

Abstract
Bystander activation, i.e., activation of T cells specific for an antigen X during an immune response against antigen Y may occur during viral infections. However, the low frequency of bystander-activated T cells has rendered it difficult to define the mechanisms and possible in vivo relevance of this nonspecific activation. This study uses transgenic mice expressing a major histocompatibility complex class I–restricted TCR specific for glycoprotein peptide 33-41 of lymphocytic choriomeningitis virus (LCMV) to overcome this limitation. CD8+ T cells from specific pathogen-free maintained, unimmunized “naive” TCR transgenic mice can differentiate into LCMV-specific cytolytic effector CTL during infections with vaccinia virus or Listeria monocytogenes in vivo or mixed lymphocyte culture in vitro. We show that in these model situations (a) nonspecifically activated CTL are able to confer antiviral protection in vivo, (b) bystander activation is largely independent of the expression of a second T cell receptor of different specificity, (c) bystander activation is not mediated by a broadly cross-reactive TCR, but rather by cytokines, (d) bystander activation can be mediated by cytokines such as IL-2, but not α/β-IFN in vitro; (e) bystander activation is, overall, a rare event, occuring in vivo in roughly 1 in 200 of the LCMV-specific CTL during infection of TCR transgenic mice with vaccinia virus; (f) bystander activation does not have a significant functional impact on nontransgenic CTL memory under the conditions tested; and (g) even in the TCR transgenic situation, where unphysiologically high numbers of T cells of a single specificity are present, bystander activation is not sufficient to cause clinically manifest autoimmune disease in a transgenic mouse model of diabetes. We conclude that although bystander activation via cytokines may generate cytolytically active CTL from naive precursors, quantitative considerations suggest that this is usually not of major biological consequence.
Specificity is one of the hallmarks of the adaptive immune system. For CTL, specific activation requires the interaction of the TCR with its nominal peptide bound to MHC class I molecules. The questions of whether, how, and to what extent CTL can also be nonspecifically activated in the absence of this cognate interaction are of obvious importance; nonspecific activation of potentially self-reactive T cells during immune responses to foreign antigens may trigger autoimmune diseases. In addition, reactivation of primed T cells by heterologous viruses or cytokines has been postulated to contribute to maintenance of immunological memory (1–5).
Unspecific polyclonal stimulation of alloreactive CTL has been described to occur during viral infections (6–8) in mice and in humans (9–11). The high precursor frequency of alloreactive CTL clones allows detection of this nonspecific stimulation in a cytotoxicity assay. Also, nonspecific activation of antiviral memory CTL, displaying an intermediate precursor frequency, has been demonstrated, mainly using limiting dilution assays (2, 3). In contrast, the frequency of naive precursor CTL against viral or self antigens is comparatively low. Whether these cells may be nonspecifically activated has, therefore, been difficult to study in functional assays.
The mechanisms postulated to be involved in nonspecific T cell activation are poorly characterized; they include the following. (a) Cross-reactivity at the level of the TCR, which recognizes MHC molecules presenting the nominal peptide, but could also bind to a nonnominal peptide with sufficient avidity for the T cell to be activated. Several examples for this “molecular mimicry” have been postulated and presented (12). (b) Activation of a given T cell by a virus-specific TCR, which could lead to effector function of this T cell via a second TCR of different (e.g., self) specificity (13). It has been shown that allelic exclusion of the α chain of the T cell receptor is incomplete (14, 15), and T cells carrying two different TCRs have been demonstrated in mice (16, 17) as well as in humans (18). (c) “Bystander activation” via cytokines secreted by antigen-responsive cells such as type I IFN (5) or combinations of other cytokines (4) which could act independently of the TCR.
When discussing the biological effects of this nonspecific T cell activation, a strict definition of “activation” is indispensable. Whereas proliferation or upregulation of certain surface markers are measurable signs of activation, the extent these phenomena reflect in vivo effector function varies between experimental systems. In the lymphocytic choriomeningitis virus (LCMV)1 model used in this study, CTLmediated antiviral effector function is almost exclusively mediated by contact-dependent perforin-mediated cytotoxicity. Within this system, the question of whether bystander activation is of biological significance in vivo, can therefore be studied by assessing whether nonspecific activation can induce cytolytically active effector CTL, which can mediate antiviral protection, immunopathology, or autoimmunity.
In this study, we analyzed the mechanisms of how TCR transgenic CD8+ cytotoxic T cells specific for an LCMVderived peptide presented by H-2Db may differentiate into LCMV-specific cytolytic effector cells during infections with unrelated pathogens in vivo or mixed lymphocyte cultures in vitro. The high precursor frequency of CTL of a defined specificity in these transgenic mice allowed us to address the following questions: (a) Are these “bystander” CTL protective against LCMV infection in vivo? (b) Which mechanisms are responsible for the nonspecific activation?, (c) How many CTL of a defined specificity are activated?, (d) Does bystander activation have a functional impact on CTL memory in nontransgenic mice?, and (e) Can bystander CTL cause autoimmune disease in a corresponding transgenic mouse model of diabetes mellitus?
Materials and Methods
Mice.
C57BL/6 and BALB/c mice were obtained from the Institut für Labortierkunde (University of Zürich, Zürich, Switzerland). The transgenic mice expressing a Vα2/Vβ8.2 T cell receptor specific for amino acids 33–41 of the LCMV glycoprotein 1 in association with H-2 Db have been described previously (19). For this study, mice of the line 327 (expressing the transgenic TCR on 85–95% of all CD8+ T cells) and for some indicated experiments of line 318 (expressing the transgenic TCR on 50–60% of CD8+ T cells) were used. TCR transgenic mice (line 318) crossed onto a RAG-2–deficient background (referred to as TCR × RAG −/− mice; 20) were bred locally (breeding pairs provided by Dr. Pamela Ohashi, Ontario Cancer Institute, Toronto, Canada). TCR transgenic mice (line 318) deficient in functional expression of the α/β-IFN receptor were generated by crossing with the appropriate gene targeted mice (21). TCR transgenic mice (line 327) expressing the LCMV glycoprotein under control of the rat insulin promoter (RIP-gp/TCR mice) have been described previously (22). All mice were kept under specific pathogen-free conditions, which included a test every 6 mo for the absence of 9 specified viruses, 21 bacteria, and 13 other pathogens such as fungi and protozoa. The mice are considered “naive” if they were not deliberately immunized.
Viruses and Bacteria.
Vaccinia virus WR was grown on BSC40 cells. Recombinant vaccinia virus expressing the LCMV glycoprotein (vacc G2) was obtained from B.H. Bishop (University of Oxford, Oxford, UK) and also grown on BSC 40 cells. LCMVWE was originally obtained from F. Lehmann-Grube (HeinrichPette-Institut für Experimentelle Virologie und Immunologie der Universitàt Hamburg, Hamburg, Germany) and was propagated on L 929 fibroblast cells. A seed of Listeria monocytogenes was originally obtained from R.V. Blanden (Australian National University, Canberra, Australia) and was maintained in a virulent state by passage in mice. A frozen (−70°C) stock culture was used to prepare a fresh 12–16 h culture in trypticase soy broth (BBL Microbiology Systems, Cockeysville, MD) for each experiment. The infective dose used was aimed at being about 3 × 103 bacteria per mouse and was assessed retrospectively by plating each inoculum.
Cytotoxicity Assays.
6 d after vaccinia virus infection or 5 d after infection with Listeria monocytogenes, effector cell suspensions were prepared from spleens of infected mice. For some experiments, mice were injected with 200 μg poly (IC) (Fluka, Chemie Ab, Buchs, Switzerland) intravenously 3 and/or 1 d before the cytotoxicity assay. MC57G (H-2b) target cells were pulsed with LCMV glycoprotein peptide 33-41 (gp33; 10−6 M) for 2 h, infected with LCMV-WE for 48 h or infected with vaccinia virus WR at a multiplicity of infection of 3 for 2.5 h; uninfected cells served as controls. 5–6 h 51Cr-release assays were performed according to standard protocols (23); for overnight (15 h) assays, EL-4 cells unlabeled or pulsed with gp33 were used as target cells. For the cold target competition assays, effector cells and 51Cr-labeled (“hot”) LCMV-WE–infected MC57G target cells were incubated at a fixed effector/target ratio and nonradiolabeled (“cold”) target cells were added at the indicated cold/hot target ratio.
For in vitro MLC, 5 × 106 responder spleen cells (H-2b) were cultured in 24-well plates with 5 × 106 irradiated (2,000 rad) allogeneic spleen cells (H-2d). Cultures were set up in a total volume of 2 ml IMDM 10% FCS. The ability of cytokines to induce cytolytic effector CTL in the absence of stimulator cells was tested by culturing responder spleen cells from TCR × RAG −/− mice in medium supplied with 500 U/ml of recombinant human IL-2 (Hoffmann-La Roche, Nutley, NJ) on days 1 and 4. Alternatively, the cells were incubated in supernatant of C57BL/6 (H-2b) × BALB/c (H-2d) MLC cultures which had been set up 1 d previously, and medium was supplied daily from the parallel MLC. After 5 d, the cultures were harvested in 500 μl MEM 2% FCS and 100 μl of a 1-, 3-, 9-, and 27-fold dilution (referred to as dilution of culture) was added to 104 51Cr-labeled target cells. MC57G (H-2b) target cells were used either unlabeled or pulsed with LCMV peptide gp33. To test for anti–H-2d alloreactivity, P815 cells were used. Specific 51Cr-release was determined after 5 h incubation.
Antiviral effector function of activated CTL in vivo was measured in adoptive transfer experiments. For this, recipient C57BL/6 mice were infected with 104 PFU LCMV-WE intravenously and 10 h later, spleen cells from various naive and acutely infected donor mice were injected intravenously in a volume of 500 μl balanced salt solution. 18 h after transfer, virus titers were determined in the spleen using a virus plaque assay on MC57G cells as described previously (24).
Induction of Diabetes.
RIP-gp/TCR double-transgenic mice were infected with 2 × 106 PFU of vaccinia WR or recombinant vaccinia G2. Blood glucose levels were then determined every 3 d by using a haemo-glucotest kit and were quantitated with reflolux II (Boehringer Mannheim, GmbH, Mannheim, Germany). Animals were considered diabetic when blood glucose levels persisted above 14 mM for at least 3 d. Immunohistochemical analysis of pancreata with antibodies directed against CD4 and CD8 was performed as descibed previously (22). The islets of Langerhans were considered infiltrated if more than 10 positive lymphocytes were detected per islet section.
Results
Activation of Naive LCMV-specific Cytotoxic T Cells by Unrelated Infections In Vivo or by Allo-antigens In Vitro.
The question of whether cytotoxic T cells specific for a defined viral antigen can be activated by nonspecific antigens in vivo was analyzed in a mouse transgenic for the Vα2/Vβ8 TCR specific for the gp33 of LCMV (19). This mouse expresses the transgenic TCR on 90% of all CD8+ CTL rendering rare bystander activation events in this CTL population more easily detectable in functional assays. Naive (as defined in Material and Methods) TCR transgenic and C57BL/6 mice were infected with 5 × 106 PFU vaccinia WR virus, which is unrelated to LCMV. 6 d later, cytotoxic activity against target cells infected with vaccinia WR (Fig. 1 a) or pulsed with LCMV peptide gp33 (Fig. 1 b) was tested in a 51Cr-release assay. Whereas spleen cells from both mouse strains showed effective lysis of vaccinia infected target cells, cytotoxic activity against LCMV peptide–loaded target cells could only be found in spleens of vaccinia WR–infected TCR transgenic but not in C57BL/6 mice, the latter exhibiting a low LCMV-specific CTL precursor frequency (Fig. 1 b). Furthermore, activation of LCMVspecific CTL could be demonstrated after infection of TCR transgenic mice with Listeria monocytogenes, a facultative intracellular bacterium (Fig. 1, d and e). Finally, when spleen cells from TCR transgenic mice (H-2b) were stimulated with allogeneic spleen cells from BALB/c mice (H-2d) in an MLC, significant LCMV-specific cytotoxicity was generated (Fig. 1 g). These data, obtained similarly with mice kept under conventional or under SPF conditions, show that naive virus-specific precursor CTL can be activated by nonspecific antigens in vitro and in vivo. In TCR transgenic mice, this bystander activation generates strong LCMVspecific cytotoxic activity, which allows further analysis of the mechanisms involved. As will be shown later, monoclonal TCR × RAG −/− mice could not be used in the in vivo experiments because the monospecific repertoire renders these mice incompetent to control and eliminate infections with vaccinia virus or Listeria (data not shown).
Figure 1.
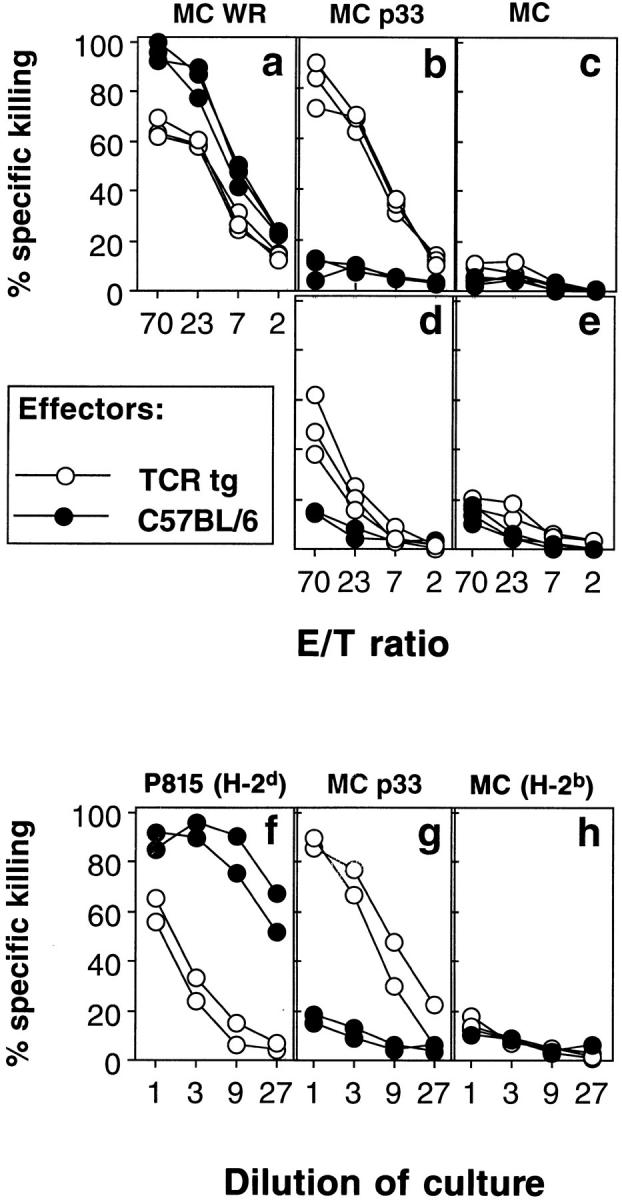
Activation of LCMV-specific TCR transgenic T cells by unrelated pathogens in vivo and by allo-antigens in vitro. (a–e) C57BL/6 mice (closed circles) and TCR transgenic mice from line 327 (open circles) were infected intravenously with 2 × 106 PFU vaccinia WR (a–c) or with 5 × 103 CFU Listeria monocytogenes (d and e). 6 and 5 d later, respectively, splenic cytotoxic activities were tested on MC57G target cells infected with vaccinia WR (a), labeled with the LCMV peptide gp33 (b and d), or left unlabeled (c and e). (f–h) Spleen cells from C57BL/6 (closed circles) and TCR trangenic mice of line 327 (both H-2b; open circles) were stimulated in a mixed lymphocyte reaction on irradiated spleen cells from BALB/c mice (H-2d). 5 d later, cytotoxic activity of the MLC cultures was tested on P815 (H-2d) (f), MC57G (g), or MC57G (H-2b) (h) cells pulsed with LCMV gp33. Spontaneous release in all assays was <24%. All experiments were performed at least two times.
Antiviral Protection In Vivo Induced by Bystander Activation of Naive Cytotoxic T Cells.
To study whether LCMV-specific CTL activated by the unrelated vaccinia virus infection are functional in vivo, we tested their ability to confer antiviral protection upon adoptive transfer. This indirect approach was chosen instead of direct LCMV challenge of the vaccinia infected mice, because LCMV does not reach significant titers even in naive TCR transgenic mice. A group of C57BL/6 mice was infected with 104 PFU LCMV-WE. 10 h later, these preinfected, but otherwise unmanipulated, recipient mice were transfused with various spleen cell populations to compare their ability to control virus replication in vivo. Donor spleen cell populations from the following groups of mice were used (Fig. 2): 1, naive C57BL/6 mice; 2, C57BL/6 mice infected with 2 × 106 PFU vaccinia WR 6 d previously; 3, naive TCR transgenic mice (negative controls); 4, TCR transgenic mice infected with 2 × 106 PFU vaccinia WR 6 d previously (experimental group); and 5 and 6, C57BL/6 mice infected with 200 PFU LCMV-WE 8 d before transfer (positive control). 20 h after transfer, the recipent mice were killed and virus titers were determined in the spleens. Fig. 2 shows that within 20 h, the transgenic CTL activated by vaccinia virus had almost completely controlled the infection with LCMV, while spleen cells from naive transgenic or vaccinia primed nontransgenic mice had no effect. These data demonstrate that in TCR transgenic mice bystander activation may induce CTL which are antivirally protective in vivo.
Figure 2.

Antiviral protection in vivo induced by bystander activation of cytotoxic T cells. C57BL/6 mice were infected with 104 PFU LCMVWE intravenously, and 10 h after infection, adoptively transfused with the indicated numbers of spleen cells from naive (groups 1 and 3), vaccinia infected (groups 2 and 4), or LCMV-infected (groups 5 and 6) C57BL/6 or TCR transgenic mice (line 327). 18 h after adoptive transfer, virus titers were determined in the spleens of recipient mice. One of three similar experiments is shown.
Bystander Activation of LCMV-specific Memory CTL Is of Little Functional Consequence.
Whereas the TCR transgenic mice have an extremely high (10−1) LCMV-specific CTLp frequency and naive C57BL/6 mice a low (10−6), LCMVinfected memory C57BL/6 mice display intermediate (10−3– 10−4) frequencies. Bystander proliferation and modulation of memory CTLp has previously been described after infection with heterologous viruses (2, 3) or injection of poly IC, a strong inducer of type I IFN (5). To test whether these nonspecific stimuli may also have functional consequences for CTL memory, we compared the cytolytic effector function of memory CTL in the presence or absence of these stimuli. Infection with 2 × 106 PFU of the heterologous vaccinia virus for 6 d (Fig. 3 a) did not improve cytolytic effector function of memory CTL as measured ex vivo in a 5 h and an overnight CTL assay, which permits assessment of even relatively minor cytotoxic activity. Similar results were obtained when 200 μg poly IC was injected 3 and 1 d before the assay (Fig. 3 c) The antiviral protective capacity of memory spleen cells 50 d after priming with LCMV upon adoptive transfer into preinfected recipients was small, but reproducible (Fig. 3 b, group 2). However, this antiviral protection was also not improved if the LCMV memory mice had been boosted with 2 × 106 PFU vaccinia 6 d before the transfer (Fig. 3 b, group 3).
Figure 3.

Functional analysis of bystander-activated LCMV-specific memory CTL with no evidence of enhancement of cytolytic CTL activity assessed in vitro and in vivo. (a) Primary ex vivo cytolytic activity of C57BL/6 mice immunized with 200 PFU LCMV-WE 50 d previously (open circles) or immunized with LCMV and boosted with 2 × 106 PFU vaccinia WR 6 d before the assay (closed circles) was analyzed in a 5-h and 15-h 51Cr-release assay on EL-4 target cells labeled with LCMV gp33. Spontaneous release was 28%. (b) C57BL/6 mice were infected with 104 PFU LCMV-WE intravenously, and 10 h after infection, adoptively transfused with the indicated numbers of spleen cells from naive (group 1), LCMV infected 50 d previously (group 2), LCMV infected and boosted with vaccinia virus 6 d before the assay (group 3), or acutely LCMV infected (group 4) C57BL/6 mice. 18 h after adoptive transfer, virus titers were determined in the spleens of recipient mice. (c) Primary ex vivo cytolytic activity of C57BL/6 mice immunized with 200 PFU LCMV-WE 150 d previously (open circles) or immunized with LCMV and injected with 200 μg poly IC 3 and 1 d before the assay (closed circles) was analyzed in a 15-h 51Cr-release assay on EL-4 target cells labeled with LCMV gp33. Spontaneous release was 28%.
Quantitative Analysis of Bystander Activation of CTL In Vivo.
To put the results obtained in a TCR transgenic mouse into a quantitative perspective, we addressed the question of how many LCMV-specific precursors needed to be present for bystander activation to become detectable in a functional CTL assay. TCR transgenic mice from line 327 (90% of CD8+ T cells express the transgenic TCR), from line 318 (50% of CD8+ T cells express the transgenic TCR), and C57BL/6 mice, the latter had been adoptively transfused with 108 or 107 spleen cells from mice of line 318 1 d previously (∼17 and 4%, respectively, of CD8+ T cells expressed the transgenic TCR as determined by blood FACS® analysis; data not shown), were infected with vaccinia WR. 6 d later, LCMV-specific cytotoxic activity was tested in an overnight 51Cr-release assay. Fig. 4 a shows that >4% of the spleen cells (corresponding to a CTLp frequency of >10−2) had to express a defined TCR for this experimental protocol to detect activation of CTL in vivo by a heterologous virus infection in a subsequent cytotoxicity assay. This readily explains the fact that in using functional assays, bystander activation was not detectable in nontransgenic mice. Although in a memory situation CTL may differ in their sensitivity to nonspecific (re-) activation stimuli (25, 26), this important quantitative consideration may help to explain why protective bystander activation could also not be observed in memory mice.
Figure 4.

Quantitative analysis of vaccinia virus–induced LCMVspecific TCR transgenic CTL. (a) TCR transgenic mice from line 327 (closed circles), line 318 (closed triangles), as well as C57BL/6 mice adoptively transfused with 108 (closed squares) or 2 × 107 (open squares) spleen cells from mice of line 318 were infected with 2 × 106 PFU vaccinia WR. 6 d later, LCMV-specific splenic cytotoxicity was assessed in a 15-h 51Cr-release assay on EL-4 target cells pulsed with LCMV gp33 or unlabeled. Spontaneous release was 29%. One of two similar experiments is shown. (b) C57BL/6 mice (closed circles) were infected with 200 PFU LCMV-WE and TCR transgenic mice from line 327 (open circles) with 2 × 106 PFU vaccinia WR. 8 and 6 d after infection, respectively, splenic cytotoxic activity was tested in a 15-h 51Cr-release assay on EL-4 target cells pulsed with LCMV gp33. Spontaneous release was 31%. One of two similar experiments is shown.
An approximate estimation of how many of the LCMVspecific naive precursor CTL actually differentiated into cytolytic effector CTL during the infection with the unrelated vaccinia virus was obtained as follows. The LCMVspecific cytolytic activity in vitro and the antiviral protective capacity in vivo of TCR transgenic spleen cells primed nonspecifically with vaccinia virus was compared to that of C57BL/6 spleen cells primed specifically with LCMV. The indirect comparison with C57BL/6 mice was chosen because in the acute phase of LCMV infection (day 8) of these mice, most LCMV-specific CTLp have differentiated into cytolytically active effector CTL; this extent can not reliably be achieved by LCMV infection of TCR transgenic mice, rendering quantitative comparisons difficult. In an overnight CTL assay, 50% lysis of LCMV gp33-labeled target cells required an E/T ratio of ∼14:1 in the vacciniaprimed TCR transgenic spleen and about 0.7:1 in the LCMVprimed C57BL/6 spleen, corresponding to a roughly 20-fold relative difference in cytotoxic activity (Fig. 4 b). A similar number was obtained when the antiviral effector function in vivo was compared. About 25 times more, i.e., 5 × 107 spleen cells from transgenic mice infected with the nonspecific virus (Fig. 2, group 4) were needed to reach the same antiviral protection as with 2 × 106 spleen cells from C57BL/6 mice activated by the appropriate virus (group 5). Thus, a spleen from TCR transgenic mice infected with vaccinia virus generates about 20-fold less LCMV-specific cytotoxic activity than a spleen from C57BL/6 mice acutely infected with LCMV. This difference in cytotoxic activity based on total spleen cell numbers increases even more if we consider that on the day of the assay, the number of LCMVspecific CTL differs by a factor of ∼10 between the two spleen cell populations; at the peak of the anti-LCMV response in the C57BL/6 spleen, 5–10% of CTL are LCMVspecific (27), whereas in the TCR transgenic spleen, 90% of CTL are specific for LCMV. On a per cell basis, it can therefore be calculated, that ∼20 × 10 = 200-fold less LCMV-specific cytotoxic activity was generated through bystander activation than after infection with the appropriate LCM virus. Within the limits of the indirect approach, this suggests that overall ∼1 in 200 LCMV-specific CTL differentiated into a cytolytic effector CTL during the infection of TCR transgenic mice with vaccinia virus.
Bystander Activation of CTL Occurs in the Absence of a Second TCR with Different Specificity and Is Likely to Be Mediated by Cytokines.
How can LCMV-specific transgenic CTL be activated during an immune response to vaccinia virus? A possible explanation would be the expression of a second TCR specific for vaccinia virus by some of the LCMVspecific CTL. FACS® analysis revealed that in naive TCR transgenic mice, ∼1.8% of the CD8+ T cells using the transgenic Vα2 chain expressed an additional Vα3 or Vα8 chain (data not shown). However, the limited range of Vαspecific antibodies available makes a more precise determination of the number of dual receptor T cells difficult and the specificities conferred by a second α chain could not be determined. We therefore chose cold target competition as a functional assay to check for dual receptor T cells. We reasoned that if lysis of 51Cr-labeled LCMV-infected target cells was mediated by CTL carrying two TCRs (one specific for vaccinia and one specific for LCMV), it should be possible to inhibit LCMV-specific lysis by adding an excess of nonlabeled (cold) targets infected with LCMV and with vaccinia virus, respectively. Fig. 5 a shows, that significant inhibition was only achieved with LCMV-infected cold targets, whereas inhibition by cold vaccinia-infected target cells did not exceed competition by cold uninfected MC57G cells. Similar results were obtained when cold target competition assays were performed with LCMV-specific effector cells generated in mixed lymphocyte cultures (Fig. 5 b).
Figure 5.

LCMV-specific cytotoxicity generated by vaccinia infection of TCR transgenic mice cannot be blocked by vaccinia virus–infected target cells. (a) TCR transgenic mice (line 327) were infected with 2 × 106 PFU vaccinia WR, and 6 d later, splenic cytotoxic activity was tested on 51Cr-labeled “hot” MC57G cells infected with LCMV-WE at a fixed effector/ target ratio of 100:1. Nonradiolabeled “cold” MC57G cells uninfected (closed bars), or infected with LCMV-WE (open bars) or with vaccinia WR (hatched bars) were added at the indicated hot/ cold target ratio. (b) Spleen cells from TCR transgenic mice (line 327; H-2b) were stimulated in an MLC with irradiated BALB/c (H-2d) spleen cells for 5 d and used as effectors in a cold target competition assay as outlined above. MC57G cells (H-2b), either uninfected (closed bars), or infected with LCMV-WE (open bars) or P815 (H-2d) cells (hatched bars) were used as cold targets. Spontaneous release was <23% in both assays. Each experiment was repeated three times.
To further dissect the mechanism of bystander activation, we used TCR transgenic mice crossed into a RAG2deficient background (TCR × RAG −/− mice). These mice are unable to rearrange and express endogenous (nontransgenic) TCR α or β chains, and therefore have only T cells of a single specificity. This renders these mice incompetent to control and eliminate infection with vaccinia virus (data not shown). Since these mice have a Sv129 genetic background and have not been sufficiently backcrossed to C57BL/6, minor histocompatibility differences did not allow the study of these cells after adoptive transfer into immunocompetent C57BL/6 mice due to cell rejection. Therefore, we were only able to study nonspecific activation of CTL from these mice in vitro. When stimulated with irradiated BALB/c spleen cells, responder spleen cells from TCR × RAG −/− mice neither generated allospecific (Fig. 6 a) nor LCMV-specific cytotoxicity (Fig. 6 b) above background. This indicates that the LCMV-specific transgenic TCR does not cross-react with allogeneic H-2d MHC antigen. However, when C57BL/6 spleen cells were added to the culture at a TCR × RAG −/−:B6 ratio of 10:1 (corresponding to the ratio of transgenic to endogenous CD8+ T cells in the TCR transgenic mice from line 327), the cultures showed both allo- and LCMV-specific cytotoxic activity similar to cultures of TCR transgenic spleen cells (Fig. 6, a–c). In the context of the experiments summarized above, these data suggested that (a) bystander activation in our experimental system occurs in the absence of a second T cell receptor, (b) it is unlikely to be mediated by a cross-reactive TCR, and (c) that the presence of cells directly responsive to the activating antigen is necessary for bystander activation, presumably via the secretion of cytokines.
Figure 6.

Nonspecific activation of TCR transgenic LCMV-specific CTL during a mixed lymphocyte reaction is mediated by cytokines and not a cross-reactive TCR. (a–c) Spleen cells from C57BL/6 (open circles), TCR transgenic line 327 (closed circles), TCR × RAG −/− mice (closed circles), and spleen cells from TCR × RAG −/− mice supplemented with 10% C57BL/6 (closed triangles) spleen cells (all H-2b) were used as responder cells in a mixed lymphocyte reaction on BALB/c (H-2d) stimulator cells. (d–f) Spleen cells from TCR × RAG −/− mice were cultured in the absence of stimulator cells either in supernatant of C57BL/6 versus BALB/c MLC (closed triangles) or in medium supplemented with 500 U/ml of IL-2 on days 1 and 4 of culture (closed circles). 51Cr-labeled P815 cells (H-2d) (a, c), or EL-4 cells (H-2b) labeled with gp33 (b, d) or unlabeled (c, f ) were used as target cells.
The role of cytokines in nonspecific CTL activation was further addressed as follows. Spleen cells from TCR × RAG −/− mice were cultured in the absence of stimulator cells in medium obtained from an MLC using BALB/c (H-2d) stimulators and C57BL/6 (H-2b) responders, which had been set up 1 d previously. The medium of the TCR × RAG −/− culture was then daily exchanged with medium of the parallel MLC. Alternatively, spleen cells from TCR × RAG −/− mice were cultured in medium supplied with 500 U/ml of recombinant human IL-2. After 5 d of culture, cytotoxic activity against target cells labeled with LCMV peptide gp33 was determined. Fig. 6, d–f shows that supernatant of an allo-specific MLC or high concentrations of IL-2 alone were sufficient for the nonspecific bystander activation of LCMV-specific CTL.
Type I interferons have recently been implicated in “bystander proliferation” of memory CTL (5). To test whether these cytokines are also involved in bystander activation of naive CTL, we performed an MLC using spleen cells from TCR transgenic mice crossed into an α/β interferon receptor–deficient background. Spleen cells from these mice generated LCMV-specific cytotoxicity similar to littermate controls (Fig. 7, a–c), suggesting that the nonspecific activation of naive CTL does not require type I interferons. Also, the injection of poly IC, a strong inducer of type I interferons 3 and/or 1 d before the cytotoxicity assay, did not lead to generation of LCMV-specific cytotoxicity, although significant lysis of NK-sensitive YAC-1 targets was observed (Fig. 7, d–f).
Figure 7.

No role for α/β-IFN in bystander activation of naive LCMVspecific TCR transgenic CTL. (a–c) Spleen cells from TCR transgenic mice (line 318) lacking the receptor for α/β-IFN (open circles) and heterozygous littermates (closed circles) (both H-2b) were used as responder cells in a mixed lymphocyte reaction on BALB/c (H-2d) stimulators. After 5 d of culture, cytotoxic activity was tested on MC57G (H-2b) target cells either unlabeled (c) or labeled with LCMV gp33 (b) and on P815 (H-2d) cells (a). Spontaneous release was <25%. One of two similar experiments is shown. (d–f) TCR transgenic mice (line 327) were injected with 200 μg poly(IC) intravenously 1 d (open circles), or 1 and 3 d (closed circles) before testing splenic cytotoxic activity on NK-sensitive YAC-1 target cells (d), MC57G target cells pulsed with LCMV gp33 (e), or unlabeled ( f ). Spontaneous release was <23%.
Bystander Activation of CTL Specific for a Self-Antigen Is Not Sufficient to Cause Autoimmune Disease in a Transgenic Mouse Model of Diabetes Mellitus.
The important question of whether bystander activation of CTL specific for a self-antigen may cause autoimmune disease, was addressed in RIP-gp mice (22). Autoimmune diabetes in RIP-gp mice can be induced by infection with LCMV in the absence of an LCMV-specific TCR transgene (22). The introduction of this second transgene amplifies the LCMV-specific CTLp ∼10,000fold, thereby significantly increasing the sensitivity of the model (28). We infected such double transgenic (RIP-gp/ TCR mice) with vaccinia virus WR and monitored the mice for development of diabetes by monitoring blood glucose; in parallel, immunohistological examination evaluated local inflammation and lymphocytic infiltrations of islets of the pancreas. All of the 10 mice observed from 6 to 60 d and 5 additional mice observed for 120 d after virus infection remained normoglycemic even though primary LCMV-specific cytotoxicity could readily be demonstrated in these mice (data not shown, but similar to Fig. 1). The histological examination revealed that ⩽50% of the islets showed infiltration by CD4+ and CD8+ lymphocytes (Table 1). This demonstrates that bystander activation in these mice generated effector CTL that were indistinguishable from CTL activated by their nominal antigen in their ability to lyse target cells and to home to inflammatory lesions in vivo. Nevertheless, the induction of a limited number of CTL leading to infiltration of less than half of the pancreatic islets appeared to be insufficient to cause clinically manifest diabetes mellitus. In contrast and as previously shown (22), infection of RIPgp/TCR mice with a vaccinia recombinant virus expressing the LCMV glycoprotein leading to activation of the CTL with their nominal antigen, induced diabetes and infiltration of >90% of the islets within 5–10 d after infection (Table 1).
Table 1.
Bystander Activation of CTL Is Not Sufficient to Cause Clinically Manifest Autoimmune Diabetes
| Virus | RIP gp/TCR mouse | Number of islets with infiltrates >10 lymphocytes/islets monitored | Onset of diabetes (day after infection) | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| d6 | d15 | d60 | ||||||||||||||
| CD4 | CD8 | CD4 | CD8 | CD4 | CD8 | |||||||||||
| Vacc WR | 1 | 1/9 | 1/6 | 3/6 | 3/6 | >120 | ||||||||||
| 2 | 1/6 | 3/9 | 0/7 | 0/8 | >120 | |||||||||||
| 3 | 0/12 | 0/13 | 5/11 | 6/12 | >120 | |||||||||||
| 4 | 6/18 | 6/17 | 8/11 | 5/12 | >120 | |||||||||||
| Vacc LCMV GP | 1 | 6/9 | 6/6 | 6 | ||||||||||||
| 2 | 11/11 | 10/10 | 8/8 | 7/7 | 10 | |||||||||||
RIP-gp/TCR double transgenic mice (line 327) were infected with 2 × 106 PFU of vaccinia WR or recombinant vaccinia G2. 6, 15, and 60 d after virus infection, mice were killed and immunohistochemical analysis of pancreata was performed with antibodies against CD4 and CD8. The islets of Langerhans were considered infiltrated if >10 positive lymphocytes were detected per islet section. Nontransgenic C57BL/6 mice showed <1 CD4+ or CD8+ cell per islet. Also indicated is the day when blood glucose levels reached levels >14 mM in four animals infected with vaccinia WR and in two animals infected with vaccinia G2.
Discussion
This study evaluated qualitative and quantitative aspects of nonspecific CTL activation in a viral model system. The use of TCR transgenic mice provided a sensitive tool to experimentally address several questions that escape detection in mice with a normal T cell repertoire. Although this model situation may limit extrapolations of our results to nontransgenic situations in some aspects, its high sensitivity may also allow reevaluation of observations made under nontransgenic conditions.
Our experiments are based on the observation that naive TCR transgenic CD8+ cytotoxic T cells specific for an LCMV-derived peptide may differentiate into LCMV-specific cytolytic effector cells during infections with unrelated pathogens in vivo or stimulation with allogenic spleen cells in vitro. Since there is some uncertainty about the definition of naive cells, it may be important to state that we consider naive cells to be from a 6–8-wk-old mouse that has been kept under strict SPF conditions and that has not undergone deliberate immunization. In a C57BL/6 mouse, such naive cytotoxic T cells stain negative for CD69, but ∼35% stain positive for CD44. LCMV-specific spleen cells of SPF-kept TCR transgenic mice are also CD69 negative and ∼9% stain positive for CD44. In TCR × RAG −/− mice, >98% of the TCR transgenic T cells are CD44 negative, which is similar to the findings of a recently published report (29). In all of these mice, CD44 expression is not associated with measurable cytolytic activity (Fig. 4), which can be readily detected in spleen cells from nontransgenic virus-infected memory mice (Fig. 3). Furthermore, it should be noted that the SPF maintenance conditions are already quite artificial in terms of exposure to environmental antigens if compared to outbred mouse or human populations in a natural environment. For the experimental purpose of this study, we, therefore, do not think that it would be useful to define an even more naive state with the use of cell surface markers. Finally, bystander activation also occurs in CD44low cells, since it can also be demonstrated using CTL from TCR × RAG −/− mice, which display a clear CD44 and CD69 negative phenotype (Fig. 6).
The finding that virus infections have the ability to polyclonally stimulate CTL has previously been reported for alloreactive CTL (7, 8) and for antigen-experienced virusspecific CTL (2, 3, 30). Our studies in TCR transgenic mice extend these findings to naive CTL specific for a viral antigen and support the conclusion that the polyclonal stimulation may comprise naive CTL clones of allo as well as conventional specificities; however, they must be present at high enough precursor frequency to be detectable in a functional assay. Moreover and more importantly, we show that this nonspecific activation of naive precursors does not only elevate virus-specific CTLp, but that it is sufficient to generate CTL that are cytolytically active. This is not only demonstrated by their ability to lyse target cells in vitro, but also by the finding that they can confer antiviral protection in vivo. This shows that they home to infected tissue and encounter infected cells in a complex environment where there are many more uninfected than infected cells and where they lyse these cells to efficiently stop further spread of virus.
What are the molecular mechanisms of this nonspecific activation of cytotoxic T cells? This may be an important question for understanding the pathogenesis of T cell– mediated autoimmune disease and it has previously been addressed in several experimental models. Three main mechanisms have been discussed. The simplest explanation may be that a given TCR not only recognizes its nominal peptide, but can also interact with MHC molecules presenting either homologous peptides derived from unrelated proteins or peptides sharing crucial anchor positions (31, 32) with sufficient avidity for the T cell to be activated. Examples of such cross-reactivity are the basis of an extended concept of molecular mimicry (12) and have also been implied in the maintenance and modulation of T cell memory (2, 3). To what extent these cross-reactivities defined in vitro are of biological relevance in vivo is unknown (33). Another possibility would be activation of a T cell of specificity X via a second TCR of specificity Y. Such dual receptor T cells have been described as occurring as a result of incomplete allelic exclusion of the TCR-α gene locus (16, 18). It was recently shown that cytolytic effector function for specificity X can be achieved after activation of a CTL through specificity Y (13). Experiments using TCR × RAG −/− mice have suggested a role for a second TCR in the generation of memory cells as defined by CD45 RB expression in the absence of specific antigen priming (29). However, for the interpretation of these results, it should be kept in mind that TCR × RAG −/− mice do not only lack the ability to rearrange and express endogenous TCR α chains, but completely lack B or T cells of other specificities. Experiments using RAG −/− mice are therefore unable to exclude that the induction of CD44 expression is a consequence of cytokine-mediated bystander activation as defined in this study. Furthermore, it is still an open question whether these cells are of biological relevance in nontransgenic mice in vivo, for example, whether activation via an antiviral specificity can lead to autoimmune disease via a second, antiself specificity.
Finally, rather than involving a single CTL reactive to two different antigens, nonspecific activation of CTL may represent bystander activation in a more strict definition of the term; cytokines secreted by antigen-responsive cells at infectious foci may directly stimulate surrounding CTL in the absence of direct triggering of the T cell receptor. In vitro studies have revealed that naive human CD4+ T cells can be activated by a combination of IL-2, TNF-α, and IL-6 (4). Furthermore, in vivo studies of antigen-experienced T cells have shown that cytokines may break staphylococcal enterotoxin B–induced T cell tolerance (34) and induce nonspecific proliferation of CD44hi T cells (5). However, (re-)activation requirements of antigen-experienced T cells may be different from those of naive CTL. Also, the latter studies have not addressed the question of whether the T cell population induced to proliferate was also activated to provide effector function in vivo.
In the model situation presented in this study, naive T cells differentiate into LCMV-specific cytolytic effector CTL after the infection of TCR transgenic mice with vaccinia virus or Listeria monocytogenes, both of which are strong inducers of cytokine responses. Cold target competition experiments and in vitro experiments with TCR × RAG −/− mice showed that this nonspecific CTL activation is largely independent of a second TCR. Also, a cross-reactive TCR appears an unlikely explanation, since it would be surprising if stimuli as different as a virus, an intracellular bacterium, and allogeneic spleen cells should all share crossreactivity with LCMV. In line with this argument, nonspecific activation of a pure population of LCMV-specific CTL in vitro was only possible in the presence of cells directly responsive to the antigen. Moreover, incubation of TCR transgenic T cells in IL-2–containing medium in the absence of stimulating spleen cells, but obviously in the presence of 10% FCS and other potential foreign antigens, was sufficient for the generation of LCMV-specific cytolytic effector CTL. Thus, while we cannot unequivocally analyze nonspecific CTL activation in vivo, our data obtained in vitro suggest that TCR-independent activation mediated by cytokines (but not type I IFN alone) is able to drive naive CTLp into cytolytic effector CTL and may therefore be responsible for bystander activation.
Whatever the mechanism of nonspecific activation of CTL, an important question is whether it is of significance in vivo. Can bystander activation induced by a viral infection or cytokines functionally improve CTL memory to a previously encountered unrelated virus? Former studies have provided evidence that these nonspecific stimuli may induce proliferation of memory CTL (5) and limiting dilution analysis has revealed that CTLp frequencies may be influenced by a factor of 2–4, rarely of 10 (2, 3). By assessing cytolytic activity in vitro and antiviral protection in vivo, we used a functional, rather than a numeric, definition of CTL memory. By these criteria, we could not observe a significant impact of a heterologous virus infection or interferon stimulation on memory CTL. These results differ from those of similar experiments published previously (2). The observed decrease in ex vivo cytolytic activity of memory spleens with increasing time after virus infection (compare Fig. 3, a and c) might explain these differences; a fair comparison of memory CTL activity is only possible at the same time after the priming infection. Thus, our quantitative analysis revealed that bystander activation is overall of low efficiency. It is therefore not surprising that in LCMV-infected C57BL/6 memory mice, where CTLp frequencies are ∼10−4–10−3, we found little, if any, consequence as assessed by cytotoxicity assays.
Can bystander activation of potentially self-reactive CTL cause autoimmune disease? We addressed this question in a diabetes model where an LCMV antigen is expressed as a transgene on pancreatic islet cells. Whereas autoimmune diabetes in these mice can be induced by infection with LCMV (28), the introduction of a second LCMV-specific TCR transgene amplifies the LCMV-specific CTLp about 10,000fold, and therefore increases the sensitivity of the model significantly (22). Infection of these mice with an unrelated third party virus generated significant LCMV-specific cytotoxicity. However, although some islet infiltration could be demonstrated, the mice did not become diabetic. Why is there insulitis but no progression to overt diabetes? Since cytolytic effector function and homing properties of bystanderactivated CTL are indistinguishable from CTL activated via their nominal antigen, it is unlikely to be due to a qualitative difference in the CTL. It can be hypothesized that the necessary LCMV-specific T cell help is not provided; although infiltration of the islets with CD4+ helper T cells was observed, these cells may not be of the appropriate specificity since the mice were primed with vaccinia virus. Lack of concurrent bystander activation of antigen-specific T help in addition to CTL could thus be a safeguard against nonspecifically induced autoimmune disease. More importantly, previous studies have shown that induction of a threshold number of self-reactive CTL is necessary for the induction of diabetes in these mice (22). We therefore performed quantitative experiments, which revealed that despite the much higher precursor frequency of LCMV-specific CTLp, the LCMV-specific cytotoxic activity per spleen was about 20-fold lower in TCR transgenic mice infected with the nonspecific vaccinia virus when compared to nontransgenic mice infected with the specific LCMV. These quantitative differences may readily explain why the nonspecific CTL activation did not suffice to cause clinically manifest autoimmune diabetes.
With respect to the pathogenesis of autoimmune disease, these data should be carefully interpreted within the limits of the experimental model used in this study. Nevertheless, since, due to the introduction of a transgenic TCR, a sensitive, strongly reactive model is used that can readily reveal antiviral effector funtion in vivo, a few aspects can be discussed in a broader context and may offer generalizable aspects. Besides negative selection, a relatively high level of specificity (within the usual limits of biological systems) of T cell–mediated immune responses is probably one of the most important safeties against autoimmune disease. However, nonspecific activation by cytokines (and thus circumventing the specificity of the TCR) may occur, even to a degree where cytolytic activity is demonstrable in vivo. For this scenario, the present study emphazises an important additional safety mechanism: that of a relatively high threshold level of total induced and activated immune cells necessary for biological effects (33).
Acknowledgments
We thank Hans Peter Pircher, Stephan Oehen, and Anne Rensing-Ehl for helpful discussions and Karin Riem for excellent technical assistance.
This work was supported by the Swiss National Science Foundation grant 31-32159.91, the Deutsche Forschungsgemeinschaft (S. Ehl), the Foundation Jeantet, and the Kanton Zürich.
Footnotes
S. Ehl and J. Hombach have contributed equally to this work.
1 Abbreviations used in this paper: gp33, glycoprotein 33-41; LCMV, lymphocytic choriomeningitis virus; RIP, rat insulin promotor; SPF, specific pathogen-free.
References
- 1.Beverly P. Is T cell memory maintained by cross-reactive stimulation? . Immunol Today. 1990;11:203–205. doi: 10.1016/0167-5699(90)90083-l. [DOI] [PubMed] [Google Scholar]
- 2.Selin S, Nahill S, Welsh R. Cross-reactivities in memory cytotoxic T lymphocyte recognition of heterologous viruses. J Exp Med. 1994;179:1933–1943. doi: 10.1084/jem.179.6.1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Selin L, Vergilis K, Welsh R, Nahill S. Reduction of otherwise remarkably stable virus-specific cytotoxic T lymphocyte memory by heterologous viral infection. J Exp Med. 1996;183:2489–2499. doi: 10.1084/jem.183.6.2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Unutmaz D, Pileri P, Abrignani S. Antigen-independent activation of naive and memory resting cells by a cytokine combination. J Exp Med. 1994;180:1159–1164. doi: 10.1084/jem.180.3.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tough D, Borrow P, Sprent J. Induction of bystander T cell proliferation by viruses and type I interferon in vivo. Science (Wash DC) 1996;272:1947–1950. doi: 10.1126/science.272.5270.1947. [DOI] [PubMed] [Google Scholar]
- 6.Nahill S, Welsh R. High frequency of cross-reactive cytotoxic T lymphocytes elicited during the virus-induced polyclonal cytotoxic T lymphocyte response. J Exp Med. 1993;177:317–327. doi: 10.1084/jem.177.2.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang H, Welsh R. Induction of alloreactive cytotoxic T cells by acute virus infection of mice. J Immunol. 1986;136:1186–1193. [PubMed] [Google Scholar]
- 8.Yang H, Dundon P, Nahill S, Welsh R. Virusinduced polyclonal cytotoxic T lymphocyte stimulation. J Immunol. 1989;142:1710–1718. [PubMed] [Google Scholar]
- 9.Schendel D, Reinhardt C, Nelson P, Maget B, Pullen L, Bornkamm G, Steinle A. Cytotoxic T lymphocytes show HLA-C–restricted recognition of EBV-bearing cells and allorecognition of HLA class I molecules presenting selfpeptides. J Immunol. 1992;149:2406–2414. [PubMed] [Google Scholar]
- 10.Strang G, Rickinson A. Multiple HLA class I–dependent cytotoxicities constitute the “non-HLA-restricted” response to infectious mononucleosis. Eur J Immunol. 1987;17:1007–1014. doi: 10.1002/eji.1830170717. [DOI] [PubMed] [Google Scholar]
- 11.Gaston J, Rickinson A, Epstein M. Cross-reactivity of self-HLA-restricted Epstein-Barr virus-specific cytotoxic T lymphocytes for allo-HLA determinants. J Exp Med. 1983;158:1804–1821. doi: 10.1084/jem.158.6.1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oldstone M. Molecular mimicry and autoimmune disease. Cell. 1987;50:819. doi: 10.1016/0092-8674(87)90507-1. [DOI] [PubMed] [Google Scholar]
- 13.Zal T, Weiss S, Mellor A, Stockinger B. Expression of a second receptor rescues self-specific T cells from thymic deletion and allows activation of autoreactive effector function. Proc Natl Acad Sci USA. 1996;93:9102–9107. doi: 10.1073/pnas.93.17.9102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Borgulya P, Kishi H, Uematsu Y, von Boehmer H. Exclusion and inclusion of α and β T cell receptor alleles. Cell. 1992;69:529–537. doi: 10.1016/0092-8674(92)90453-j. [DOI] [PubMed] [Google Scholar]
- 15.Casanova J, Romero P, Widman C, Kourilsky P, Maryanski J. T cell receptor genes in a series of class I major histocompatibility complex–restricted cytotoxic T lymphocyte clones specific for a plasmodium berghei nonapeptide: implications for T cell allelic exclusion and antigen-specific repertoire. J Exp Med. 1991;174:1371–1383. doi: 10.1084/jem.174.6.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heath W, Miller J. Expression of two α chains on the surface of T cells in T cell receptor transgenic mice. J Exp Med. 1993;178:1807–1811. doi: 10.1084/jem.178.5.1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heath W, Carbone F, Bertolino P, Kelly J, Cose S, Miller J. Expression of two T cell receptor α chains on the surface of normal murine T cells. Eur J Immunol. 1995;25:1617–1623. doi: 10.1002/eji.1830250622. [DOI] [PubMed] [Google Scholar]
- 18.Padovan E, Casorati G, Dellabona P, Meyer S, Brockhaus M, Lanzavecchia A. Expression of two T cell receptor α chains: dual receptor T cells. Science (Wash DC) 1993;262:422–424. doi: 10.1126/science.8211163. [DOI] [PubMed] [Google Scholar]
- 19.Pircher H, Bürki K, Lang R, Hengartner H, Zinkernagel R. Tolerance induction in double specific T-cell receptor transgenic mice varies with antigen. Nature (Lond) 1989;342:559–562. doi: 10.1038/342559a0. [DOI] [PubMed] [Google Scholar]
- 20.Shinkai Y, Rathbun G, Lam K, Oltz E, Stewart V, Mendelsohn M, Charron J, Datta M, Young F, Stall A, Alt F. RAG-2–deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangements. Cell. 1992;68:855–867. doi: 10.1016/0092-8674(92)90029-c. [DOI] [PubMed] [Google Scholar]
- 21.Müller U, Steinhoff U, Reis L, Hemmi S, Pavlovic J, Zinkernagel R, Aguet M. Functional role of type I and type II interferons in antiviral defense. Science (Wash DC) 1994;264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- 22.Ohashi PS, Oehen S, Aichele P, Pircher H, Odermatt B, Herrera P, Higuchi Y, Buerki K, Hengartner H, Zinkernagel RM. Induction of diabetes is influenced by the infectious virus and local expression of MHC class I and tumor necrosis factor-α. J Immunol. 1993;150:5185. [PubMed] [Google Scholar]
- 23.Zinkernagel R, Haenseler E, Leist T, Cerny A, Hengartner H, Althage A. T cell–mediated hepatitis in mice infected with lymphocytic choriomeningitis virus. J Exp Med. 1986;164:1075–1092. doi: 10.1084/jem.164.4.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Battegay M, Cooper S, Althage A, Bänziger J, Hengartner H, Zinkernagel R. Quantification of lymphocytic choriomeningitis virus with an immunological focus assay in 24- or 96-well plates. J Virol Methods. 1991;33:191–198. doi: 10.1016/0166-0934(91)90018-u. [DOI] [PubMed] [Google Scholar]
- 25.Tough D, Sprent J. Turnover of naive- and memory-phenotype T cells. J Exp Med. 1994;179:1127–1135. doi: 10.1084/jem.179.4.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Akbar A, Salmon M, Janossy G. The synergy between naive and memory T cells during activation. Immunol Today. 1991;12:184–188. doi: 10.1016/0167-5699(91)90050-4. [DOI] [PubMed] [Google Scholar]
- 27.Moskophidis D, Assmann-Wischer U, Simon M, Lehmann-Grube F. The immune response of the mouse to lymphocytic choriomeningitis virus. V. High numbers of cytolytic T lymphocytes are generated in the spleen during acute infection. Eur J Immunol. 1987;17:937–942. doi: 10.1002/eji.1830170707. [DOI] [PubMed] [Google Scholar]
- 28.Ohashi P, Oehen S, Bürki K, Pircher H, Ohashi C, Odermatt B, Malissen B, Zinkernagel R, Hengartner H. Ablation of “tolerance” and induction of diabetes by virus infection in viral antigen transgenic mice. Cell. 1991;65:305–317. doi: 10.1016/0092-8674(91)90164-t. [DOI] [PubMed] [Google Scholar]
- 29.Lee W, Cole-Calkins J, Street N. Memory T cell development in the absence of specific antigen priming. J Immunol. 1996;157:5300–5307. [PubMed] [Google Scholar]
- 30.Selin L, Welsh R. Specificity and editing by apoptosis of virus-induced cytotoxic T lymphocytes. Curr Opin Immunol. 1994;6:553–559. doi: 10.1016/0952-7915(94)90140-6. [DOI] [PubMed] [Google Scholar]
- 31.Rammensee H, Falk K, Rötzschke O. MHC molecules as peptide receptors. Curr Biol. 1993;5:35–44. doi: 10.1016/0952-7915(93)90078-7. [DOI] [PubMed] [Google Scholar]
- 32.Gautam A, Lock C, Smilek D, Pearson C, Steinmann L, McDevitt H. Minimal structural requirements for peptide presentation by major histocompatibility class II molecules: implications in induction of autoimmunity. Proc Natl Acad Sci USA. 1994;91:767–771. doi: 10.1073/pnas.91.2.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aichele P, Bachmann M, Hengartner H, Zinkernagel R. Immunopathology or organ-specific autoimmunity as a consequence of virus infection. Immunol Rev. 1996;152:21–45. doi: 10.1111/j.1600-065x.1996.tb00909.x. [DOI] [PubMed] [Google Scholar]
- 34.Röcken M, Urban J, Shevack E. Infection breaks T-cell tolerance. Nature (Lond) 1992;359:79–82. doi: 10.1038/359079a0. [DOI] [PubMed] [Google Scholar]