains suspensions failed to elicit detectable inflammatory responses, NK cell cytotoxicity or significant increases in cytokine levels (data not shown). Soluble tachyzoite antigen (STAg) was prepared as described previously (24).
In Vivo Assessment of Acute and Chronic Infection.
Acute tachyzoite growth was assessed using cytocentrifuge smears of cells from infected animals (22). Samples were prepared from 1.5 × 105 peritoneal exudate cells in a Cytospin (Shandon Lipshaw, Pittsburgh, PA) set for 5 min at 1,000 RPM. Slide preparations were fixed in absolute methanol for 5 min and then stained with Diff-Quik (Baxter Healthcare Corporation, McGaw Park, IL), a modified Wright-Giemsa stain, as specified by the manufacturer. Differential analyses, including assessment of the number of infected cells, were performed on 400–500 cells using an oil immersion (100× objective). In the experiments indicated, the presence of parasites in heart, lung, liver, spleen, and peritoneum was assessed by microscopic examination of impression smears made from tissues at 5–6 d after infection.
To assess chronic disease progression animals were killed by CO2 or cervical dislocation. Brain tissue was removed aseptically and homogenized in 2 ml of PBS. The total number of cysts was determined by enumerating the organisms in a 10-μl suspension and multiplying by 200. Parallel quantitation of cysts was performed on periodic acid–Schiff's stained, sagittal sections of brain, heart, lung, liver, and spleen tissues. Brain sections were also examined microscopically for histopathological changes.
In Vitro Assessment of Tachyzoite Killing.
Resident macrophages and inflammatory macrophages were harvested from animals which were untreated or inoculated i.p. 4–5 d previously with either 1.5 ml of 3% thioglycollate (Sigma) or 20 cysts of the ME49 strain. Cells were harvested by injecting cold RPMI into the peritoneal cavity and plated at 2 × 105 per well in 96-well plates for 2 h in the presence or absence of rMuIFN-γ 100 U/ml (generously provided by Genentech, Inc., San Francisco, CA). Cultures were incubated overnight in the presence of medium alone or RH tachyzoites (0.2 or 1.0 per cell). At this time, an aliquot of supernatant was harvested from the cultures for measurement of nitrite (NO2−) levels (see below) and the remaining cells were pulsed with Uracil-[5,6-3H] (ICN Pharmaceuticals, Inc., Irvine, CA) at 0.5 μCi/well for an additional 12 h to measure T. gondii proliferation (38, 39). An incubation period of 24 h followed by a 12–18-h pulse with uracil-[5,6-3H] was found to be optimal in our assay. The incorporation of radioactive uracil was determined by liquid scintillation counting. In indicated experiments NGmonomethyl-l-arginine (l-NMMA; Calbiochem-Novabiochem Corporation, LaJolla, CA) was added (1 mM) during the initial IFN-γ activation period. The percentage of killing was determined by the following calculation:
1 − [(IFN-γ(infected)) − (IFN-γ(no parasites)) / (media(infected)) − (media(no parasites))] × 100.
Parasite growth was also measured microscopically by a method adapted from previous studies (40, 41). In brief, 4 × 106 cells were cultured in 6 ml, polypropylene tubes in 1 ml of media in the presence or absence of IFN-γ (100 U/ml) and/or l-NMMA (2.5 mM). After a 2-h incubation at 37°C, cells were infected at 1 RH tachyzoite per well for 2 h. Extracellular T. gondii were then removed by adding room temperature media followed by low-speed centrifugation (10 min at 800 RPM) two times. At this time point (zero) an aliquot of cells were removed, cytocentrifuge smears prepared, and both the number of infected cells, and tachyzoites per infected cell determined microscopically. The remaining tubes were then retreated with the appropriate additives (media, IFN-γ, or IFN-γ + l-NMMA) and a second set of slides was prepared 48 h later. Essentially identical results were observed using both the proliferation and microscopic assays.
Cell Cultures and Serum Preparation.
Single cell suspensions were prepared from spleen and peritoneal cells harvested at various time points post infection. Peritoneal cells were cultured at 4 × 105 cells and spleen cells at 8 × 105 per well in a total volume of 200 μl in a medium consisting of RPMI-1640 (Bio Whittaker) supplemented with 10% FCS, penicillin (100 U/ml), streptomycin (100 μg/ml), l-glutamine (2 mM), Hepes (10 mM), and 2-mercaptoethanol (5 × 10−5 M) in the presence or absence of STAg (5 μg/ml). Supernatants were harvested 72 h later for IFN-γ, IL-12, and nitrite determinations.
Blood was collected from mice at the time of death and allowed to clot at room temperature for 2 h. Serum was then separated from the individual samples after a 5-min centrifugation at 5,000 RPM and assayed for cytokine content.
NO, IFN-γ, and IL-12 Measurements.
Nitrite (NO2−) levels were used as an indicator of reactive nitrogen intermediates in samples and were measured by the Griess assay (42). In brief, 100-μl aliquots of supernatant were added to 96-well plates followed by a 100 μl of a 1:1 mixture of 1% sulfanilamide dihydrochloride (Sigma) in 2.5% H3PO4 and 0.1% naphthylethylenediamide dihydrochloride (Sigma) in 2.5% H3PO4. After a 10-min incubation at room temperature, the absorbance of the samples (A550) was read spectrophotometrically and units of nitrite (range of sensitivity: 4–250 μM) determined by comparison with a standard curve generated with sodium nitrite (NaNO2) (Sigma).Levels of IFN-γ and IL-12 were assayed by 2-site ELISA as previously described (22). Cytokine levels were quantitated by reference to standard curves generated with rIFN-γ (Genentech) or rIL-12 (provided by Genetics Institute, Cambridge, MA).
In Vivo Anti-Granulocyte, Anti–IFN-γ, Anti–IL-5, and Anti– IL-12 treatments.
For cytokine depletion, mice were treated 1 d before infection with 1 mg anti–IFN-γ mAb (rat IgG1, XMG6 [43]), 2 mg anti–IL-5 mAb (rat IgG1, TRFK-5 [44]), or 1 mg anti–IL-12 mAb (C17.8, rat IgG2a [45]). The anti-granulocyte IgG2b mAb, RB6-8C5, originally derived by Robert Coffman, was administered initially on d 0 at 0.5 mg and subsequently on day 2 and 4 at 0.25 mg per mouse. The ascites employed was produced by Harlan Bioproducts for Science, Inc. (Indianapolis, IN) from nude mice inoculated with the hybridomas and partially purified by ammonium sulfate precipitation. Normal rat Ig (Sigma) was used as a control.
Statistical Analyses.
Statistical determinations of the difference between means of experimental groups was determined using an unpaired, two tailed Student's t test.
Results
iNOS ko Mice Survive Acute Infection with T. gondii.
We and others have previously demonstrated that mice with impaired IFN-γ function succumb to acute infection with T. gondii (21, 22). Loss of NO synthesis is one striking immune defect that is apparent in these mice and could contribute to the observed lack of parasite control. To assess the role for RNI in host resistance against this pathogen, iNOS ko mice were infected with 20 cysts of the ME49 strain and their survival compared with that of IFN-γ ko and C57BL/6 control mice. As previously reported, IFN-γ ko mice succumbed to i.p. infection within 9 d of parasite inoculation, whereas 100% of the control animals remained alive for the 40 d of the experiment (Fig. 1 A). Similarly, mice with a targeted disruption of the IL-12 p40 subunit (IL-12 p40−/−) that are also defective in both IFN-γ (36) and NO synthesis (Fig. 2 A) failed to survive the acute stage of infection (Fig. 1 A). In striking contrast, iNOS ko animals infected under the same conditions survived for 19–24 d after exposure and thus displayed an intermediate pattern of susceptibility. It was formally possible that the difference in survival between iNOS−/− and C57BL/6 animals might be due to the different genetic backgrounds of the two strains. However, this appears extremely unlikely since both parental strains, C57BL/6 and 129, are known to control avirulent T. gondii infection for at least 4 wk longer than the iNOS-deficient animals (34, 35). Moreover, the analysis performed here of survival rates in C57BL/6 and 129 × C57BL/6 F1 mice receiving the same ME49 challenge as the iNOS animals failed to reveal a difference in mortality over the first 90 d of infection (Fig. 1 A, inset).
Figure 2.
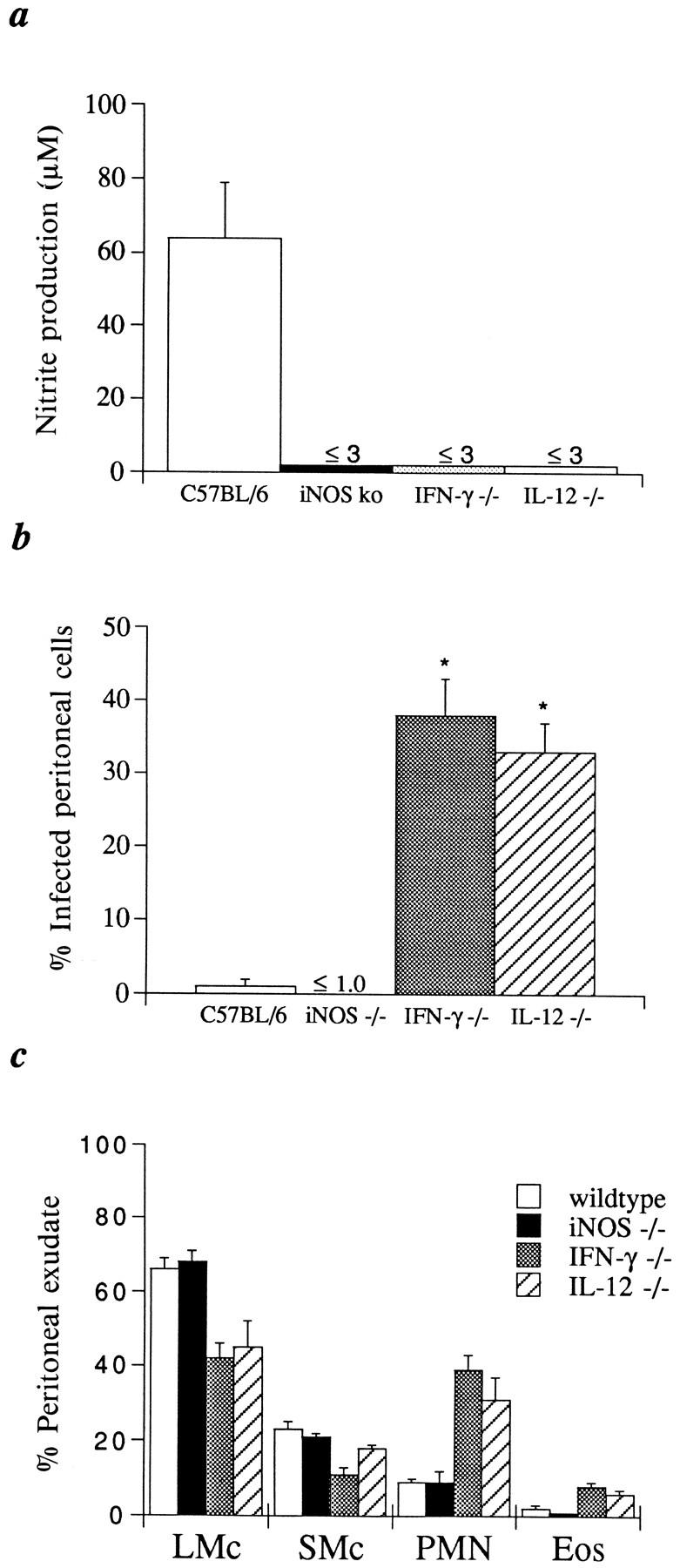
iNOS ko mice control acute tachyzoite replication effectively in the peritoneal cavity. (A) C57BL/6, iNOS ko, IL-12 p40−/− , and IFN-γ ko mice were infected i.p. with 20 cysts each and peritoneal cells harvested 5 d later. Nitrite levels were measured by the Griess reaction in 72-h supernatants of the cultured cell populations. The values shown are the mean ± SE of data points pooled from three different experiments involving a total of 6–12 animals per group. (B) The percentage of infected cells determined microscopically from cytospin preparations from the same animals studied in A, as detailed in Materials and Methods. (C) Differential counts of peritoneal exudate cells from the same animals studied in A and B. The mean ± SE cell recovery from the infected animals was: C57BL/6 (5.5 ± 0.8 × 106), iNOS ko (7.5 ± 2.1 × 106), IL-12 p40−/− (10.1 ± 1.8 × 106), IFN-γ ko (10.9 ± 1.5 × 106). Uninfected mice of each of the four strains did not differ significantly in the composition of their peritoneal cells (75–85% LMC, 10–20% SMC, 1–5% granulocytes) or cell yields (⩽1.5 × 106).
One concern raised by our data was that i.p. inoculation of the parasites does not induce the same mechanisms of immunity as the natural, p.o. route of infection. However, as shown in Fig. 1 B, p.o. infection with ME49 cysts led to a mortality pattern similar to that observed after i.p. inoculation (Fig 1 A). Thus, control animals exhibited no mortality during the observation period whereas IFN-γ ko mice succumbed within 1 wk and iNOS mice at ∼3 wk after parasite challenge.
Previous studies have demonstrated that T. gondii stimulates NO production in infected animals and have suggested that iNOS (NOS2) rather than NOS1 or NOS3 is the primary enzyme involved in its induction. However, it was formally possible that the latter isoforms might compensate for the deficiency in the iNOS ko animals. Measurement of the NO derivative, nitrite (N02 −) in supernatants of peritoneal cells harvested from 5-d infected animals revealed synthesis of NO in C57BL/6 but not iNOS ko cultures and thus failed to demonstrate a compensatory mechanism (Fig. 2 A). A similar deficit in NO synthesis was also observed in cultured spleen cells from these animals (C57BL/6 animals: naive, ⩽3 μM, 5-day infected = 29 ± 6 μM; 5-d infected iNOS ko mice, ⩽3 μM). Interestingly, peritoneal and spleen cells from IL-12 p40−/− and IFN-γ ko animals were also unable to synthesize significant levels of nitrite even after stimulation with STAg. The latter findings are likely to reflect the impaired IFN-γ synthesis in these animals.
Based on the observed time of death, we postulated that iNOS ko mice effectively restrict growth of the acute tachyzoite form. To test this hypothesis, peritoneal cells were recovered from mice at 5 d or brain tissue at 15 and 20 d after parasite challenge and the number of intracellular tachyzoites or cysts determined. As shown in Fig. 2 B, the percentage of infected exudate cells recovered from iNOS and C57BL/6 was comparable and considerably less than that detected in samples from IFN-γ ko or IL-12 p40–deficient mice (1–2% versus 30–40%, respectively) indicating that tachyzoite growth is restricted during the acute stage in the former mouse strains. Consistent with this result, differential counts of the peritoneal exudate cells from the infected mice revealed a comparable inflammatory response in C57BL/6 and iNOS ko animals (Fig. 2 C). In contrast, corresponding exudates from both IL-12 p40−/− and IFN-γ ko mice contained over twice the number of cells (data not shown) and significantly more granulocytes than either the iNOS ko or C57BL/6 samples that were equivalent (Fig. 2 C). Since we clearly observed an induction of nitrite in the C57BL/6-derived PEC and spleen cell cultures, but not in comparable cultures of iNOS ko cells (Fig. 2 A and text), these data indicate that the control of parasites in the acute stage of infection is not dependent upon the microbicidal activity of nitric oxide. In support of this conclusion, impression smears of heart, liver, lung, and spleen tissues revealed detectable tachyzoite replication in 5 d infected IFN-γ ko but not iNOS ko mice (data not shown) arguing against the possibility that loss of parasite control occurs in the iNOS-deficient animals but at a location distinct from the peritoneal inoculation site.
Mortality of iNOS ko Animals Is Associated with Defective Control of Parasite Growth in the CNS.
The mortality of iNOS ko animals at 3–4 wk after infection (Fig. 1) suggested that the control of parasite replication in the CNS, particularly the brain, might be impaired in these mice. To investigate this hypothesis, cysts were quantitated in brain homogenates or in tissue sections from infected iNOS ko mice as well as the parental C57BL/6 and C57BL/6 × 129 F1 strains. As shown in Fig. 3, more than twice as many cysts were apparent in the brains of iNOS ko as compared to C57BL/6 animals at days 12 and 21 after infection. In contrast, the parental strains, C57BL/6 and C57BL/6 × 129, displayed comparable cyst counts (1,600 ± 368 versus 1360 ± 305 cysts per brain, respectively, [n = 5]) as late as 30 d after infection. Moreover, inflammation was clearly more extensive in brain sections of infected iNOS ko as compared to C57BL/6 animals (Fig. 3, C–F). In addition, the sections from the iNOS ko, as opposed to control mice, displayed numerous necrotizing lesions (Fig. 3 E) that in many cases were associated with active tachyzoite replication. Nonetheless, immediately before developing an obvious moribund state, peripheral tissues (lung, liver, and spleen) from infected iNOS ko and C57BL/6 controls displayed indistinguishable histologic changes consisting of moderate peribronchial and periarterial inflammation in the lung, slight periportal inflammation, granulomatous hepatitis with poorly formed granulomas and occasional necrotic hepatocytes in the liver, and extramedullary hematopoiesis in the spleen. Closer to the time of death, the iNOS ko animals were severely depressed, demonstrated a weakness in all four limbs with the rear limbs more strongly affected, and had closed eyes. These symptoms are consistent with a severe necrotizing encephalitis leading to mortality.
Figure 3.
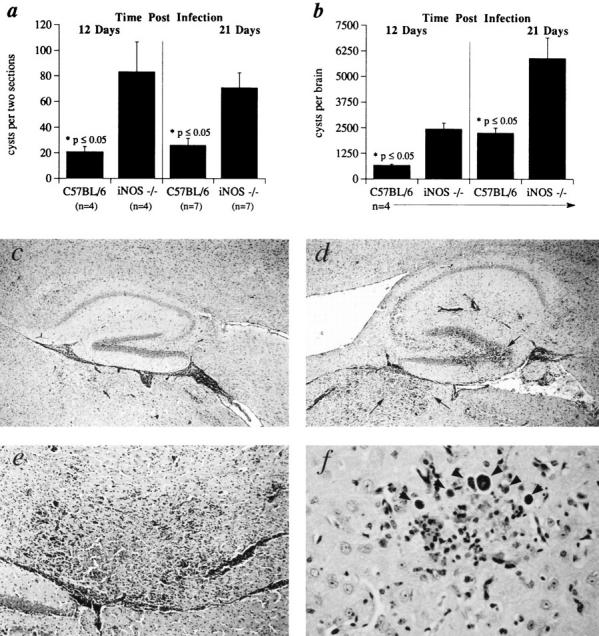
Brains of iNOS ko mice show increased cyst burdens as well as necrotizing encephalitis. C57BL/6 and iNOS ko mice were infected i.p. with 20 cysts each and brains harvested 12 and 21 d later for cyst enumeration in histological sections (A) or tissue homogenates (B). The mean cyst count and SE are shown for each group consisting of the number of animals indicated. (C–F) Photomicrographs of periodic acid–Schiff's stained, sagittal sections of brains from iNOS (D–F) and C57BL/6 (C) mice at 21 d after infection. Hippocampal regions of the brain in C57BL/6 (C) and iNOS ko (D) mice. Large areas of inflammation and necrosis seen in the iNOS brain (D) but not in C57BL/6 brain (C) are indicated by arrows. E shows a higher magnification of the same necrotizing lesion in D. (F) Brain section from iNOS ko mouse with numerous cysts (arrowheads) arranged in a satellite-like array, a feature frequently observed in these animals. Original magnification: (C and D) ×50; (E) ×200; (F) ×400.
Macrophages from iNOS ko Mice Fail to Control In Vitro Replication of T. gondii Tachyzoites.
The low frequency of tachyzoites in peritoneal exudates at 5 d after parasite challenge indicated that iNOS ko animals control T. gondii replication in this site during the acute stage of infection. From the latter observation and the predominance of large mononuclear cells in the peritoneal cavity, it might be predicted that iNOS ko macrophages are capable of intracellular parasite killing. To test this hypothesis, we evaluated the toxoplasmacidal activity of elicited and resident peritoneal cell populations from C57BL/6 and iNOS ko mice. In our initial experiments, inflammatory cells were collected from iNOS ko and C57BL/6 animals that were i.p. inoculated with thioglycollate (Table 1). As previously described (46), differential counts revealed that macrophage/monocytes were the primary cell type in the exudates from both strains (80– 90% macrophage, 10% lymphocyte, 5% eosinophil, 2% mast cell). Tachyzoites were added to the cultures and the amount of parasite growth monitored by measurement of 3H-uracil incorporation. As expected, unactivated cultures from either iNOS ko or C57BL/6 mice exhibited a 70– 100-fold increase in nucleotide incorporation after the addition of tachyzoites, suggesting that parasite replication had occurred. Microscopic evaluation confirmed the latter assumption: at 48 h after the addition of tachyzoites, the number of parasites per cell increased from 1.7 to 6.3 in C57BL/6 and from 2.1 to 4.5 in iNOS ko-derived cultures. Tachyzoite proliferation in the cells from C57BL/6 mice was dramatically reduced when the cells were pretreated with IFN-γ as measured by the 3H-Uracil assay (Table 1, >90% reduction ) or microscopic examination (t = 0, 1.7; t = 48 h, 1.5 tachyzoites per cell) and, in agreement with previous studies, this inhibition was reduced to less than 3% by the inclusion in the assay of l-NMMA, an established nitric oxide antagonist (9, 10, 29). Consistent with the postulated toxoplasmacidal role for RNI, IFN-γ– treated macrophages from iNOS ko animals were markedly impaired in the ability to kill exogenously added parasites (Table 1). The mean killing in activated cells from iNOS ko animals was 13.9 ± 5.8% versus 91.7 ± 2.6% in cultures from C57BL/6 mice (P ⩽0.001, n = 10).
Table 1.
Intracellular Killing of T. gondii Tachzyoites by Thioglycollate Elicited, Peritoneal Cells from Noninfected C57BL/6 and iNOS ko Mice
| In vitro stimulus | Tachyzoite/cell ratio | C57BL/6 | iNOS KO | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cpm | % killing | cpm | % killing | |||||||||
| I | None | 0 | 88 ± 4 | 130 ± 20 | ||||||||
| None | 1 | 8602 ± 2774 | 7078 ± 697 | |||||||||
| IFN-γ | 1 | 883 ± 61 | 90.7% | 5943 ± 1217 | 16.3% | |||||||
| II | None | 0 | 61 ± 36 | 31 ± 3 | ||||||||
| None | 0.2 | 4078 ± 706 | 3288 ± 472 | |||||||||
| IFN-γ | 0.2 | 335 ± 51 | 94.1% | 3780 ± 378 | 0.0% | |||||||
| IFN-γ l-NMMA | 0.2 | 4352 ± 469 | 2.3% | 4525 ± 375 | 2.8% | |||||||
| None | 1 | 10764 ± 1691 | 8117 ± 1084 | |||||||||
| IFN-γ | 1 | 389 ± 112 | 97.3% | 6849 ± 405 | 16.0% | |||||||
| IFN-γ l-NMMA | 1 | 9078 ± 660 | 0.0% | 7923 ± 341 | 21.0% | |||||||
Mice (3–5 per group) were injected intraperitoneally with sterile thioglycollate solution and 4–5 d later, the animals were killed and their peritoneal cells were isolated and pooled. The populations were then pretreated for 2 h with murine IFN-γ (100 U/ml) in the presence or absence of 2.5 mM l-NMMA. Cultures were subsequently infected with RH tachyzoites and 24 h later pulsed with 3H-uracil. Incorporated radioactivity was next determined and expressed as mean CPM ± SE for triplicate cultures. Percentage killing was then calculated using the formula indicated in Materials and Methods. The experiments shown are representative of five performed.
We have previously reported that i.p. challenge with T. gondii induces an influx of macrophages, neutrophils and lymphocytes into the peritoneal cavity and that the cellular composition of exudates from naive and 5-d infected animals is significantly different. One interpretation of our data is that the cells required to kill T. gondii in vivo are recruited to the site of infection and are thus not normally present in either resident or thioglycollate elicited populations. To test this hypothesis we assessed the microbicidal activity of resident and T. gondii elicited cell populations against exogenously added tachyzoites (Table 2). Cells from naive C57BL/6 mice, although initially unable to kill T. gondii, efficiently limited parasite replication when IFN-γ was added to the cultures. As expected, cells from infected C57BL/6 mice limited tachyzoite growth even in the absence of exogenously added IFN-γ, a finding consistent with the substantial endogenous production of this cytokine by PEC from infected animals (47) (Table 3). Neither resident nor T. gondii elicited cells from iNOS ko animals mediated appreciable parasite killing in the presence or absence of exogenously added cytokine. In these experiments, the mean killing in iNOS ko cultures was 13.2 ± 5.2% versus 88.1 ± 3.7% for C57BL/6 cells (P ⩽0.01, n = 10). Together, our data indicate that both in vitro and ex vivo macrophages from iNOS ko mice are defective in their ability to kill the parasite although the same animals clearly control early infection in vivo.
Table 2.
In Vitro Control of T. gondii Replication by Peritoneal Exudate Cells Harvested from Naive and 5-d Infected iNOS ko or C57BL/6 Mice
| In vitro stimulus | Tachyzoite/cell ratio | Normal resident cells | T. gondii elicited cells | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C57BL/6 | iNOS ko | C57BL/6 | iNOS ko | |||||||||||||||
| cpm | % killing | cpm | % killing | cpm | % killing | cpm | % killing | |||||||||||
| None | 0 | 43 ± 22 | 74 ± 53 | 294 ± 49 | 181 ± 21 | |||||||||||||
| None | 0.2 | 3146 ± 478 | 2746 ± 660 | 225 ± 74 | 1866 ± 44 | |||||||||||||
| None | 1 | 7554 ± 202 | 8654 ± 1489 | 226 ± 39 | 4882 ± 815 | |||||||||||||
| IFN-γ | 0 | 58 ± 15 | 54 ± 20 | 125 ± 26 | 97 ± 9 | |||||||||||||
| IFN-γ | 0.2 | 300 ± 33 | 92.2 | 1827 ± 317 | 31.0 | 205 ± 90 | 0 | 3031 ± 88 | 0 | |||||||||
| IFN-γ | 1 | 1180 ± 126 | 85.1 | 5997 ± 892 | 29.9 | 188 ± 35 | 0 | 5023 ± 822 | 0 | |||||||||
Mice (3–5 per group) were infected with 20 ME49 cysts each and 5 d later the animals were killed and peritoneal cells were isolated and pooled. The cells were next precultured in the presence or absence of IFN-γ, infected and harvested as described in Table 1. Percentage killing of tachyzoites was then calculated. The experiment shown is representative of four performed.
Table 3.
IL-12 and IFN-γ Synthesis by Cells Harvested from iNOS ko and C57BL/6 Mice at 5 d after i.p. Challenge with T. gondii
| Peritoneal exudate | Spleen | Serum | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean ± SE | n | Range | Mean ± SE | n | Range | Mean ± SE | n | Range | ||||||||||||||
| IL-12p40 | C57BL/6 | bkg | 1.4 ± 0.5 | 6 | 0–3.5 | 5.4 ± 1.8 | 7 | 2.3–12.7 | 17.1 ± 3.9 | 3 | 15.6–23.2 | |||||||||||
| STAg | 10.9 ± 3.3 | 6 | 0.3–19.5 | 35.4 ± 6.0 | 7 | 20.7–53.5 | ||||||||||||||||
| iNOS ko | bkg | 1.9 ± 0.4 | 6 | 1.2–4.1 | 10.3 ± 2.8 | 9 | 3.6–17.6 | 19.5 ± 9 | 3 | 13.7–27.3 | ||||||||||||
| STAg | 11.9 ± 4.3 | 6 | 4.3 ± 25.9 | 56.4 ± 9.1 | 9 | 38.0–93.6 | ||||||||||||||||
| IFN-γ | C57BL/6 | bkg | 0.1 ± 0.06 | 6 | 0–0.3 | 0.6 ± 0.5 | 7 | 0–3.1 | 5.1 ± 0.5 | 3 | 4.4–5.8 | |||||||||||
| STAg | 1.0 ± 0.3 | 6 | 0.2–2.1 | 8.3 ± 2.1 | 7 | 1.3–17.9 | ||||||||||||||||
| iNOS ko | bkg | 6.0 ± 2.9 | 6 | 0.3–15.4 | 1.1 ± 0.7 | 9 | 0–3.9 | 3.4 ± 0.3 | 3 | 2.9–3.8 | ||||||||||||
| STAg | 8.2 ± 4.0 | 6 | 0.8–2.13 | 10.4 ± 2.7 | 9 | 1.8–19.2 | ||||||||||||||||
Mice were infected with 20 ME49 cysts each, killed 5 d later, and then peritoneal and spleen cells were isolated. Cell suspensions from each animal were cultured for 72 h in medium alone or in the presence of STAG (5 μg/ml). Supernatants were then assayed by ELISA for IL-12p40 and IFN-γ as described in Materials and Methods. The data shown are pooled from three experiments employing the number of animals indicated.
The Control of Acute T. gondii Replication in INOS ko Mice Involves an IL-12/IFN-γ–dependent Mechanism.
We and others have previously demonstrated that control of acute T. gondii infection in normal hosts is highly dependent upon the induction of IL-12 and IFN-γ (22, 47–49). To determine whether the same effector mechanism operates in the absence of iNOS, we first assessed the production of these cytokines during early infection with the ME49 strain (Table 3). IL-12 p40 levels measured in sera or cell cultures (peritoneal or spleen) were not significantly different in 5-d infected iNOS ko and C57BL/6 animals. Similarly, levels of IFN-γ were comparable in sera and splenic cell supernatants of cultures from knockout and control mouse strains. Nevertheless, peritoneal cell cultures from iNOS ko animals produced higher levels of IFN-γ than cultures from control mice. The latter results suggest that the iNOS deficiency may have a localized rather than systemic effect on the regulation of IFN-γ synthesis. In vitro restimulation of spleen and peritoneal cell cultures substantially augmented production of both cytokines but failed to reveal further differences between the knockout and control animals (Table 3).
As might be predicted by the intact production of both IL-12 p40 and IFN-γ, these cytokines were found to play a critical role in the control of acute T. gondii infection by the iNOS ko animals. Thus, treatment with neutralizing mAb against IL-12 or IFN-γ led to enhanced mortality of the infected knockout mice with a kinetics comparable to that previously observed in antibody treated wild-type animals (Fig. 4). Taken together, the above experiments argue that iNOS-deficient mice survive acute infection as a result of an IL-12/IFN-γ–dependent mechanism indistinguishable from that arising in conventional mice and not because of the induction of a normally inactive pathway of host resistance.
Figure 4.

Innate resistance to T. gondii in iNOS ko mice is IL-12– and IFN-γ–dependent. iNOS ko animals were injected i.p. with 1 mg of either normal rat IgG (n = 8), anti–IFN-γ mAb XMG-6 (n = 7), or anti– IL-12 mAb C17.8 (n = 5) 1 d before infection with 20 cysts. Survival was monitored as in Fig. 1. The experiment shown is representative of three performed.
Neutrophils Contribute to Acute Resistance against T. gondii in both iNOS ko and Control Mice.
The ability of iNOS ko mice to control T. gondii infection in vivo despite their impaired macrophage-toxoplasmacidal function suggested that effector cells other than activated macrophages/monocytes may be crucial in mediating host resistance at the acute stage. One alternative effector cell we considered is the PMN since purified human PMN have been shown to restrict T. gondii replication in vitro (50) and these cells are rapidly recruited to the site of infection in mice (22, 51). To test the proposed contribution of PMN to acute resistance, both iNOS ko and C57BL/6 mice were treated with a mAb (RB6-8C5) against the GR-1 antigen, a protein which is expressed at high levels on murine neutrophils and eosinophils and at much lower levels on cells of the myeloid lineage (52). In vivo administration of the RB6-8C5 mAb significantly reduced neutrophil infiltration in the peritoneal cavity of both iNOS ko and C57BL/6 animals at 5 and 7 d after parasite challenge (Fig. 5 A). More importantly, a majority of the animals injected with the RB68C5 mAb succumbed during the acute phase of T. gondii infection. Thus, by 14 d after infection 75% of mAb treated iNOS ko and 40% of treated C57BL/6 mice had succumbed to infection with the ME49 strain (Fig. 5, B and C). In contrast, in vivo administration of the IL-5 neutralizing mAb, TRFK-5, ablated the low level eosinophil response in T. gondii infected C57BL/6 and iNOS ko strains but failed to affect mortality (data not shown). Taken together these experiments suggest that granulocytes, and in particular neutrophils, contribute to acute resistance against the parasite and may account for the ability of iNOS ko animals to control infection in the apparent absence of macrophage killing function.
Figure 5.
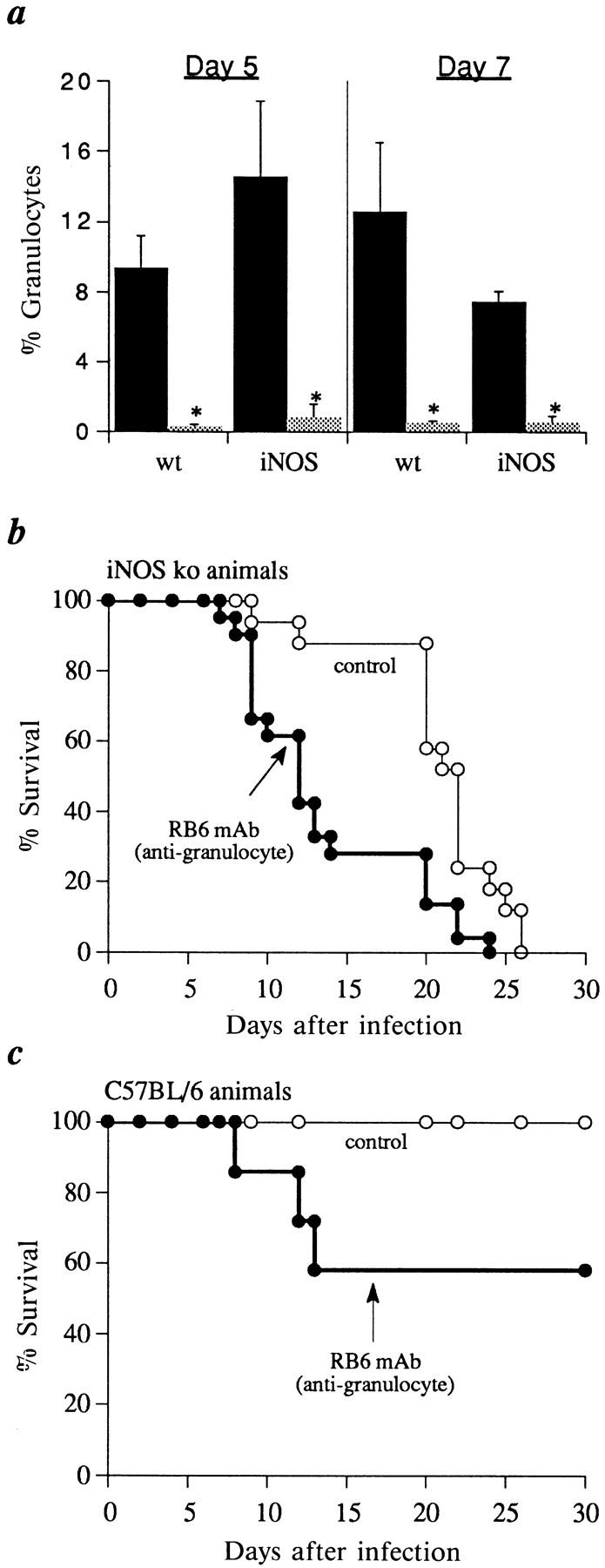
Treatment with anti-granulocyte mAb diminishes innate resistance to T. gondii in both iNOS and C57BL/6 (wt) mice. Groups of iNOS ko (n = 17–22, B) or C57BL/6 mice (n = 7–13, C) were treated at day 0, 2, and 4 with either the RB6-8C5 mAb or PBS as described in Materials and Methods. Differential counts (mean ± SE) were performed on peritoneal cells from animals killed at day 5 and 7 after infection (A, black bars, PBS treated; shaded bars, RB6-treated mice) and survival of the remaining animals determined as in Fig. 1 (B and C). The asterisks (*) indicate statistically significant differences (P ⩽0.05) in granulocyte composition. The experiment shown is representative of three performed.
Discussion
IFN-γ is known to be a critical mediator of innate resistance to T. gondii infection in vivo (20–22). Based on its readily demonstrated function in activating macrophages to kill tachyzoites in vitro (27–30), IFN-γ has been assumed to restrict parasite growth in the host primarily through the action of this effector cell. Nevertheless, it is clear that T. gondii can productively infect a wide range of different nucleated host cells (18) and therefore, in contrast to pathogens such as Leishmania, need not be controlled through contact with macrophages. In the present study, we have addressed this issue by examining the development of innate resistance to Toxoplasma in iNOS-deficient animals that exhibit markedly impaired macrophage toxoplasmacidal activity in vitro as a result of their inability to generate inducible NO. Surprisingly, these mice displayed normal control of acute T. gondii infection arguing that NO-dependent killing by macrophages is not an essential mechanism of innate resistance against the parasite.
A trivial explanation for the observed results is that differences in background genes, rather than the absence of iNOS, are responsible for the failure of the knockout mice to die during the acute phase or, alternatively for their early death relative to C57BL/6 controls. However, several lines of evidence argue against this possibility. First, with regard to the acute stage of infection, we know of no examples, from our own experience or the literature, in which background genes rescue or protect mice from T. gondii induced, acute mortality. For example, IFN-γ ko mice succumb with the same rapid kinetics whether they are back-crossed on to a C57BL/6 or BALB/c background (22) although the same backgrounds have a major influence on survival during the chronic stage of infection. Moreover, in direct contrast to the iNOS ko animals, ICSBP ko mice (lacking the interferon consensus sequence-binding protein) at a similar 129 × C57BL/6 back-cross generation rapidly succumb to acute infection with a phenotype virtually indistinguishable from infected IFN-γ ko mice (Scharton-Kersten, T., A. Sher, and K. Ozato, manuscript in preparation). Similarly, it is unlikely that contaminating 129 genes are responsible for the earlier CNS associated death of iNOS ko relative to the C57BL/6 mice since C57BL/6 × 129 F1 animals show enhanced, rather than decreased survival when compared to C57BL/6 mice (Fig. 1, inset). Thus, although it would have been preferable to use more fully back-crossed iNOS ko animals for these experiments, it is difficult to escape the conclusion that the observed phenotype in the knockout mice is a direct effect of the absence of iNOS rather than a difference in genetic background.
A central role for NO as the primary toxoplasmacidal mediator of activated macrophages has been established from in vitro studies employing NO synthase inhibitors (9, 29, 30). Our results strongly support this concept by demonstrating that peritoneal macrophages harvested from naive, thioglycollate injected, or ME49 infected iNOS ko mice are grossly impaired in their ability to control parasite replication in vitro even after IFN-γ activation (Tables 1 and 2). Nevertheless, iNOS ko mice were clearly able to control early infection such that few, if any, tachyzoites were apparent in peritoneal macrophages obtained from the animals during the acute stage (Fig. 2 B). One interpretation of this major discrepancy between the in vitro and in vivo findings is that activated macrophages are not crucial for innate resistance against T. gondii. Instead, other IFN-γ–dependent effector mechanisms may prevent parasite growth before productive infection of macrophages can occur. Alternatively, it is possible that macrophages are indeed the major effector cells of tachyzoite killing but use a microbicidal mechanism which is operative in vivo but not in vitro. For example, macrophages in vitro may not receive the appropriate costimuli or other accessory factors needed to induce NO-independent parasite control. Given that peritoneal cells, freshly isolated ex vivo, from infected iNOS ko animals show the same defect in tachyzoite killing as resident or elicited macrophages from naive donors, the latter explanation seems unlikely. A final possibility not ruled out by our experiments is that macrophages are indeed the major effector cells of innate resistance but that the relevant subset is NO independent and thus, distinct from the NOdependent peritoneal population assayed in our experiments.
Regardless of the particular effector cell involved, it is clear from the antibody neutralization experiments (Fig. 4) that the mechanism of innate resistance in both wild-type and iNOS-deficient animals is IFN-γ as well as IL-12 dependent. In addition to RNI-mediated killing by macrophages, three additional tachyzoite killing functions have been identified that are IFN-γ dependent and thus could explain the acute resistance of the iNOS ko mice. First, reactive oxygen intermediates (ROI; e.g., H2O2 and O2−) have been implicated in the toxoplasmacidal activity of certain murine and human macrophage populations in vitro (53, 54). However, the physiologic significance of the ROI pathway remains controversial since other laboratories have reported that T. gondii tachyzoites fail to trigger the oxidative burst in the same cells (55) or that the parasites are resistant to the metabolites produced (56). We have recently addressed this issue by studying T. gondii infection in p47 phox-deficient animals, which lack an inducible oxidative burst (57). These knockout mice were found to efficiently control both acute and chronic infection in vivo and macrophages from the mutant animals were fully capable of limiting tachyzoite replication in vitro arguing against a crucial role for the ROI in host resistance (Scharton-Kersten, T., S. Jackson, and S. Holland, unpublished observations). A second alternative control mechanism is tryptophan starvation of the parasite, an IFN-γ–induced pathway used by human fibroblasts and macrophage populations to inhibit T. gondii replication in vitro (58, 59). Although clearly functional in the human immune response, this microbicidal mechanism cannot be demonstrated in mice (60). Finally, an IFN-γ–dependent toxoplasmacidal activity, which is independent of RNI, ROI, or tryptophan starvation, has recently been described in human endothelial cells (61). Again however, a murine equivalent of this uncharacterized human effector mechanism has not yet been identified.
In addition to IFN-γ–activated macrophages, fibroblasts and/or endothelial cells, we have also considered the possible contribution of granulocytes to parasite control since human peripheral blood PMN have been shown to kill intracellular tachyzoites in vitro (50). Moreover, neutrophils have recently been shown to play an important role in innate resistance to both L. monocytogenes (62, 63) and Candida albicans (64) in murine models. Depletion of granulocytes by treatment with RB6-8C5 resulted in enhanced mortality of ME49 infected iNOS ko as well as wild-type animals. Since depletion of eosinophils by neutralization of IL-5 failed to affect host resistance, the neutrophil is likely to be the relevant effector cell in the granulocyte population. Nevertheless, because peritoneal cell populations from infected iNOS ko animals do not display significant levels of microbicidal activity, it has so far been difficult to demonstrate a direct effect of murine neutrophils on parasite survival in vitro. It is possible however, that the role of neutrophils in host resistance does not involve direct lysis of tachyzoites but rather, indirect anti-microbial functions such as scavenging infected cells (65), secretion of toxic products leading to metabolic poisoning of the parasite, or the production of chemokines required for recruitment of other effector cell populations (66).
A critical issue raised by our findings is whether the control of acute T. gondii infection in iNOS ko animals is due to a normally occurring, but previously unrecognized, effector mechanism or instead reflects the induction of an aberrant compensatory host response. Numerous instances of the expression of such compensatory mechanisms have been documented in knockout mice. For example, mice lacking the β2-microglobulin chain of the MHC class I complex develop an abnormal expansion of NK 1.1+, IFN-γ–producing effector cells following T. gondii infection (67). We believe that such an interpretation is an unlikely explanation for the behavior of iNOS ko animals described here. Thus, in almost every parameter examined, infected iNOS and wild-type mice were indistinguishable during the acute stage of infection. For instance, control and ko mouse strains exhibited essentially identical local inflammatory responses (Fig. 2 C) as well as systemic IL-12 and IFN-γ synthesis (Table 3). Moreover, the effect of granulocyte depletion, although more dramatic in iNOS ko mice, was apparent in both mutant and wild-type strains (Fig. 5). A minor difference noted was the elevated, local production of IFN-γ in infected knockout mice, an alteration which was not evident in either spleens or sera of the same animals and which may reflect the absence of NO mediated suppression of lymphocyte function (68). A final argument is that iNOS ko mice clearly show defective resistance to other intracellular pathogens such as L. major and L. monocytogenes indicating the absence of compensatory immune responses at least in these models (32, 33). We therefore postulate that the mechanism of innate immunity operating in iNOS ko animals is the same as that induced in conventional mice and thus represents an as yet unappreciated pathway of host resistance against the parasite.
Although iNOS ko animals were clearly able to control acute infection, they did eventually succumb to T. gondii at 3–4 wk after inoculation (Fig. 1) and, when compared to control mice, harbored significantly higher numbers of brain cysts at 12 and 21 d (Fig. 3, A and B). Histopathological examination of the brains of infected iNOS ko mice on d 20 and beyond revealed the development of severe necrotizing lesions in the CNS (Fig. 3, C–F) and the presence of unchecked tachyzoite replication in the affected areas. These features were reminiscent of the reactivation-associated toxoplasmic encephalitis observed in chronically infected mice treated with neutralizing mAb against TNFα (34) or IFN-γ (23) and, to some extent, in animals treated with the nitric oxide synthase inhibitor, aminoguanidine (11). In the mouse, IFN-γ–activated microglial cells, but not astrocytes, have been shown to inhibit tachyzoite replication by means of NO-dependent mechanisms (29). In addition to microglial cells, macrophages also infiltrate the CNS and may exhibit NO-mediated toxoplasmacidal activity. Our results suggest that these NO-dependent effector cells are essential for controlling tachyzoite replication and dissemination in the CNS. This is in direct contrast to the situation in the periphery, where as discussed above, other NO-independent effector cells or mechanisms play the dominant role in mediating resistance. One explanation for why inducible NO is more critical for the control of toxoplasma in the CNS is that the NO-independent mechanism operative in the periphery is excluded from or cannot function within nervous tissue. Further studies in the iNOS ko infection model described here should be useful in defining the basis of this stage specificity in effector function as well as the uncharacterized mechanism which limits parasite growth during acute infection.
Acknowledgments
We are grateful to Dr. Allen Cheever for his expert evaluation of histopathologic sections. We also thank Pat Caspar, Ricardo Dreyfus, and Sara Hieny for technical assistance, Drs. John MacMicking, Carl Nathan (Cornell University Medical College, New York, NY) and John Mudgett(Merck Research Laboratories, Rahway, NJ) for providing breeding pairs of the iNOS ko animals, and Drs. Stephanie James, Warren Leonard, and Tom Wynn for critical reading of the manuscript.
Footnotes
1 Abbreviations used in this paper: CNS, central nervous system; iNOS, inducible nitric oxide synthase; IRF, interferon response factor; ko, knockout; LMC, large mononuclear cell; l-NMMA, NG-monomethyl-l-arginine; NO, nitric oxide; p.o., peroral; RNI, reactive nitrogen intermediates; ROI, reactive oxygen intermediates; SMC, small mononuclear cell; STAg, soluble tachyzoite antigen.
Dr. Scharton-Kersten and Dr. Yap made equally significant contributions.
References
- 1.James S. Role of nitric oxide in parasitic infections. Microb Rev. 1995;59:533–547. doi: 10.1128/mr.59.4.533-547.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nathan C, Xie Q. Nitric oxide synthases: roles, tolls and controls. Cell. 1994;78:915–918. doi: 10.1016/0092-8674(94)90266-6. [DOI] [PubMed] [Google Scholar]
- 3.James SL, Glaven J. Macrophage cytotoxicity against schistosomula of Schistosoma mansoniinvolves arginine-dependent production of reactive nitrogen intermediates. J Immunol. 1989;143:4208–4212. [PubMed] [Google Scholar]
- 4.Green SJ, Meltzer JB, Hibbs JB, Jr, Nacy CA. Activated macrophages destroy intracellular Leishmania majoramastigotes by an L-arginine dependent killing mechanism. J Immunol. 1990;144:278–283. [PubMed] [Google Scholar]
- 5.Liew FY, Yun L, Millott S. TNF-alpha synergizes with IFN-gamma in mediating killing of Leishmania majorthrough the induction of nitric oxide. J Immunol. 1990;145:4306–4310. [PubMed] [Google Scholar]
- 6.Mauel J, Ransijn A, Buchmuller-Rouiller Y. Killing of Leishmaniaparasites in activated murine macrophages is based on an L-arginine-dependent process that produces nitrogen derivatives. J Leukocyte Biol. 1991;49:73–82. doi: 10.1002/jlb.49.1.73. [DOI] [PubMed] [Google Scholar]
- 7.Liew FY, Millott S, Parkinson C, Palmer RMJ, Moncada S. Macrophage killing of Leishmaniaparasites in vivo is mediated by nitric oxide from L-arginine. J Immunol. 1990;144:4794–4797. [PubMed] [Google Scholar]
- 8.Stenger S, Donhauser N, Thuring H, Rollinghoff M, Bodgan C. Reactivation of latent leishmaniasis by inhibition of inducible nitric oxide synthase. J Exp Med. 1996;183:1501–1514. doi: 10.1084/jem.183.4.1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adams LB, Hibbs JB, Jr, Taintor RR, Krahenbuhl JS. Microbistatic effect of murine activated macrophages for Toxoplasma gondii. Role for synthesis of inorganic nitrogen oxides from L-arginine. J Immunol. 1990;144:2725–2729. [PubMed] [Google Scholar]
- 10.Langermans JAM, Van Der Hulst MEB, Nibbering PH, Hiemstra PS, Fransen L, Van Hurth R. IFNgamma-induced L-arginine-dependent toxoplasmastatic activity in murine peritoneal macrophages is mediated by endogenous tumor necrosis factor. J Immunol. 1992;148:568–574. [PubMed] [Google Scholar]
- 11.Hayashi S, Chan C, Gazzinelli R, Roberge FG. Contribution of nitric oxide to the host parasite equilibrium in toxoplasmosis. J Immunol. 1996;156:1476–1481. [PubMed] [Google Scholar]
- 12.Gazzinelli RT, Oswald IP, Hieny S, James SL, Sher A. The microbicidal activity of interferon-gammatreated macrophages against Trypanosoma cruziinvolves an L-arginine-dependent, nitrogen oxide-mediated mechanism inhibitable by interleukin-10 and transforming growth factorbeta. Eur J Immunol. 1992;22:2501–2506. doi: 10.1002/eji.1830221006. [DOI] [PubMed] [Google Scholar]
- 13.Vespa GN, Cunha FQ, Silva JS. Nitric oxide is involved in control of Trypanosoma cruzi-induced parasitemia and directly kills the parasite in vitro. Infect Immun. 1994;62:5177–5182. doi: 10.1128/iai.62.11.5177-5182.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Munoz-Fernandez MA, Fernandez MA, Fresno M. Synergism between tumor necrosis factor-alpha and interferon-gamma on macrophage activation for the killing of Trypanosoma cruzithrough a nitric oxide dependent mechansim. Eur J Immunol. 1992;22:301–307. doi: 10.1002/eji.1830220203. [DOI] [PubMed] [Google Scholar]
- 15.Beckerman KP, Rogers HW, Corbett JA, Schreiber RD, McDaniel ML, Unanue ER. Release of ntiric oxide during the T-cell independent pathway of macrophage activation: its role in resistance to Listeria monocytogenes . J Immunol. 1993;150:888–895. [PubMed] [Google Scholar]
- 16.Bookvar KS, Granger DL, Poston RM, Maybodi M, Washington MK, Hibbs J, Jr, Kurlander RL. Nitric oxide produced during murine listeriosis is protective. Infect Immun. 1994;62:1089–1100. doi: 10.1128/iai.62.3.1089-1100.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gazzinelli, R.T., D. Amchay, T., Scharton-Kersten, E. Grunwald, J.M. Farber, and A. Sher. 1996. Role of marophage derived cytokines in the induction and regulation of cell-mediated immunity to Toxoplasma gondii In Toxoplasma gondii. U. Gross, editor. Springer, Berlin. 127–139. [DOI] [PubMed]
- 18.Werk R. How does Toxoplasma gondii enter host cells? . Rev Infect Dis. 1985;7:449–457. doi: 10.1093/clinids/7.4.449. [DOI] [PubMed] [Google Scholar]
- 19.Kasper, L.H., and J.C. Boothroyd. 1992. Toxoplasma gondii: immunology and molecular biology. In Immunology of Parasitic Infections. K. Warren and N. Agabian, editors. Oxford, UK. 269–301.
- 20.Suzuki Y, Orellana MA, Schreiber RD, Remington JS. Interferon-gamma: the major mediator of resistance against Toxoplasma gondii . Science (Wash DC) 1988;240:516–518. doi: 10.1126/science.3128869. [DOI] [PubMed] [Google Scholar]
- 21.Johnson LL, Sayles PC. Strong cytolytic activity of natural killer cells is neither necessary nor sufficient for preimmune resistance to Toxoplasma gondiiinfection. Nat Immun. 1995;14:209–215. [PubMed] [Google Scholar]
- 22.Scharton-Kersten TM, Wynn TA, Denkers EY, Bala S, Grunvald E, Hieny S, Gazzinelli RT, Sher A. In the absence of endogenous interferon-gamma mice develop unimpaired IL-12 responses to Toxoplasma gondiiwhile failing to control acute infection. J Immunol. 1996;157:4045–4054. [PubMed] [Google Scholar]
- 23.Suzuki Y, Conley FK, Remington JS. Importance of endogenous IFN-gamma for prevention of toxoplasmic encephalitis in mice. J Immunol. 1989;143:2045–2050. [PubMed] [Google Scholar]
- 24.Gazzinelli R, Xu Y, Hieny S, Cheever A, Sher A. Simultaneous depletion of CD4+ and CD8+ T lymphocytes is required to reactivate chronic infection with Toxoplasma gondii . J Immunol. 1992;149:175–180. [PubMed] [Google Scholar]
- 25.McCabe RE, Luft BJ, Remington JS. Effect of murine interferon gamma in murine toxoplasmosis. J Infect Dis. 1984;150:961–962. doi: 10.1093/infdis/150.6.961. [DOI] [PubMed] [Google Scholar]
- 26.Suzuki Y, Conley FK, Remington JS. Treatment of toxoplasmic encephalitis with recombinant gamma interferon. Infect Immun. 1990;58:3050–3055. doi: 10.1128/iai.58.9.3050-3055.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Remington JS, Krahenbuhl JL, Mendenhall JW. A role for activated macrophages in resistance to infection with Toxoplasma . Infect Immun. 1972;6:829–834. doi: 10.1128/iai.6.5.829-834.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nathan CF, Murray HW, Wiebe ME, Rubin BY. Identification of interferon-gamma as the lymphokine that activates human macrophage oxidative metabolism and antimicrobial activity. J Exp Med. 1983;158:670–689. doi: 10.1084/jem.158.3.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chao CC, Anderson WR, Hu S, Gekker G, Martella A, Peterson PK. Activated microglia inhibit multiplication of Toxoplasma gondiivia a nitric oxide mechanism. Clin Immun Immunopath. 1993;67:178–183. doi: 10.1006/clin.1993.1062. [DOI] [PubMed] [Google Scholar]
- 30.Bohne W, Heesemann J, Gross U. Reduced replication of Toxoplasma gondiiis necessary for induction of bradyzoite-specific antigens: a possible role for nitric oxide in triggering stage conversion. Infect Immun. 1994;62:1761–1767. doi: 10.1128/iai.62.5.1761-1767.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Khan IA, Ma11tsuura T, Fonseka S, Kasper L. Production of nitric oxide (NO) is not essential for protection against acute Toxoplasma gondiiinfection in IRF-1−/− mice. J Immunol. 1996;156:636–643. [PubMed] [Google Scholar]
- 32.MacMicking JD, Nathan C, Hom G, Chartrain N, Fletcher DS, Trumbauer M, Stevens K, Xie Q, Sokol K, Hutchinson N, et al. Altered responses to bacterial infection and endotoxic shock in mice lacking inducible nitric oxide synthase. Cell. 1995;81:641–650. doi: 10.1016/0092-8674(95)90085-3. [DOI] [PubMed] [Google Scholar]
- 33.Wei X, Charles IG, Smith A, Ure J, Feng G, Huang F, Xu D, Muller W, Moncada S, Liew FY. Altered immune responses in mice lacking inducible nitric oxide synthase. Nature (Lond) 1995;375:408–411. doi: 10.1038/375408a0. [DOI] [PubMed] [Google Scholar]
- 34.Gazzinelli RT, Eltoum I, Wynn TA, Sher A. Acute cerebral toxoplasmosis is induced by in vivo neutralization of TNF-alpha and correlates with the down-regulated expression of inducible nitric oxide synthase and other markers of macrophage activation. J Immunol. 1993;151:3672–3681. [PubMed] [Google Scholar]
- 35.Suzuki Y, Yang Y, Yang S, Nguyen N, Lim S, Liesenfeld O, Kojima T, Remington JS. IL-4 is protective against development of Toxoplasmicencephalitis. J Immunol. 1996;157:2564–2569. [PubMed] [Google Scholar]
- 36.Magram J, Connaughton SE, Warrier RR, Carvajal DM, Wu C, Ferrante J, Stewart C, Sarmiento U, Faherty DA, Gately MK. IL-12-deficient mice are defective in interferon-gamma production and type 1 cytokine responses. Immunity. 1996;4:471–481. doi: 10.1016/s1074-7613(00)80413-6. [DOI] [PubMed] [Google Scholar]
- 37.Dalton DK, Pitts-Meek S, Keshav S, Figari IS, Bradley A, Stewart TA. Multiple defects of immune cell function in mice with disrupted interferon-gamma genes. Science (Wash DC) 1993;259:1739–1742. doi: 10.1126/science.8456300. [DOI] [PubMed] [Google Scholar]
- 38.McLeod R, Remington JS. A method to evaluate the capacity of monocytes and macrophages to inhibit multiplication of an intracellular pathogen. J Immunol Methods. 1979;27:19–29. doi: 10.1016/0022-1759(79)90235-7. [DOI] [PubMed] [Google Scholar]
- 39.Pfefferkorn ER, Pfefferkorn LC. Specific labeling of intracellular Toxoplasma gondiiwith uracil. J Protozool. 1977;24:449–453. doi: 10.1111/j.1550-7408.1977.tb04774.x. [DOI] [PubMed] [Google Scholar]
- 40.Murray HW, Rubin BY, Carriero SM, Harris AM, Jaffee EA. Human mononuclear phagocyte anti-protozoal mechanism: oxygen-dependent, vs. oxygen-independent activity against intracellular Toxoplasma gondii . J Immunol. 1985;134:1982–1988. [PubMed] [Google Scholar]
- 41.Catterall JR, Black CM, Leventhal JP, Rizk JP, Wachtel JS, Remington JS. Non-oxidative microbicidal activity in normal human alveolar and peritoneal macrophages. Infect Immun. 1987;55:1635–1640. doi: 10.1128/iai.55.7.1635-1640.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Green LCD, Wagner DDA, Glogowski J, Skepper PL, Sishnok JS, Tannenbaum SR. Analysis of nitrate, nitrite and (15N) in biological fluids. Anal Biochem. 1982;126:131–138. doi: 10.1016/0003-2697(82)90118-x. [DOI] [PubMed] [Google Scholar]
- 43.Cherwinski HM, Schumacher JH, Brown KD, Mosmann TR. Two types of mouse T cell clone: further differences in lymphokine synthesis between Th1 and Th2 clones revealed by RNA hybridization, functionally monospecific bioassays and secreted proteins. J Exp Med. 1987;166:1229–1239. doi: 10.1084/jem.166.5.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schumacher JH, O'Garra A, Schrader B, Van Kimmenade A, Bond MW, Mosmann TR, Coffman RL. The characterization of four monoclonal antibodies specific for mouse IL-5 and development of mouse and human IL-5 ELISA. J Immunol. 1988;141:1576–1581. [PubMed] [Google Scholar]
- 45.Wysocka M, Kubin M, Vieira LQ, Ozmen L, Garotta G, Scott P, Trinchieri G. Interleukin-12 is required for interferon-gamma production and lethality in lipopolysaccharide-induced shock in mice. Eur J Immunol. 1995;25:672–676. doi: 10.1002/eji.1830250307. [DOI] [PubMed] [Google Scholar]
- 46.Czuprynski CJ, Henson PM, Campbell PA. Killing of Listeria monocytogenesby inflammatory neutrophils and mononuclear phagocytes from immune and non-immune mice. J Leukocyte Biol. 1984;35:193–208. doi: 10.1002/jlb.35.2.193. [DOI] [PubMed] [Google Scholar]
- 47.Gazzinelli RT, Wysocka M, Hayashi S, Denkers EY, Hieny S, Caspar P, Trinchieri G, Sher A. Parasiteinduced IL-12 stimulates early IFN-gamma synthesis and resistance during acute infection with Toxoplasma gondii . J Immunol. 1994;153:2533–2543. [PubMed] [Google Scholar]
- 48.Hunter CA, Remington JS. The role of IL12 in toxoplasmosis. Res Immunol. 1995;146:546–552. doi: 10.1016/0923-2494(96)83030-6. [DOI] [PubMed] [Google Scholar]
- 49.Khan IA, Matsuura T, Fonseka S, Kasper LH. Interleukin-12 enhances murine survival against acute toxoplasmosis. Infect Immun. 1994;62:1639–1645. doi: 10.1128/iai.62.5.1639-1642.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wilson CB, Remington JS. Activity of human blood leukocytes against Toxoplasma gondii . J Infect Dis. 1979;140:890–895. doi: 10.1093/infdis/140.6.890. [DOI] [PubMed] [Google Scholar]
- 51.Khavkin T. Histological and ultrastructural studies of the interaction of Toxoplasma gondiitachyzoites with mouse omentum in experimental infection. J Protozool. 1981;28:317–325. doi: 10.1111/j.1550-7408.1981.tb02858.x. [DOI] [PubMed] [Google Scholar]
- 52.Hestdal K, Ruscetti FW, Ihle JN, Jacobsen SEW, DuBois CM, Kopp WC, Longo DL, Keller JR. Characterization and regulation of RB6-8C5 antigen expression on murine bone marrow cells. J Immunol. 1991;147:22–28. [PubMed] [Google Scholar]
- 53.Murray HW, Cohn ZA. Macrophage oxygendependent anti-microbicidal activity. I. Susceptibility of Toxoplasma gondiito oxygen intermediates. J Exp Med. 1979;150:938–949. doi: 10.1084/jem.150.4.938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Murray HW, Juangbhanich CW, Nathan CF, Cohn ZA. Macrophage oxygen-dependent anti-microbicidal activity. II. The role of oxygen intermediates. J Exp Med. 1979;150:950–964. doi: 10.1084/jem.150.4.950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wilson CB, Tsai V, Remington JS. Failure to trigger the oxidative metabolic burst by normal macrophages. J Exp Med. 1980;151:328–346. doi: 10.1084/jem.151.2.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chang HR, Pechere JC. Macrophage oxidative metabolism and intracellular Toxoplasma gondii . Microb Pathogen. 1989;7:37–44. doi: 10.1016/0882-4010(89)90109-5. [DOI] [PubMed] [Google Scholar]
- 57.Jackson SH, Gallin JI, Holland SM. The p47 phox mouse knock-out model of chronic granulomatous disease. J Exp Med. 1995;182:751–758. doi: 10.1084/jem.182.3.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pfefferkorn ER. Interferon gamma blocks the growth of T. gondiiin human fibroblasts by inducing the host cells to degrade tryptophan. Proc Natl Acad Sci USA. 1984;81:908–912. doi: 10.1073/pnas.81.3.908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Murray HW, Szuro-Sudol A, Wellner D, Oca MJ, Granger AM, Libby DM, Rothermel CD, Rubin BY. Role of tryptophan degradation in respiratory burstindependent anti-microbial activity of gamma interferon stimulated human macrophages. Infect Immun. 1989;57:845–849. doi: 10.1128/iai.57.3.845-849.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schwartzman JD, Gonias SL, Pfefferkorn ER. Murine gamma interferon fails to inhibit Toxoplasma gondiigrowth in murine fibroblasts. Infect Immun. 1990;58:833–834. doi: 10.1128/iai.58.3.833-834.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Woodman JP, Dimier IH, Bout DT. Human endothelial cells are activated by IFN-gamma to inhibit Toxoplasma gondiireplication. J Immunol. 1991;147:2019–2023. [PubMed] [Google Scholar]
- 62.Conlan JW, North RJ. Neutrophil-mediated dissolution of infected host cells as a defense strategy against a facultative intracellular bacterium. J Exp Med. 1991;174:741–743. doi: 10.1084/jem.174.3.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rogers HW, Unanue ER. Neutrophils are involved in acute, nonspecific resistance to Listeria monocytogenesin mice. Infect Immun. 1993;61:5090–5096. doi: 10.1128/iai.61.12.5090-5096.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Romani L, Mencacci A, Cenci E, Spaccapelo R, Toniatti C, Puccetti P, Bistoni F, Poli V. Impaired neutrophil response and CD4+ T helper cell 1 development in interleukin 6-deficient mice infected with Candida albicans . J Exp Med. 1996;183:1345–1355. doi: 10.1084/jem.183.4.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rogers HW, Callery MP, Deck B, Unanue ER. Listeria monocytogenesinduces apoptosis of infected hepatocytes. J Immunol. 1996;156:679–684. [PubMed] [Google Scholar]
- 66.Kasama T, Strieter RM, Lukacs NW, Lincoln PM, Burdick MD, Kunkel SL. Interferon gamma modulates the expression of neutrophil derived chemokines. J Invest Med. 1995;43:58–67. [PubMed] [Google Scholar]
- 67.Denkers EY, Gazzinelli RT, Martin D, Sher A. Emergence of NK 1.1+ cells as effectors of immunity to Toxoplasma gondiiin MHC class I–deficient mice. J Exp Med. 1993;178:1465–1472. doi: 10.1084/jem.178.5.1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Candolfi E, Hunter CA, Remington JS. Mitogen- and antigen specific proliferation of T cells in murine toxoplasmosis is inhibited by reactive nitrogen intermediates. Infect Immun. 1994;62:1995–2001. doi: 10.1128/iai.62.5.1995-2001.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]