

Abstract
The inflammatory response involves sequential adhesive interactions between cell adhesion molecules of leukocytes and the endothelium. Unlike the several adhesive steps that precede it, transendothelial migration (diapedesis), the step in which leukocytes migrate between apposed endothelial cells, appears to involve primarily one adhesion molecule, platelet–endothelial cell adhesion molecule (PECAM, CD31). Therefore, we have focused on PECAM as a target for antiinflammatory therapy. We demonstrate that soluble chimeras made of the entire extracellular portion of PECAM, or of only the first immunoglobulin domain of PECAM, fused to the Fc portion of IgG, block diapedesis in vitro and in vivo. Furthermore, the truncated form of the PECAM-IgG chimera does not bind stably to its cellular ligand. This raises the possibility of selective anti-PECAM therapies that would not have the untoward opsonic or cell-activating properties of antibodies directed against PECAM.
The emigration of leukocytes from the bloodstream into a site of inflammation involves a series of interactions between cell adhesion molecules (CAMs)1 on the leukocyte and the venular endothelium. This phenomenon has been dissected into discrete steps of rolling, activation, tight adhesion, transmigration, and migration across the basement membrane (1–4). If a relevant leukocyte or endothelial CAM is inhibited, leukocytes do not proceed to the next step. During transendothelial migration (TEM), the leukocytes squeeze between tightly apposed endothelial cells. This process involves the function of platelet–endothelial cell adhesion molecule (PECAM, CD31), a member of the immunoglobulin gene superfamily, which is expressed on the surfaces of monocytes (Mo), granulocytes, NK cells, some T cell subsets, and concentrated at the borders between endothelial cells (5–7).
In the presence of appropriate anti-PECAM mAbs, leukocytes can bind tightly to endothelial monolayers and migrate to the endothelial junctions, but they do not proceed through the junctions (8, 9). This process is reversible since diapedesis resumes shortly after removing the blocking mAb (8). Transmigration appears to involve homophilic interaction of PECAM on the leukocyte with PECAM on the endothelial cell. Blocking the PECAM on either cell is sufficient to maximally block TEM in vitro; blocking PECAM on both cells has no additional effect (8).
Depending on the leukocyte type and the inflammatory stimulus, more than one CAM can participate in each of the steps of rolling, activation, and tight adhesion (1, 10). Thus, it is difficult to block inflammation using individual reagents directed at the particular molecules involved in these steps. In contrast, PECAM mediates a common final step in emigration for many leukocyte types activated by a variety of stimuli. In addition, PECAM has no other known function in vivo. Most of the other CAMs important in emigration of leukocytes have other roles in the immune system (1, 11), the blockade of which could lead to untoward consequences.
Therefore, PECAM is an attractive target molecule for antiinflammatory therapy. In fact, mAbs (12) and polyclonal antibodies (13–15) against PECAM block acute inflammation in response to a variety of stimuli. However, xenogeneic mAb has the potential to opsonize leukocytes, leading to leukopenia, as well as to stimulate production of neutralizing antibodies by the host, making it a poor agent for chronic therapy. Moreover, engagement of CAMs by high affinity mAbs can activate cells, especially leukocytes. Ligation of leukocyte PECAM by a variety of mAbs can trigger an adhesion cascade resulting in the upregulation of leukocyte integrin binding activity on T cells (16), PMN, Mo (17), and NK cells (9, 18).
To avoid these potential problems, we fused portions of the extracellular region of autologous PECAM to the human Fc chain. These soluble chimeras competitively inhibit TEM in vitro and in vivo. A chimera containing only PECAM domain 1, which is incapable of binding stably to cellular PECAM, blocks emigration of both PMN and monocytes into the inflamed peritoneal cavity. This is the first demonstration that a portion of a CAM with no stable binding activity itself can block inflammation in vivo.
Materials and Methods
Cell Culture
Human umbilical vein endothelial cells (HUVEC) were isolated from fresh umbilical veins and cultured in medium 199 (M199; GIBCO BRL, Gaithersburg, MD) + 20% normal human serum on hydrated collagen gels as described previously (5). Cells were used at passage two. For experiments involving FACS® (Becton Dickinson, San Jose, CA) analysis of chimeric proteins bearing the human IgG Fc region, HUVEC were cultured in 20% fetal bovine serum (LPS-free; Hyclone Labs., Logan, UT).
Monocyte-selective Transendothelial Migration Assay
The details of this assay have been previously published (8, 19). Transendothelial migration was quantitated by Nomarski optics as described previously (4, 8). In some experiments, transmigration was also quantitated on cross sections of paraffin-embedded monolayers. These specimens were prepared by carefully removing replicate sample monolayers and placing the endothelial surfaces against each other with the collagen gel sides facing outward. This avoided mechanical dislodgement of cells during the embedding process. After substitution in wax, the specimens were bisected so that cuts through the specimen produced cross sections of four monolayer samples (two different portions of each of the two monolayers). Quantitation was performed on three levels of such specimens separated by at least 50 μm so that different areas of the specimen would be sampled and the same cells would not be counted twice.
Construction and Production of Chimeric Ig Fusion Proteins
Truncated Human PECAM-IgG.
Construction of the set of human PECAM-IgG chimeras has been described previously (4). The novel human PECAM-IgG chimera consisting of domains 3–6 was made using a similar PCR strategy. The sequences of the primer pair used in generating the DNA fragment corresponding to domains 3–6 were: 5′-TAG ATC GAT ATC GAA GGA GCT CAG CTC-3′ and 5′-TAG AAT ATC GCG GCC GCT TTC TTC CAT-3′, with the EcoRV and NotI sequences indicated in bold.
Murine PECAM-IgG Proteins.
A full-length soluble PECAMIgG cDNA was constructed by ligating the cDNA encoding the extracellular portion of murine PECAM (20) with a cDNA encoding the human IgG1 Fc domain, in a similar fashion to the construction of human PECAM-IgG (4). This was subcloned into pcDNAI/neo (Invitrogen, San Diego, CA) at the SalI and XbaI sites. A PCR cloning strategy similar to that used to make the human truncated PECAM-IgGs (4) was used to construct the murine counterparts with the full-length PECAM-IgG cDNA used as the template. The sequence of the 5′ PCR primer was 5′-TCA GAA GCT TCC ACC ATG CTC CTG-3′. The HindIII restriction sequence is in bold; the initiation codon is underlined. The sequence of the 3′ primer for producing the first domain of murine PECAM was 5′-TAG AAT ATC GCG GCC GCT TCT GTC ACC TCC TT-3′. The NotI restriction site is in bold.
CD14-IgG cDNA Fusion Plasmid.
The CD14-IgG cDNA fusion plasmid was a gift of Dr. Henri Lichenstein (Amgen, Inc., Boulder, CO). The CD14-IgG insert (21) was retrieved from the original pSPORT vector by XbaI and SalI digestions, bluntended, and transferred to the EcoRV site of the selectable mammalian expression vector, pcDNAI/neo.
PECAM-IgM Fusion Plasmid.
Construction of the PECAM-IgM fusion plasmid was based on a patented vector, pm2CD2IgM, gpt (American Type Culture Collection accession No. 68280, provided by Dr. M.F. Concino of Procept, Inc., Cambridge, MA). This vector contains the Ch2 + Ch3 + Ch4 domains of human IgM fused to the extracellular portion of CD2 (22). The IgM portion was generated from the vector by PCR using the following pair of frame-retaining oligonucleotide primers containing NotI and XbaI restriction sequences: 5′-AAT ACA TAG AGG CCG CCA GTG ATT GCT GAG CTG-3′ and 5′-GGG TTT CTA GAA GCC ACT-3′. The NotI and XbaI restriction sequences, respectively, are printed in bold; the first five codons of the IgM are underlined. PECAM-IgM constructs were made from the corresponding PECAM-IgG pcDNAI/Neo vectors by replacing the IgG portion with IgM cDNA at the NotI and XbaI sites.
To produce soluble fusion proteins, L cell fibroblasts were stably transfected by electroporation (23) with cDNAs encoding each of the constructs and selected in 0.5 mg/ml G418. Clones were picked and transplanted to 96-well trays. Supernates from these clones were tested by ELISA for expression of human IgG or IgM. Positive clones were expanded, subcloned, and tested for production of the appropriate PECAM domains by mAb-binding ELISA (4), and for size by Western blotting.
Cultures were weaned to growth in 1.5% FCS and expanded to roller bottle culture. Three to four liters of conditioned medium from transfectant cultures were pooled. PECAM-IgG was purified by affinity chromatography on protein A–Sepharose; PECAMIgM was purified by affinity chromatography on hec7-Sepharose. Proteins were eluted with 0.1 M glycine buffer, pH 2.5, neutralized, and dialyzed against phosphate-buffered saline. When necessary, purified protein was concentrated using centricon membranes (Amicon, Beverly, MA). Chimeric proteins were filter sterilized. All mAb and chimeric protein reagents used in these experiments were free of detectable endotoxin (<0.1 endotoxin units) by limulus amebocyte lysate assay (BioWhittaker, Inc., Walkersville, MD).
FACS® Analysis.
FACS® analysis was performed with Consort 30 software. Nonenzymatically resuspended HUVEC or freshly isolated PBMC were incubated with mAb or PECAM-IgG chimeras on ice for 30 min, washed gently, and then incubated with F(ab′)2 fragments of FITC-labeled rabbit anti–mouse IgG or FITC-labeled goat anti–human IgG for 30 min on ice. Cells were then washed and analyzed. At least 10,000 cells were collected for each sample. PBMC were preincubated with mAb IV.3 and 3G8 (Medarex, Inc., Annandale, NJ) against FcγR II and III, respectively. Monocytes were selectively analyzed in the PBMC samples using appropriate forward- and side-scatter gates, which were confirmed using monocyte-specific markers. Graphs were produced using WinList software for curve smoothing.
Thioglycollate Broth-induced Peritonitis.
These studies were performed and analyzed as previously described (12), except that thioglycollate was injected 1 h after intravenous administration of control or anti-PECAM reagents. Measurements (animal weight; peritoneal lavage volume, cell density, and differential count; peripheral blood count and smear; general autopsy) were performed at 4, 18, or 24 h, as indicated in the figures. Representative sections of bowel and mesentery were submitted for histologic sections, which were stained with hematoxylin and eosin, and scored in a blinded manner for leukocyte adhesion to the walls of postcapillary venules as previously described (12).
Statistics.
The figures show representative experiments from the many of each type performed. The bars give the mean ± standard error for five to six replicates of each variable tested. Since the data involved nonparametric independent samples, statistical significance was tested by the Mann-Whitney U test.
Results
PECAM-IgG Binds Homophilically to Endothelial Cell PECAM.
The extracellular portion of PECAM is composed of six immunoglobulin domains (6). Full-length PECAMIgG (i.e., domains 1–6 fused to IgG) and truncated versions all migrated on SDS-PAGE at the appropriate relative molecular mass values for dimers of the expected size, and all were recognized by CD31 mAb whose epitopes were included in their sequence, but not by CD31 mAb with epitopes on domains not included in the constructs (reference 4 and data not shown.) Full-length PECAM-IgG bound stably enough to HUVEC to be detectable by FACScan® analysis (Fig. 1). As had been demonstrated previously (24), this binding represents homophilic adhesion to PECAM on the HUVEC, since it was inhibitable by hec7 Fab, which binds to PECAM domains 1 and/or 2, but not by mAb P1.2, which binds to PECAM domain 6 or mAb 7E3 (25), a blocking mAb against αvβ3 (Fig. 1 a). Binding to monocytes, which express an order of magnitude less PECAM than HUVEC (26), was low (three times background; Fig. 1 c) to undetectable above background in seven separate experiments with seven different blood donors.
Figure 1.
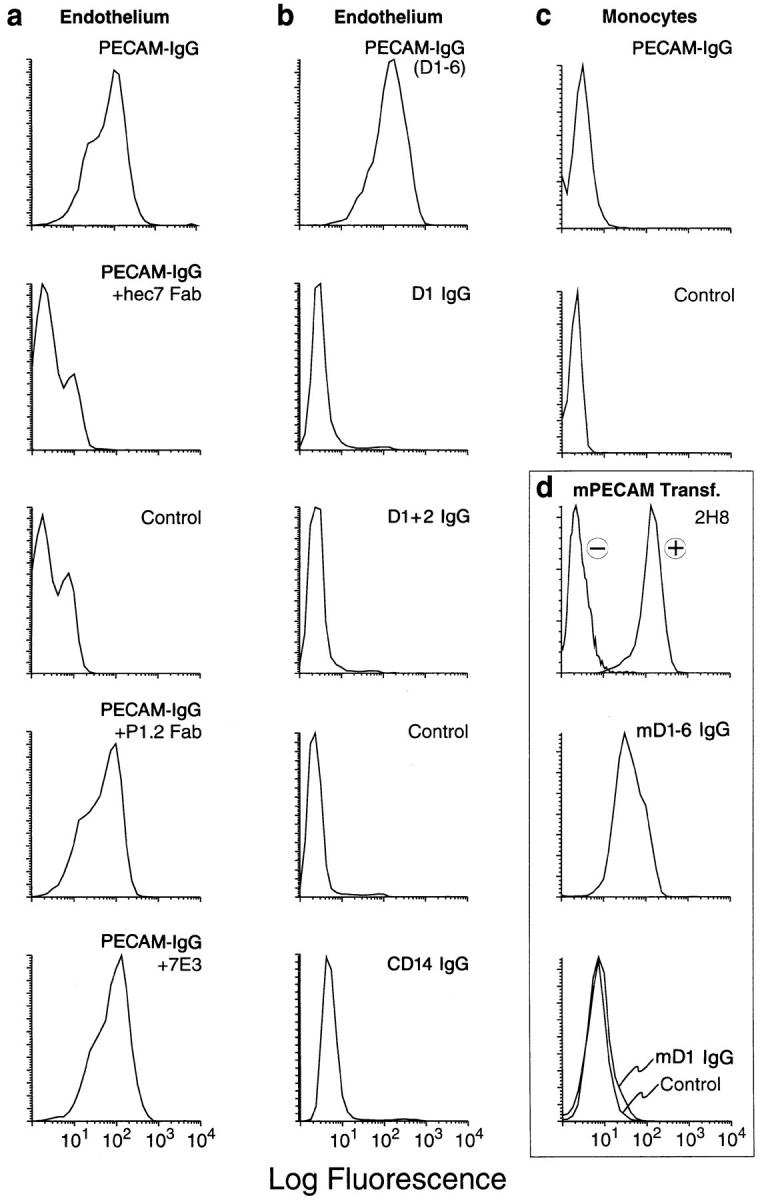
FACS® binding profiles of PECAM-IgG chimeras. Nonenzymatically resuspended HUVEC or PBMC were processed for FACScan® analysis as described in Materials and Methods. The data of this figure are arranged in vertical columns. (a) PECAM-IgG binds homophilically to PECAM on HUVEC. PECAM-IgG (100 nM) bound well to HUVEC. When binding was carried out in the presence of 100 μg/ml Fab fragments of anti-PECAM mAb hec7 (+hec7 Fab), binding was reduced to control levels seen without any primary reagent. In contrast, 100 μg/ml Fab fragments of mAb P1.2 (+P1.2 Fab), which binds to PECAM domain 6, and has been shown not to be involved in homophilic adhesion or diapedesis (4, 24), has no significant effect on PECAM-IgG binding. In addition, 7E3 (25), a blocking mAb against αvβ3, which has been reported to be a heterophilic ligand for PECAM (39, 40) did not affect PECAM-IgG binding to HUVEC (+7E3). (b) Truncated PECAM-IgG chimeras do not bind to HUVEC. While full-length PECAM-IgG (D1-6) bound well to HUVEC, chimeras consisting of domain 1 (D1 IgG) or domains 1 and 2 (D1+2 IgG) of PECAM, or a full-length CD14 chimera (CD14 IgG) did not bind significantly above background (Control). (c) When fulllength PECAM-IgG chimera (20 μg/ml, 100 nM) was incubated with monocytes under the same conditions as for HUVEC, there was no significant binding above background. (d) L cells transfected with murine PECAM (20; mPECAM Transf.) were incubated with mAb 2H8 and detected with a FITC-labeled goat anti–hamster mAb (+) or with fulllength mPECAM-IgG (mD1-6 IgG), murine domain 1-IgG (mD1 IgG), or nonbinding human IgG (Control), washed, and then incubated with FITC-labeled goat anti–human IgG. Only the mAb and full-length chimera bind to the transfectants. (d, top) −, mAb 2H8 binding to nontransfected L cells.
In contrast, none of the truncated PECAM-IgG chimeric molecules could be detected bound to either monocytes or endothelial cells by this method (Fig. 1), even at concentrations that maximally inhibited transendothelial migration (see Fig. 2). This does not mean that PECAM-IgG does not bind to the leukocyte PECAM. The off-rate of these interactions is likely too fast for detectable levels to remain bound after the multiple washings and incubations of the FACS® protocol (27).
Figure 2.

Soluble domain 1 of PECAM is sufficient to block TEM. Freshly isolated PBMC were suspended to 2 × 106/ml in medium M199 (M199), hec7 anti-PECAM mAb (20 μg/ml, 133 nM), or the indicated purified PECAM-IgG constructs at a final concentration of 100 nM. D1 IgG indicates a chimeric molecule comprised of domain 1 of human PECAM fused with the Ch2 + Ch3 domains of human IgG1. D1-6 IgM indicates a chimeric molecule comprised of full-length human PECAM fused with the Ch2 + Ch3 + Ch4 domains of human IgM. The cells were co-cultured for 1 h at 37°C to allow transendothelial migration, and the monolayers were then washed briefly in EGTA and DPBS before fixation and quantitation of transmigration as described previously (4, 8). The data are expressed as the mean ± standard error of five replicates for each sample. All chimeric proteins containing domain 1 of PECAM significantly blocked transmigration. Asterisks (*) indicate P <0.02. The results shown are from a representative experiment of seven such experiments.
Transendothelial Migration In Vitro Requires Domain 1 and/ or 2 of Endothelial Cell PECAM.
We tested the effects of PECAM-IgG chimeras on the migration of Mo across HUVEC monolayers. This system has been predictive of results obtained in vivo. We consistently found that all PECAM-IgG chimeras containing at least domain 1 blocked TEM as well as the full-length molecule D1-6 IgG (Fig. 2). The 60–80% block in TEM was equivalent to the block obtained with hec7 mAb at 20 μg/ml (133 nM.) Several important controls demonstrated that the inhibition of TEM was due to the presence of the soluble PECAM molecule and not an artifact of the human IgG tail (Fig. 2). (a) A chimeric protein consisting of an unrelated molecule, CD14, fused to the same IgG tail, had no effect on TEM. (b) Soluble PECAM chimeras were made in which either the entire extracellular portion of PECAM or only domains 1 and 2 were fused to the Ch2 + Ch3 + Ch4 domains of IgM, an immunoglobulin for which monocytes and HUVEC have no receptors. These PECAM chimeras, produced in bivalent form by L cells (which lack the ability to make J chain), blocked TEM as well as the PECAM-IgG did. (c) A form of PECAM-IgG lacking domains 1 and 2 (D3-6 IgG) did not block TEM. This last control also demonstrates the requirement for the NH2-terminal domain(s) in this process. Consistent with the hypothesis that soluble domain 1 is sufficient to block TEM, D1 IgG and D1-2 IgG as well as fulllength PECAM-IgG blocked at all concentrations tested. Some inhibition was seen at 5 nM, while maximum blocking was seen at 50 nM (Fig. 3).
Figure 3.

Domain 1-IgG blocks TEM as efficiently as full-length PECAM-IgG or mAb hec7. Freshly isolated PBMC were resuspended in the indicated concentrations of nonblocking binding control mAb W6/32 (anti–class I MHC), blocking anti-PECAM mAb hec7, or human PECAMIgG chimeras. The transmigration assay was carried out as in Fig. 2. For the mAb 133 nM is ∼20 μg/ml; for full-length PECAM-IgG, 100 nM is ∼20 μg/ml. The data shown are the mean and standard errors of three experiments with six replicates per variable in each experiment. Asterisks indicate P <0.001 compared to W6/32.
When hec7 mAb was prebound to Mo in suspension and the unbound antibody washed off, TEM was blocked for at least an hour (Fig. 4, Preincubate with monocytes, and reference 8). The same treatment performed with PECAM-IgG at 100 nM (20 μg/ml), did not block TEM, consistent with the inability of PECAM-IgG to bind stably to Mo (Fig. 1). However, PECAM-IgG molecules did inhibit TEM efficiently and in a long-lasting manner when they were retained in the fluid surrounding the Mo (Fig. 2; Fig. 4, Added at t0). Taken together, these data suggest that the low affinity interaction of PECAM-IgG chimeras with leukocytes is capable of blocking TEM when sufficient local concentration is maintained. No block in TEM was seen when either mAb or PECAM-IgG were added to the apical surface of the HUVEC monolayers for 1 h and then removed before adding the monocytes (Fig. 4, Preincubate with endothelium). Additionally, PECAM-IgG did not block TEM if preincubated separately with both monocytes and endothelium before washing and combining (data not shown). Because hec7 and D1-6 IgG would stably bind if they had access to endothelial PECAM molecules in the junctions (Fig. 1 and reference 8), it appears that the added reagents were not effectively retained by the endothelial monolayer (Fig. 4). Thus, the block mediated by either mAb or PECAM-IgG added to the monocytes in suspension above the HUVEC monolayer (Added at t0) must be due to interaction of the reagents with the monocytes. It then seems reasonable to assume that PECAM-IgG is mimicking endothelial PECAM in these interactions.
Figure 4.

PECAM-IgG chimeras interfere with leukocytes, not endothelial cells in TEM. Freshly isolated PBMC were preincubated on ice for 30 min in hec7 mAb or PECAM domain 1-2 IgG or full-length PECAMIgG all at a final concentration of 100 nM (about 20 μg/ml by IgG ELISA) before being washed free of unbound reagent, resuspended to 2 × 106/ml in M199, and added to HUVEC monolayers (Preincubate with monocytes). Alternatively, the reagents were added to the apical surfaces of HUVEC monolayers for 1 h at 37°C before washing and adding untreated PBMC (Preincubate with endothelium). The third group of samples consisted of the same PBMC resuspended to the same density in the same concentrations of reagents and added together to untreated HUVEC monolayers (Added at t0). Control PBMC were resuspended in M199 alone. Transmigration proceeded for 60 min. Data were quantitated as in Fig. 2 as well as by evaluation of cross sections as described in Materials and Methods. The data shown are the mean ± standard error of six replicates for each sample. Asterisks indicate P <0.05. The experiment was performed four times with similar results.
Domain 1 of Murine PECAM Blocks Inflammation In Vivo.
To test the relevance of the above studies to inflammation in vivo, we produced murine PECAM-IgG chimeras in a manner similar to the human PECAM-IgG chimeras (4). L cells stably transfected with cDNAs encoding either the entire extracellular portion of murine PECAM or domain 1 of murine PECAM fused to human IgG secreted the appropriate sized proteins, which were reactive with antibodies to both domain 1 of murine PECAM and human IgG (data not shown). Analogous to the behavior of their human PECAM counterparts, full-length murine PECAMIgG (mD1-6 IgG) bound stably to transfectants bearing murine PECAM, whereas the chimera containing only murine PECAM domain 1 did not (Fig. 1 d). These murine PECAM fusion proteins (mPECAM-IgG) were tested for their ability to block leukocyte emigration in the thioglycollate broth peritonitis model.
Figs. 5 and 6 show data from two experiments representative of five. The peritoneal cavities of unstimulated mice contain negligible numbers of PMN. Thioglycollate induced an acute inflammatory response in the peritoneal cavity of these mice, which could be blocked by mPECAM-IgG at both 4 and 24 h, reducing the numbers of emigrated PMN to 47 and 25% of control, respectively. This inhibition was equivalent to that produced by optimal concentrations of the blocking antimurine PECAM mAb 2H8 and similar to that produced by anti-CD11b mAb 5C6, which have been demonstrated to block in this model (12, 28) and served as our positive controls (Fig. 5). A quantitatively similar block has been produced in a rat model of acute peritonitis using a cross-reacting rabbit anti–human PECAM antibody (13).
Figure 5.

PECAM-IgG chimera blocks neutrophil emigration in vivo. Female mice of the CD2F1 strain received a single injection via tail vein (i.v.) of PBS, or 100 μg of either 2H8 anti–mouse PECAM mAb, 5C6 anti–mouse CD11b mAb, or murine PECAM-IgG chimera (mPECAMIgG) in a volume of 100 μl. 1 h later they received an intraperitoneal injection (i.p.) of 1 ml of PBS or 4% thioglycollate broth (Thio). Mice were killed at 4 h (light-shaded bars) or 24 h (dark-shaded bars) after intraperitoneal injection. Peritoneal neutrophils were collected and enumerated. The data are expressed as the mean and standard error of groups of three mice. All treatment groups are significantly different from the PBS/Thio group taken at the same time with a P value of <0.05.
Figure 6.
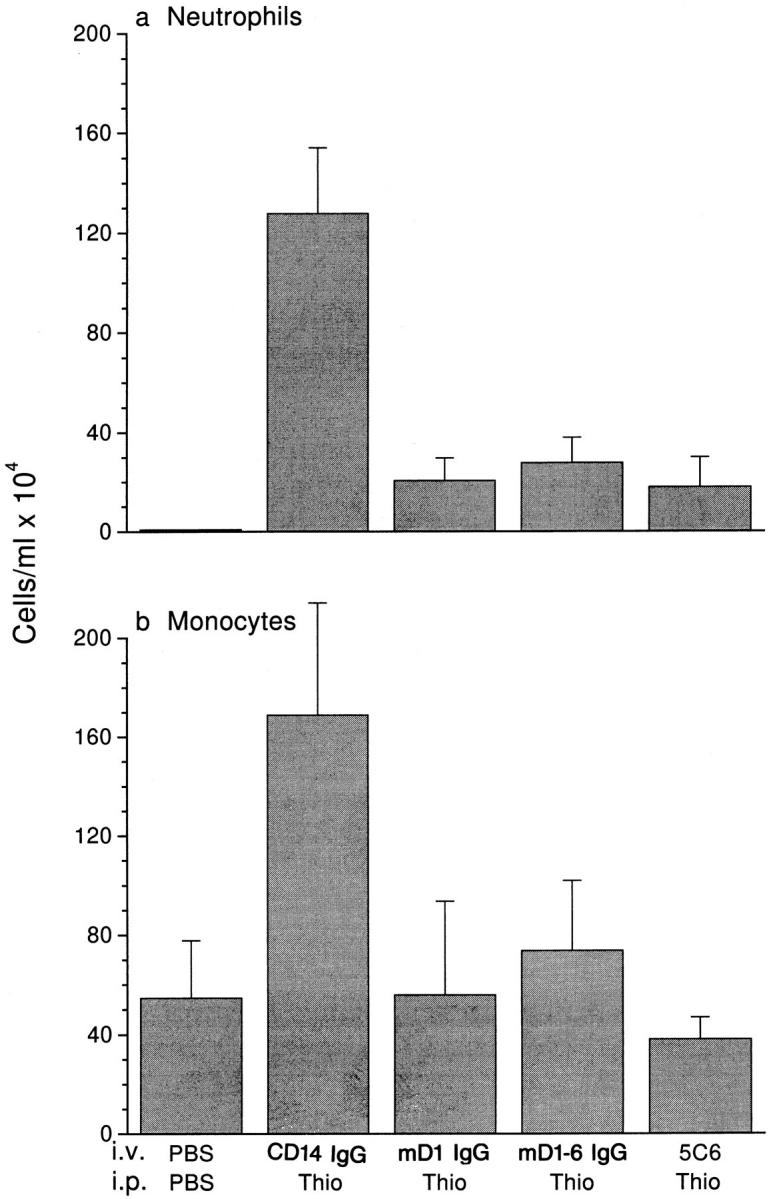
Domain 1 of PECAM blocks leukocyte emigration in vivo. Male mice of the FVB/N strain (five per group) received 100 μg of CD14-IgG chimera as a negative control, a chimeric protein consisting of domain 1 of murine PECAM (mD1 IgG), full-length murine PECAM (mD1–6 IgG), or anti-CD11b (5C6) as a positive control 1 h before thioglycollate injection. Mice were killed 18 h later and the concentration of neutrophils (a) and Mo (b) in 5 ml of peritoneal lavage fluid was determined. Data are mean and standard error. All treatment groups are significantly different from the CD14-IgG with P <0.05.
Several experiments were performed in a different strain of mice at 18 h after intraperitoneal injection to study the effects of mPECAM-IgG chimeras on the influx of both PMN and Mo. When human CD14-IgG was administered intravenously before thioglycollate, there was no decrease in PMN influx (Fig. 6), as expected from the in vitro studies. However, when either mPECAM domain 1-IgG or 1-6 IgG were administered (100 μg), the number of PMN recovered from the peritoneal cavity was reduced by ∼80%. In four experiments, mPECAM domain 1-IgG blocked PMN influx by 86 ± 8%.
The unstimulated peritoneal cavity in the FVB/N strain contains ∼5 × 105 mononuclear phagocytes/ml. In the experiment shown in Fig. 6, the number of Mo recruited into the peritoneal cavity had already risen to over 1.5 × 106/ ml (∼8 × 106 total) by 18 h after thioglycollate stimulation in mice that received the control fusion protein CD14IgG. In contrast, those mice treated with either full-length or domain 1 mPECAM-IgG or mAb 5C6 had basal levels of mononuclear phagocytes in their peritoneal cavities.
The number of circulating leukocytes was similar in all experimental groups that received thioglycollate stimulation (data not shown). Thus, the decrease in leukocytes entering the peritoneal cavity was not due to their sequestration or destruction as a consequence of treatment.
Histologic sections of the peritoneal viscera of these mice were examined. In the venules from mice treated with mPECAM-IgG (both full-length and domain 1 only) a large proportion of the leukocytes in the profile were noted to be in apparent contact with the lumenal endothelium (Fig. 7, a–c). This was not seen in the venules of mice treated with control CD14-IgG fusion protein nor anti-CD11b mAb (Fig. 7, d and e).
Figure 7.

PECAM-IgG chimeras block leukocyte efflux at the venular lumenal surface. Representative photomicrographs from mesenteric venules of the mice in the experiment depicted in Fig. 6. (a) Venule from mouse treated with domain 1 chimera showing multiple leukocytes (some indicated by arrowheads) in apparent contact with the endothelial surface. The neutrophil at 12 o'clock is shown at higher magnification in the inset. Arrows point to endothelial cell nuclei. (b) Image from another mouse treated with domain 1 construct showing a mononuclear cell (upper) and PMN (lower) in apparent contact with the endothelial cell surface. (c) A similar image from mouse treated with full-length mPECAM-IgG with two PMN. In contrast, leukocytes were only rarely seen attached to venules of mice treated with the nonblocking CD14-IgG chimera (d) or the adhesion blocking anti-CD11b mAb 5C6 (e). Bars: (a, d, and e) 50 μm; and (b and c) 20 μm.
Quantitation of these sections (see Materials and Methods) affirmed that blockade of inflammation with mPECAMIgG resulted in a significant increase in leukocytes in apparent contact with the endothelium. Other treatments, both blocking and nonblocking, were not associated with such an increase (Fig. 8). This was found whether the data were analyzed in terms of the percentage of intravascular leukocytes in contact with the vascular lumen or in terms of the percentage of venular profiles with more than one leukocyte in contact.
Figure 8.

PECAM-IgG chimeras lead to increase in apparent contact of leukocytes with venule wall. The first 10 venules of appropriate size for each of the mice in the experiment of Fig. 7 were scored in a blinded fashion for leukocytes free within the lumen and those in apparent contact with the venular wall. Data are expressed as the percent of total leukocytes adherent to the wall (top) or the percent of vascular profiles with more than one attached leukocyte (bottom). The data are expressed as the mean and standard error for all mice in the group. Asterisks indicate that data are significantly different from any of the other groups with a P <0.025. Reagent, the intravenous treatment; Thio, whether or not mice received intraperitoneal injection of thioglycollate (+) or PBS (−).
Discussion
Our data provide the first evidence that a soluble molecule corresponding to a fragment of a CAM, which is incapable of stable binding to its ligand, can nonetheless block inflammation. We show that PECAM domain 1-IgG is sufficient to block the contribution of PECAM to transendothelial migration of leukocytes. This truncated form of PECAM is as efficient as a blocking mAb or full-length PECAM-IgG at blocking TEM of monocytes in vitro and of both neutrophils and monocytes in vivo. Furthermore, a PECAM construct lacking domains 1 and 2 was without effect. This demonstrates the importance of the NH2 terminal domains of PECAM in these reactions.
PECAM-IgG Chimeric Proteins.
Other investigators have used chimeras of adhesion molecules fused with IgG to study adhesive interactions in inflammation (29–32). The bivalent nature of the molecules increases the binding affinity, and the immunoglobulin chain prolongs the biological half-life in vivo (33). While there are some well-known examples involving selectins (29, 32, 34), there are few reports on the use of CAMs from other molecular families fused to IgG to block inflammation in vivo (33, 35). This may be because most CAMs have a rather low affinity for interaction with their ligands on a molecule-to-molecule basis (27). Many immunoglobulin superfamily–Ig chimeras only function when they are immobilized to a surface, allowing multivalent interactions to occur (30, 31).
Full-length PECAM-IgG chimeras bind stably to HUVEC, but not to Mo, which bear 10-fold less PECAM (26). Consistent with this, high surface expression is apparently required for homophilic PECAM cell–cell adhesion (reference 36 and our unpublished data). Truncated forms of PECAM do not bind stably enough even to HUVEC to be detected by FACS® (Fig. 1). Sun et al. (24) demonstrated that domains 1 and 2 were necessary for homophilic binding under these conditions. They are sufficient to mediate homophilic binding, but only when expressed on a fulllength Ig superfamily backbone. Fawcett et al. also found that only full-length PECAM molecules supported stable adhesion (37).
Several controls demonstrated that the effects we observed with our PECAM-IgG chimeras were not due to the interaction of the IgG portion of the molecule with leukocyte Fc receptors; a full-length CD14 molecule and the PECAM domain 3–6 construct had no effect on TEM when fused to the same IgG molecule. On the other hand, both full-length and domain 1 + 2 of PECAM fused to the human IgM COOH tail blocked TEM. Furthermore, no Fc-mediated binding of the truncated PECAM-IgGs or the CD14-IgG to monocytes was detected by FACScan® (Fig. 1). There was no evidence that infusion of murine PECAM- IgG chimeras resulted in opsonization of leukocytes, consistent with previous experience using other CAM-IgG chimeras in vivo (29, 34). For therapeutic purposes, Fcmediated interactions could be further precluded by design of a chimera with the opsonic portions of the Fc chain deleted (38).
Identifying the Domains of PECAM Used by Endothelial Cells.
As expected from previous studies using mAbs (8), no block in TEM was seen when either mAb or PECAMIgG were added to the apical surface of the HUVEC monolayers for 1 h before washing the monolayer surface (Fig. 4). Since both hec7 mAb and D1-6 IgG are capable of binding tightly to endothelial PECAM, this observation suggests that these reagents were not accessible to PECAM sequestered in the junctions of the endothelial monolayer. Therefore, the block mediated by these reagents when added to the monocytes in suspension above the HUVEC monolayer must be due to interaction of the reagents with the monocytes. If we then reasonably assume that PECAMIgG, including D1-IgG, is mimicking endothelial PECAM in these interactions, these data provide evidence that domain 1 and/or 2 are crucial for the role of endothelial cell PECAM in transmigration.
Since we have previously demonstrated that domains 1 and/or 2 of monocyte PECAM are required for TEM (4), it seems most likely that domain 1 and/or 2 on both leukocyte and endothelial PECAM interact with each other in a homophilic manner during TEM. In support of this, the effects of blocking these domains on both the endothelial cell and the Mo simultaneously, are not additive (reference 8 and data not shown).
The Site of PECAM Blockade.
The block in TEM obtained with PECAM-IgGs both in vitro and in vivo resembles the block obtained with mAbs against PECAM both quantitatively and qualitatively. In our culture system, Mo were seen to be tightly bound to the apical surface of HUVEC monolayers over the junctions as in reference 8, while in the murine venules in the inflamed mesentery showed leukocytes in contact with the endothelial cell lining, as if arrested before diapedesis (Figs. 7 and 8), as we had previously seen with mAb 2H8 (12). It is notable that the epitope for mAb 2H8 is in domain 1 of murine PECAM.
Thus, soluble domain 1 of PECAM, which is incapable of high affinity binding to cellular PECAM, can mimic the effects of a blocking antibody without the potential complications associated with immune complex formation. This work defines domain 1 of PECAM as a target for therapeutic intervention.
Acknowledgments
We wish to thank Ahalya Nava and Elizabeth Polizzi for excellent technical assistance, and Judy Adams for preparation of the figures. We are indebted to Drs. Mark Zukowski and Henri Lichenstein (Amgen, Inc.) for the human IgG cDNA and the CD14-IgG construct, respectively; Dr. Michael Concino (Procept, Inc.) for the CD2-IgM cDNA; Dr. Barry Coller (Mt. Sinai School of Medicine, New York) for mAb 7E3; the staff of the Labor and Delivery department of Mt. Sinai Hospital and the New York Blood Center cord blood study for saving umbilical cords for these studies; the technicians of the New York Hospital Department of Pathology for excellent preparation of hematoxylin and eosin slides; and to Drs. Ralph Steinman, Gwen Randolph, and Joan Muller for critical review of this manuscript.
Supported by National Institutes of Health grant RO1 HL46849 and an Established Investigator Award from the American Heart Association to W.A. Muller.
Footnotes
1 Abbreviations used in this paper: CAM, cell adhesion molecule; HUVEC, human umbilical vein endothelial cells; Mo, monocytes; PECAM, platelet–endothelial cell adhesion molecule, CD31; TEM, transendothelial migration.
References
- 1.Carlos TM, Harlan JM. Leukocyte–endothelial cell adhesion molecules. Blood. 1994;84:2068–2101. [PubMed] [Google Scholar]
- 2.Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76:301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 3.Muller WA. Migration of leukocytes across the vascular intima. Molecules and mechanisms. Trends Cardiovasc Med. 1995;5:15–20. doi: 10.1016/1050-1738(94)00028-T. [DOI] [PubMed] [Google Scholar]
- 4.Liao F, Huynh HK, Eiroa A, Greene T, Polizzi E, Muller WA. Migration of monocytes across endothelium and passage through extracellular matrix involve separate molecular domains of PECAM-1. J Exp Med. 1995;182:1337–1343. doi: 10.1084/jem.182.5.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Muller WA, Ratti CM, McDonnell SL, Cohn ZA. A human endothelial cell-restricted, externally disposed plasmalemmal protein enriched in intercellular junctions. J Exp Med. 1989;170:399–414. doi: 10.1084/jem.170.2.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Newman PJ, Berndt MC, Gorski J, White GC, II, Lyman S, Paddock C, Muller WA. PECAM-1 (CD31) cloning and relation to adhesion molecules of the immunoglobulin gene superfamily. Science (Wash DC) 1990;247:1219–1222. doi: 10.1126/science.1690453. [DOI] [PubMed] [Google Scholar]
- 7.Delisser HM, Newman PJ, Albelda SM. Molecular and functional aspects of PECAM-1/CD31. Immunol Today. 1994;15:490–495. doi: 10.1016/0167-5699(94)90195-3. [DOI] [PubMed] [Google Scholar]
- 8.Muller WA, Weigl SA, Deng X, Phillips DM. PECAM-1 is required for transendothelial migration of leukocytes. J Exp Med. 1993;178:449–460. doi: 10.1084/jem.178.2.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berman ME, Xie Y, Muller WA. Roles of platelet/endothelial cell adhesion molecule-1 (PECAM-1, CD31) in natural killer cell transendothelial migration and beta 2 integrin activation. J Immunol. 1996;156:1515–1524. [PubMed] [Google Scholar]
- 10.Berlin C, Bargatze RF, Campbell JJ, von Andrian UH, Szabo MC, Hasslen SR, Nelson RD, Berg EL, Erlandsen SL, Butcher EC. α4integrins mediate lymphocyte attachment and rolling under physiologic flow. Cell. 1995;80:413–422. doi: 10.1016/0092-8674(95)90491-3. [DOI] [PubMed] [Google Scholar]
- 11.Springer TA. Adhesion receptors of the immune system. Nature (Lond) 1990;346:425–434. doi: 10.1038/346425a0. [DOI] [PubMed] [Google Scholar]
- 12.Bogen S, Pak J, Garifallou M, Deng X, Muller WA. Monoclonal antibody to murine PECAM-1 (CD31) blocks acute inflammation in vivo. J Exp Med. 1994;179:1059–1064. doi: 10.1084/jem.179.3.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vaporciyan AA, Delisser HM, Yan H-C, Mendiguren II, Thom SR, Jones ML, Ward PA, Albelda SM. Involvement of platelet-endothelial cell adhesion molecule-1 in neutrophil recruitment in vivo. Science (Wash DC) 1993;262:1580–1582. doi: 10.1126/science.8248808. [DOI] [PubMed] [Google Scholar]
- 14.Wakelin MW, Sanz M-J, Dewar A, Albelda SM, Larkin SW, Boughton-Smith N, Williams TJ, Nourshargh S. An anti–platelet-endothelial cell adhesion molecule-1 antibody inhibits leukocyte extravasation from mesenteric microvessels in vivo by blocking the passage through basement membrane. J Exp Med. 1996;184:229–239. doi: 10.1084/jem.184.1.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murohara T, Delyani JA, Albelda SM, Lefer AM. Blockade of platelet endothelial cell adhesion molecule–1 protects against myocardial ischemia and reperfusion injury in cats. J Immunol. 1996;156:3550–3557. [PubMed] [Google Scholar]
- 16.Tanaka Y, Albelda SM, Horgan KJ, Van Seventer GA, Shimizu Y, Newman W, Hallam J, Newman PJ, Buck CA, Shaw S. CD31 expressed on distinctive T cell subsets is a preferential amplifier of β1 integrin-mediated adhesion. J Exp Med. 1992;176:245–253. doi: 10.1084/jem.176.1.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berman ME, Muller WA. Ligation of platelet– endothelial cell adhesion molecule 1 (PECAM-1/CD31) on monocytes and neutrophils increases binding capacity of leukocyte CR3 (CD11b/CD18) J Immunol. 1995;154:299–307. [PubMed] [Google Scholar]
- 18.Piali L, Albelda SM, Baldwin HS, Hammel P, Gisler RH, Imhof BA. Murine platelet endothelial cell adhesion molecule (PECAM-1/CD31) modulates β2 integrins on lymphokine-activated killer cells. Eur J Immunol. 1993;23:2464–2471. doi: 10.1002/eji.1830231013. [DOI] [PubMed] [Google Scholar]
- 19.Muller WA, Weigl S. Monocyte-selective transendothelial migration: dissection of the binding and transmigration phases by an in vitro assay. J Exp Med. 1992;176:819–828. doi: 10.1084/jem.176.3.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xie Y, Muller WA. Molecular cloning and adhesive properties of murine platelet/endothelial cell adhesion molecule–1. Proc Natl Acad Sci USA. 1993;90:5569–5573. doi: 10.1073/pnas.90.12.5569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hailman E, Lichenstein HS, Wurfel MM, Miller DS, Johnson DA, Kelley M, Busse LA, Zukowski MM, Wright SD. Lipopolysaccharide (LPS)-binding protein accelerates the binding of LPS to CD14. J Exp Med. 1994;179:269–277. doi: 10.1084/jem.179.1.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arulanandam ARN, Moingeon P, Concino MF, Recny MA, Kato K, Yagita H, Koyasu S, Reinherz EL. A soluble multimeric recombinant CD2 protein identifies CD48 as a low affinity ligand for human CD2: divergence of CD2 ligands during the evolution of humans and mice. J Exp Med. 1993;177:1439–1450. doi: 10.1084/jem.177.5.1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muller WA, Berman ME, Newman PJ, Delisser HM, Albelda SM. A heterophilic adhesion mechanism for platelet/endothelial cell adhesion molecule 1 (CD31) J Exp Med. 1992;175:1401–1404. doi: 10.1084/jem.175.5.1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun Q-H, Delisser HM, Zukowski MM, Paddock C, Albelda SM, Newman PJ. Individually distinct Ig homology domains in PECAM-1 regulate homophilic binding and modulate receptor affinity. J Biol Chem. 1996;271:11090–11098. doi: 10.1074/jbc.271.19.11090. [DOI] [PubMed] [Google Scholar]
- 25.Coller BS, Cheresh DA, Asch E, Seligsohn U. Platelet vitronectin receptor expression differentiates IraqiJewish from Arab patients with Glanzmann thrombasthenia in Israel. Blood. 1991;77:75–83. [PubMed] [Google Scholar]
- 26.Newman PJ, Albelda SM. Cellular and molecular aspects of PECAM-1. Nouv Rev Fr Hematol. 1992;34:9–13. [PubMed] [Google Scholar]
- 27.van der Merwe PA, Barclay AN. Transient intercellular adhesion: the importance of weak protein–protein interactions. Trends Biochem Sci. 1994;19:354–358. doi: 10.1016/0968-0004(94)90109-0. [DOI] [PubMed] [Google Scholar]
- 28.Rosen H, Gordon S. Monoclonal antibody to the murine type 3 complement receptor inhibits adhesion of myelomonocytic cells in vitro and inflammatory cell recruitment in vivo. J Exp Med. 1987;166:1685–1701. doi: 10.1084/jem.166.6.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Watson SR, Fennie C, Lasky LA. Neutrophil influx into an inflammatory site inhibited by a soluble homing receptor–IgG chimaera. Nature (Lond) 1991;349:164–167. doi: 10.1038/349164a0. [DOI] [PubMed] [Google Scholar]
- 30.Damle NK, Aruffo A. Vascular cell adhesion molecule 1 induces T-cell antigen receptor–dependent activation of CD4+T lymphocytes. Proc Natl Acad Sci USA. 1991;88:6403–6407. doi: 10.1073/pnas.88.15.6403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Damle NK, Klussman K, Linsley PS, Aruffo A. Differential costimulatory effects of adhesion molecules B7, ICAM-1, LFA-3, and VCAM-1 on resting and antigenprimed CD4+T lymphocytes. J Immunol. 1992;148:1985–1992. [PubMed] [Google Scholar]
- 32.Watson SR, Imai Y, Fennie C, Geoffroy JS, Rosen SD, Lasky LA. A homing receptor–IgG chimera as a probe for adhesive ligands of lymph node high endothelial venules. J Cell Biol. 1990;110:2221–2229. doi: 10.1083/jcb.110.6.2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Capon D, Chamow S, Mordenti J, Marsters T, Gregory T, Mitsuya H, Byrn R, Lucas C, Wurm F, Groopman J, et al. Designing CD4 immunoadhesins for AIDS therapy. Nature (Lond) 1989;337:525–531. doi: 10.1038/337525a0. [DOI] [PubMed] [Google Scholar]
- 34.Lee WP, Gribling P, DeGuzman L, Ehsani N, Watson SR. A P-selectin–immunoglobulin G chimera is protective in a rabbit ear model of ischemia-reperfusion. Surgery (St Louis) 1995;117:458–465. doi: 10.1016/s0039-6060(05)80068-6. [DOI] [PubMed] [Google Scholar]
- 35.Doherty P, Williams E, Walsh FS. A soluble chimeric form of the L1 glycoprotein stimulates neurite outgrowth. Neuron. 1994;14:57–66. doi: 10.1016/0896-6273(95)90240-6. [DOI] [PubMed] [Google Scholar]
- 36.Sun J, Williams J, Yan H, Amin KM, Albelda SM, Delisser HM. Platelet/endothelial cell adhesion molecule-1 (PECAM-1) homophilic adhesion is mediated by immunoglobulin-like domains 1 and 2 and depends on the cytoplasmic domain and the level of surface expression. J Biol Chem. 1996;271:18561–18570. doi: 10.1074/jbc.271.31.18561. [DOI] [PubMed] [Google Scholar]
- 37.Fawcett J, Buckley C, Holness CL, Bird IN, Spragg JH, Saunders J, Harris A, Simmons DL. Mapping the homotypic binding sites in CD31 and the role of CD31 adhesion in the formation of interendothelial cell contacts. J Cell Biol. 1995;128:1229–1241. doi: 10.1083/jcb.128.6.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zheng XX, Steele AW, Nickerson PW, Steurer W, Steiger J, Strom TB. Administration of noncytolytic IL-10/Fc in murine models of lipopolysaccharideinduced septic shock and allogeneic islet transplantation. J Immunol. 1995;154:5590–5600. [PubMed] [Google Scholar]
- 39.Piali L, Hammel P, Uherek C, Bachmann F, Gisler RH, Dunon D, Imhof BA. CD31/PECAM-1 is a ligand for αvβ3integrin involved in adhesion of leukocytes to endothelium. J Cell Biol. 1995;130:451–460. doi: 10.1083/jcb.130.2.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Buckley CD, Doyonnas R, Newton JP, Blystone SD, Brown EJ, Watt SM, Simmons DL. Identification of αv β3as a heterotypic ligand for CD31/PECAM-1. J Cell Sci. 1996;109:437–445. doi: 10.1242/jcs.109.2.437. [DOI] [PubMed] [Google Scholar]