

Abstract
Early infection with murine cytomegalovirus (MCMV) induces circulating levels of interleukin (IL)-12, interferon (IFN)-γ, and tumor necrosis factor (TNF). Studies presented here further characterize these responses by defining kinetics and extending evaluation to include IL-1, IL-6, and glucocorticoids. IL-12 p40, IFN-γ, TNF, IL-1α, and IL-6 were shown to be increased, but IL-1β was undetectable, in serum of MCMV-infected mice. The IL-12 p40, IFN-γ, TNF, and IL-6 responses were dramatic with peak levels reaching >150–10,000 pg/ml at 32–40 h after infection and rapidly declining thereafter. Glucocorticoid induction, peaking at 36 h and reaching 30-fold increases above control values, accompanied the cytokine responses. Mice with cytokine deficiencies or neutralized cytokine function demonstrated that IL-6 was the pivotal mediator of the glucocorticoid response, with IL-1 contributing to IL-6 production. The IL-6 requirement appeared to be specific for virus-type stimuli as the synthetic analogue of viral nucleic acid, polyinosinic-polycytidylic acid, also induced IL-6–dependent glucocorticoid release, but treatments with the bacterial product lipopolysaccharide and a non-immune physical restraint stressor elicited IL-6–independent responses. Collectively, the results identify IL-6 as a primary mediator of glucocorticoid induction, and elucidate specific pathways of interactions between immune and neuroendocrine systems during viral infection.
The cytokines IL-12, IFN-γ, TNF, IL-1, and IL-6 are induced under conditions of sepsis with gram-negative bacteria and in response to administration of the gram-negative bacterial product endotoxin, i.e., LPS (1, 2). High levels of these cytokines contribute to pathologies characterized as endotoxin-induced shock with wasting, thymic atrophy, and life-threatening states (1–3). Circulating TNF, IL-1, and/or IL-6, elicited as a cascade after exposure to LPS (4–6) or after administration of purified cytokines (7), induce the steroid hormones glucocorticoids. Induction is largely a result of hypothalamic-pituitary-adrenal (HPA)1 axis activation through stimulating hypothalamus production of corticotropin-releasing hormone (CRH), which induces pituitary release of adrenocorticotropin hormone (ACTH) for stimulation of adrenal gland glucocorticoid production. Glucocorticoids can suppress multiple immune functions, including cytokine production and T cell responses (4, 8, 9). Thus, the immune and neuroendocrine systems can communicate to provide feedback inhibition mechanisms limiting immune responses.
In addition to stimulation as a result of cytokine responses to bacterial LPS, glucocorticoid release through the HPA axis occurs as part of circadian rhythm (10) and is induced by a variety of other stimuli including physical or cognitive stress (11), a synthetic analogue for viral nucleic acids, i.e., polyinosinic-polycytidylic acid (poly I:C) (12), and turpentine induction of inflammation (3, 13). The precise pathways for HPA axis activation under each of these conditions has yet to be fully elucidated, and little is known about endogenous induction in response to infections. If glucocorticoids are elicited during challenge with pathogens, they may shape or modulate down-stream T cell functions as well as control acute detrimental cytokinemediated pathologies (4, 8, 9).
This laboratory has been examining cytokine responses and functions during viral infections (14–16). In particular, responses to infections of mice with the cytopathic (14–18) murine cytomegalovirus (MCMV) are being investigated. Our group has shown that systemic levels of IL-12, IFN-γ, and TNF are induced at early times after infection (15, 16). The present studies were undertaken to more precisely define the early MCMV-elicited cytokine responses, to extend characterization to IL-1 and IL-6, and to determine the effects of cytokine expression on endogenous glucocorticoid responses. Our results demonstrate that MCMV stimulates dramatic and tightly regulated early IL-12, IFN-γ, TNF, IL-1, and IL-6 responses with an accompanying glucocorticoid response. The key mediator of glucocorticoid induction is shown to be IL-6 with IL-1 acting to stimulate IL-6 production. These studies define an IL-6–dependent pathway for endogenous glucocorticoid induction. Moreover, they suggest that distinct pathways are in place for communication between immune and neuroendocrine systems during infections with different types of pathogens.
Materials and Methods
Mice.
5–15-wk-old, male, C57BL/6 (C57BL/6NTacfBR; Taconic, Germantown, NY), IL-6–deficient (B6,129-IL6<tm1koe >; Jackson Laboratory, Bar Harbor, ME), wild-type control for IL-6– deficient ([B6129F1/J-A<W-J>/A<w> F × B6129F1/J-A <w-J >/A<w>M]F2, Jackson Laboratory), and IFN-γ–deficient C57BL/6 (a kind gift from Joan Stein-Streilein, Massachusetts General Hospital, Boston, MA) mice were housed in the Brown University Animal Care Facility for at least 1 wk before use, and kept on a 12 h light/dark cycle with lights on at 6:00 AM. Except for designated experimental manipulations, mice were minimally disturbed. Protocols were in accordance with institutional guidelines for animal care and use.
Virus and Viral Plaque Assays.
Stocks of salivary gland–extracted MCMV, Smith strain, were generated as previously described (15). Mice were intraperitoneally infected with MCMV in 100 μl or control injected with 100 μl of vehicle alone (1× media 199; GIBCO BRL, Gaithersburg, MD) between 7:00 and 8:00 PM. To determine optimal doses, ranges of virus between 1 × 105 and 6.3 × 103 PFU/mouse were tested. Cytokines were detectable 36 h after infection with doses as low as 6.3 × 103. As peak cytokine production and 100% long-term survival was obtained with 1 × 105 or 5 × 104 PFU/mouse, these MCMV doses were used. Hepatic viral titers were assessed with plaque assays on NIH-3T3 fibroblasts (gift of Ann Campbell, Eastern Virginia Medical School, Norfolk, VA). To enumerate plaques, cells were fixed with 10% buffered formalin (Fisher Scientific, Pittsburgh, PA) and stained with 0.1% crystal violet. Titers were quantitated as PFU/gram of tissue.
In Vivo Treatments.
Rat anti–mouse IFN-γ mAb (XMG1.2) and control IgG (Sigma Chem. Co., St. Louis, MO), 1 mg/mouse, were injected intraperitoneally 12 h before infection. Anti–IFN-γ antibody treatments eliminated detectable serum IFN-γ, i.e., >99% effectiveness, as tested by ELISA. Human IL-1 receptor antagonist (IL-1ra) (gift from Synergen, Boulder, CO) was administered by continuous infusion from subcutaneous pumps (ALZET, Palo Alto, CA) with a delivery rate of 125 μg/h or 3 mg/d for 7 d. Control mice received pumps delivering PBS. Pumps were implanted 3 d before injections. Serum IL-1ra levels in IL-1ra–treated mice were between 12 and 36 μg/ml, as determined using commercial ELISA kits (R&D Systems, Inc., Minneapolis, MN). Levels were sufficient to block >95% of IL-1 activity (19), i.e., >1,000 times in excess of maximum serum IL-1α detected in normal infected mice (see Results). Control mice had IL-1ra levels at or below limit of detection (47 pg/ml).
LPS from Salmonella enteritidis (Sigma Chemicals) and poly I:C (Sigma) were administered intraperitoneally at 50 μg/mouse and 100 μg/mouse, respectively, in 100 μl volume of PBS. Based on other studies of peak cytokine responses (12, 13; Cousens L.P., and C.A. Biron, manuscript in preparation), serum was collected 2 h after injection. Restraint was used as a non-immune stressor and administered by placing mice into a vented conical tube for 30 min before serum collection (20).
Serum and Organ Collection.
For serum corticosterone and ACTH measurements, blood was collected from mice under low stress conditions, i.e., within 2 or 4 min of handling. Mice were retroorbitally bled under methoxyflurane anesthesia (Metofane, Pitman-Moore, Mundelein, IL) into heparinized tubes. As there was always some clotting of samples, the fluid was identified as serum. Samples were centrifuged to pellet cells, collected and stored at −80°C. Lateral lobes of livers were harvested, placed into media and stored at −80°C for use in viral plaque assays.
Cytokine and Corticosterone Measurements.
Serum cytokine levels were determined by standard sandwich ELISA. Assays for IL-6 used different purified capture and biotinylated detection anti–IL-6 monoclonal antibodies (PharMingen, San Diego, CA). IL-1α and IL-1β levels were measured using ELISA kits (IL-1α; Genzyme, Cambridge, MA; IL-1β; R&D Systems). Assays were performed according to manufacturer protocols, and limits of detection were 160, 60, and 15 pg/ml, respectively. Assays for TNF, IFN-γ, and IL-12 p40 were performed as previously described (15, 16). The TNF assay was specific for TNF-α. Colorimetric changes of enzyme substrates were detected using a Dynatech MR 4000 reader (Chantilly, VA). Limits of detection for IL-12, IFN-γ, and TNF were 40 pg/ml. Commercially available assay kits were used for determinations of plasma corticosterone (Immunochem™, ICN Biomedicals, Costa Mesa, CA) and ACTH (Allegro HS-ACTH, Nichols Institute, San Juan Capistrano, CA). Limits of detection were 5 ng/ml and 5 pg/ml, respectively.
Statistical Analysis.
Where indicated, p-values were obtained by comparing groups using a student's t test. For experiments examining corticosterone responses to LPS and poly I:C in IL-6–deficient mice, a two-way ANOVA was used to evaluate group (deficient versus normal) as well as treatment effects (LPS versus poly I:C). The Student-Newman-Keuls test was used to assess for differences between specific pairs of group means.
Results
Kinetics of Cytokine Responses to MCMV Infection.
This laboratory's previous studies, of daily sampling, have shown induction of detectable IL-12, IFN-γ, and TNF production on day 2 of MCMV infection (15, 16). Experiments were undertaken with more frequent serum sampling to further characterize kinetics and peak responses of these cytokines, and to extend evaluation to IL-1α, IL-1β, and IL-6. Consistent with the earlier work, it was not possible to detect IL-12 p40, IFN-γ, or TNF in serum samples, from 4 C57BL/6 mice per group, receiving control vehicle injections or taken at 12-h intervals at, or before, 24 and after 72 h of MCMV infection. However, the three cytokines were observed at both 36 and 48 h after infection. At 36 h after infection with 1 × 105 PFU/mouse, serum values reached the following pg/ml (± SE): IL-12, 1431 ± 54.4; IFN-γ, 8733 ± 481.1; and TNF, 436 ± 9.5. By 48 h of infection, peak levels had decreased by >70% for IL-12 or IFN-γ and by 50% for TNF. Because cytokine values had sharp rises and falls within 12 h intervals, levels were determined in samples taken at 2 to 4 h intervals during the first 24–48 h of MCMV infection (Fig. 1). Based on preliminary experiments with a variety of doses (see Materials and Methods), infections were initiated with 5 × 104 PFU of virus. Serum IL-12, IFN-γ, and TNF levels were found to crest between 36 and 40 h of infection with peak values of IL-12 p40 at ∼500 pg/ml (Fig. 1 A), IFN-γ in excess of 10,000 pg/ml (Fig. 1 B), and TNF in excess of 150 pg/ml (Fig. 1 C). The kinetics of serum IL-12 and IFN-γ responses were rapid and sharp with peak IL-12 occurring slightly before peak IFN-γ and both responses dramatically subsiding within the next 4–10 h (Fig. 1, A and B). Peak TNF levels were achieved at ∼4 h preceding peak IL-12 and maintained for longer periods of time (Fig. 1 C). Measurements of IL-6 showed that this cytokine had kinetics reflecting IL-12 and IFN-γ; IL-6 reached peak levels of >5,000 pg/ml at 38 h and subsided by 48 h after infection (Fig. 1 D). These results demonstrate that MCMV induces significant and tightly regulated levels of serum IL-12, IFN-γ, TNF, and IL-6.
Figure 1.

Serum cytokine levels between 24 and 48 h after MCMV infection. IL-12 p40 (A), IFN-γ (B), TNF (C), IL-6 (D), and IL-1α (E) were assessed in sera collected from MCMV-infected (5 × 104 PFU/mouse) and vehicle-treated C57BL/6 mice at various times between 24 and 48 h of infection by ELISA assays as described in Materials and Methods. IL-12 p40, IFN-γ, TNF, and IL-6 values are from two mice at each time point and are expressed as means ± SD. IL-1α values are plotted from individual mice at the indicated times after infection with geometric means indicated by short lines. The IL-1α samples were taken from two different experiments. Uninfected samples for all cytokines were from mice receiving control vehicle injections.
Both isoforms of IL-1, i.e., IL-1α and IL-1β, were measured. It was not possible to detect IL-1β in any serum samples from four different experiments, with 2 to 6 C57BL/6 mice per group, and sampling at the following times after MCMV infection: (a) days 0, 2, 5 or 7, (b) 12-h intervals from 36 to 60 h, or (c) 4-h intervals from 12 to 36 h (data not shown). In contrast, IL-1α was detected in all serum samples tested, including those from uninfected mice receiving control vehicle injections (Fig. 1 E). Although there was more variability and a wider range of values than those observed for the other cytokines, IL-1α levels were elevated at 18–36 h after MCMV infection (Fig. 1 E). To compare MCMV induction of IL-1α and IL-1β to other known inducers of IL-1, serum samples were obtained from mice treated with bacterial LPS or with the synthetic double stranded analogue for viral nucleic acid poly I:C. IL-1α was detectable in either LPS (122.7 ± 10 pg/ml) or poly I:Ctreated (66.6 ± 14 pg/ml) mice, with LPS inducing relatively high (173.5 ± 21 pg/ml), but poly I:C inducing low (25.9 ± 14 pg/ml), levels of serum IL-1β (all values are means ± SD of 2 mice per group). Thus, in contrast to the clear induction of IL-1β by LPS, MCMV appears to have a unique selective induction, and poly I:C a preferential induction, of IL-1α as compared to IL-1β.
Glucocorticoid and ACTH Responses during MCMV Infection.
Because certain of these cytokines can induce glucocorticoid production as a result of activation of the HPA axis, and because ACTH is the upstream HPA axis component mediating stimulation of glucocorticoids, serum levels of corticosterone, the natural murine glucocorticoid, and ACTH were examined. Under low stress conditions, AM values for corticosterone (± SE) in uninfected mice averaged 5.5 ± 1.3 ng/ml. The diurnal rise resulted in average peak PM values of 120 ± 14 ng/ml. Serum corticosterone levels, initially measured at 12 h intervals (Fig. 2 A), were induced at 24, 36, and 48 h after MCMV infection. Maximal increases, compared to uninfected controls, were observed at the AM time point 36 h after infection, i.e., 150 ng/ml, a 30-fold increase (Fig. 2 A). The PM values were increased to 250 ng/ml, however, as control corticosterone levels had diurnal increases, net inductions at these times were only twofold. Examination of additional time points (Fig. 2 B) demonstrated that peak corticosterone responses were at 36 h. This MCMV induction of glucocorticoids at 36 h has been observed in >10 independent experiments with groups of 2–6 mice. In other experiments, ACTH levels were shown to be significantly increased at 28 and 32 h after MCMV infection (Fig. 2 C). Thus, dramatic endogenous glucocorticoid and ACTH responses accompany circulating cytokine induction during MCMV infections.
Figure 2.
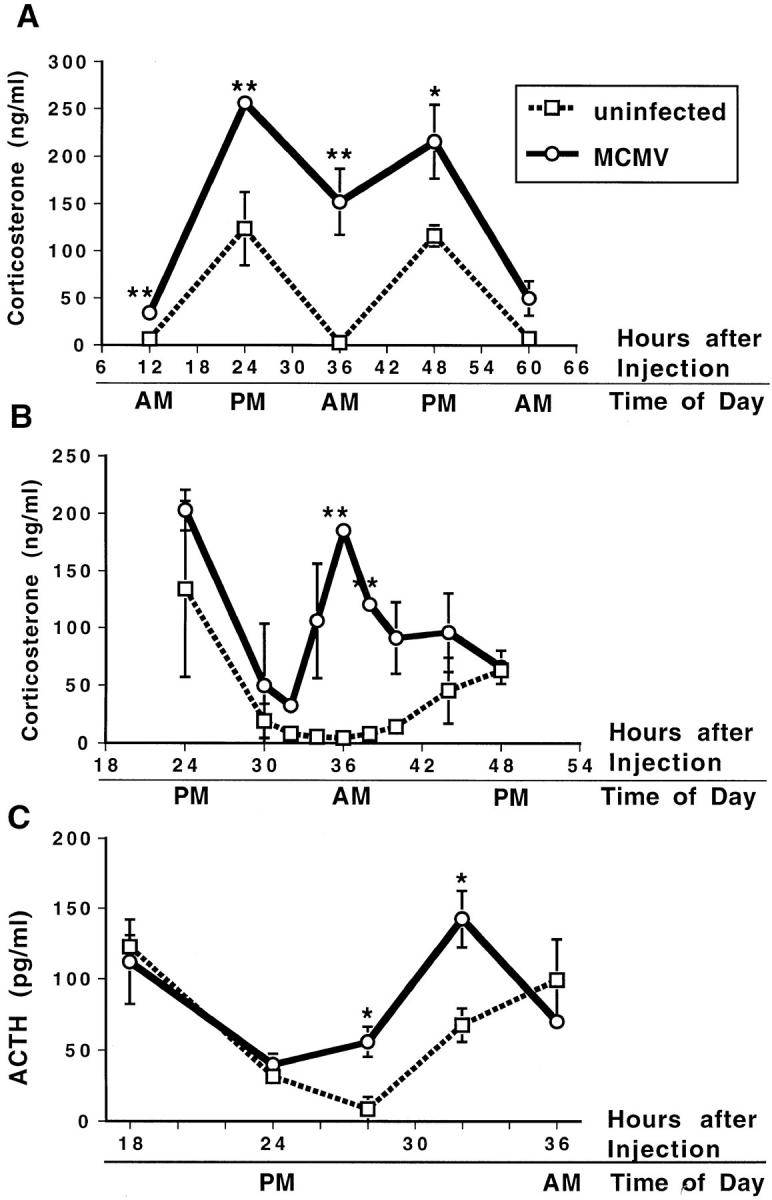
Serum corticosterone and ACTH levels after MCMV infection. Corticosterone and ACTH levels were measured in serum samples collected from C57BL/6 mice under low stress conditions (mice were bled within 4 min of handling for corticosterone measurement and within 2 min of handling for ACTH measurement). Serum corticosterone was measured at 12-h intervals between 12 and 72 h (A) or 2–4-h intervals between 24 and 48 h (B) and serum ACTH was measured at 4–6-h intervals between 18 and 36 h after MCMV (5 × 104 PFU/mouse) infection (open circles) or vehicle injection (open squares). In A, data are mean ± SE of four mice per time point. In B, data are means ± SD of two mice per time point. For C, data are means ± SE of three mice per time point. *P <0.05, **P <0.01.
Role of Cytokines in MCMV-Induced Glucocorticoid Responses.
To characterize the contribution of early cytokine production to induction of glucocorticoid responses, serum corticosterone levels were examined in mice with specifically neutralized endogenous cytokine functions. Individual cytokine functions were evaluated in mice with targeted disruptions of the IL-6 or IFN-γ genes and in normal mice treated with neutralizing anti–IFN-γ antibodies or IL-1 receptor antagonist (IL-1ra). MCMV-induced corticosterone levels were not modified significantly by the presence or absence of IFN-γ; infected IFN-γ–deficient (Fig. 3) and anti–IFN-γ–treated (data not shown) mice had serum corticosterone values similar to those in respective infected control mice. In contrast, MCMV-infected IL-6–deficient and IL-1ra–treated mice demonstrated dramatically reduced corticosterone levels in comparison to respective infected control mice; serum corticosterone levels were decreased by up to 75% in IL-6–deficient mice and by >50% in IL-1ra– treated mice (Fig. 3). Reductions resulting from IL-6 deficiencies were observed in three independent experiments with each experiment containing three mice/group, and those resulting from blocking IL-1 function were observed in two independent experiments with each containing three mice/group. Statistical significance was increased by combination of results from multiple experiments such that the IL-1ra effects were to P <0.05 and the IL-6 deficiency effects were to P <0.01. The cytokine effects on glucocorticoid induction were not a consequence of changes in response kinetics as, in addition to the shown dramatically reduced glucocorticoid levels at 36 h, MCMV-infected IL-6–deficient mice also lacked induced glucocorticoid levels at 24, 48, and 60 h after infection (data not shown). Thus, blocks in endogenous IL-6 or IL-1 functions dramatically attenuate glucocorticoid responses to MCMV infection.
Figure 3.

Corticosterone levels in mice with neutralized cytokine function. Corticosterone levels in MCMV-infected (5 × 104 PFU/ mouse) (filled bar) or vehicle-injected (striped bar) IL-6–deficient (IL-6–), IFN-γ–deficient (IFN-γ−), IL-1ra–treated (IL-1ra) mice or respective control mice (IL-6+, IFN-γ+, PBS). Treatments were as described in Materials and Methods. Corticosterone levels were measured in serum samples collected from mice under low stress conditions (bled within 4 min of handling) 36 h after infection. Results from one of two to three experiments are shown. Data are means ± SE of three mice per group. **P <0.01.
Studies were carried out to determine whether modulation of pathogen burdens or other endogenous cytokines may have contributed to reduced corticosterone levels. At the 36 h infection time point, log viral titers ranged between 4.3 and 5.1 PFU/g of liver. Effects on glucocorticoids were independent of modified viral burden as, at this time, there were no significant differences in any experimental or treatment groups, compared to respective controls, including IL-6–deficient and IL-1ra–treated (data not shown). They were also independent of IFN-γ changes; this cytokine was not detectable in mice with a mutated IFN-γ gene or those treated with antibody neutralizing IFN-γ and was not dramatically inhibited in mice having had other cytokines blocked (data not shown). TNF levels also did not appear to be significantly altered in any of the cytokine-deficient or -blocked mice. Compared to wild-type controls, IL-1α levels were reduced in IFN-γ–deficient mice [113 ± 37 versus 41 ± 5 pg/ml (mean ± SE of 3 mice)], but increased in IL-6–deficient mice [92 ± 32 versus 211 ± 53 pg/ml (mean ± SE of 3 mice)]. In contrast to the increased IL-1α in IL-6–deficient mice, IL-6 levels were dramatically reduced in IL-1ra–treated mice by up to 70%. Serum IL-6 values in IL-1ra–treated mice were 369 ± 126 pg/ml as compared to PBS-treated levels of 1,500 ± 467 pg/ml (values are mean ± SE of three mice per group). Upon combination of results from multiple experiments, IL-1ra effects on IL-6 were statistically significant to P <0.05. Thus, although cytokine neutralization or deficiency did not affect viral load or production of most cytokines during MCMV infection, IL-1ra treatment decreased serum IL-6 levels. Taken together with the demonstrated IL-1α expression in the absence of IL-6, these data indicate that IL-6 is a critical and required cytokine for induction of peak glucocorticoids during MCMV infection, and that IL-1α is playing a secondary role by contributing to IL-6 induction.
Glucocorticoid Responses to Non-Viral Stimuli in IL-6–deficient Mice.
To characterize the role of endogenous IL-6 for glucocorticoid induction by other non-viral and replication-independent stimulators of the HPA axis, responses to LPS, poly I:C, and restraint stress were evaluated in IL-6– deficient mice. Modest, but statistically significant, reductions of corticosterone levels were observed in response to LPS treatment of IL-6–deficient as compared to similarly treated wild-type mice (Fig. 4 A). In contrast, IL-6–deficient mice treated with poly I:C demonstrated significantly and profoundly decreased, i.e., up to 85%, glucocorticoid responses compared to wild-type poly I:C–treated mice (Fig. 4 A). Two way ANOVA also revealed significant main effects of both group (IL-6–deficient versus wild-type control) (F[1,23] = 78.2, P <0.0001) and treatment condition (PBS versus poly I:C versus LPS) (F[2,23] = 84.5, P <0.0001) on corticosterone concentrations. This analysis demonstrated that the poly I:C–treated group of IL-6–deficient mice was significantly different from both the poly I:C–treated wildtype group and the LPS-treated IL-6–deficient group. In contrast, corticosterone levels were not significantly different between PBS-treated IL-6–deficient and poly I:C–treated IL-6–deficient mice. Activation of the HPA axis by the non-immune physical restraint stressor was unaffected in IL-6–deficient mice compared to wild-type controls (Fig. 4 B). These results demonstrate that glucocorticoid responses to LPS or restraint stress are largely IL-6 independent, but, as with MCMV infection, poly I:C–induced responses are IL-6–dependent.
Figure 4.

Serum corticosterone levels after LPS, poly I:C, or restraint stress administration in IL-6–deficient and wild-type mice. (A) Corticosterone levels were measured in plasma collected from IL-6–deficient (IL-6−) (filled bar) and wild-type (IL-6+) (dotted bar) mice under low stress conditions (less than 4 min of handling) 2 h after injection with PBS, 50 μg LPS, or 100 μg poly I:C. (B) Corticosterone levels were measured in serum samples collected from IL-6–deficient (IL-6−) and wild-type (IL-6+) mice bled under low stress conditions (within 4 min of handling) or after 30 min of restraint. Data are means ± SE of four mice per group. **P <0.01.
Discussion
The present studies demonstrate that, in addition to IL-12, TNF, and IFN-γ, MCMV infections of mice induce detectable IL-1α and IL-6, but not IL-1β, production. The IL-12, TNF, IFN-γ, and IL-1α cytokines all rapidly reach high peak levels in the circulation and decline shortly thereafter. Moreover, cytokine activation of the HPA axis appears to be engaged as endogenous serum corticosterone and ACTH responses also are induced. IL-6 is the critical and required factor leading to peak induction of endogenous glucocorticoids, and IL-1α is a necessary factor for optimal IL-6 production. This pathway to glucocorticoid release through IL-1α and IL-6 is clearly delineated because IL-1β is undetectable in MCMV-infected mice and because IL-1α levels are similar or greater in MCMV- infected IL-6–deficient mice as compared to infected wildtype controls. The IL-6 requirement for glucocorticoid responses is specific to viral infections and virus-type stimuli because, although the synthetic double stranded analogue for viral nucleic acid, poly I:C, the bacterial product, LPS, and restraint stress all induce glucocorticoid responses in normal mice, only peak responses to MCMV and poly I:C are exquisitely dependent upon endogenous IL-6. Taken together, these results identify: (a) the kinetics and magnitudes of early circulating cytokine induction during MCMV infection, (b) a unique IL-6–dependent glucocorticoid response to viral infection and virus-type, non-replicating stimuli, and (c) a specific IL-1α to IL-6 induction pathway. Furthermore, as they present evidence for a distinct cytokine dependency for HPA axis activation, the studies begin to define precise communication pathways between immune and neuroendocrine systems under conditions of different microbial challenges.
IL-12, IFN-γ, and TNF are important for antiviral defense (15, 16) and, as IL-1 and IL-6 have known proinflammatory functions (19, 21), these factors may also promote defense against MCMV. However, at high systemic levels, the cytokines can cause significant and life-threatening pathologies (1, 2, Orange, J.S., T.P Salazar-Mather, and C.A. Biron, manuscript in preparation). As glucocorticoids can decrease production of multiple cytokines, including IL-6, IFN-γ, IL-1, and TNF (8), induction of these endogenous steroid hormones by high virus-induced IL-6 levels may provide feedback inhibition to limit cytokine responses and protect the host from cytokine-mediated disease (22). In support of this hypothesis, another acute viral infection, lymphocytic choriomeningitis virus (LCMV), does not induce detectable systemic cytokines (15; Ruzek, M.C., A.H. Miller, B.D. Pearce, and C.A. Biron, unpublished observation) or significant levels of glucocorticoids (12, Ruzek, M.C., A.H. Miller, B.D. Pearce, and C.A. Biron, data not shown) at early times. As a consequence, immune responses to MCMV are shaped and/or limited by factors in addition to those influencing responses to LCMV. It is noteworthy that in comparison to LCMV, MCMV infections induce relatively weak later T cell responses (14). Thus, glucocorticoids may be specifically elicited during certain viral infections initiating a sequence of events with the potential to result in cytokine-mediated detrimental effects, i.e., inducing extremely high levels of cytokines, but not during viral infections failing to do so, and these hormones may shape additional down-stream immune responses to these viruses.
The IL-1α–induced IL-6 requirement for optimal glucocorticoid stimulation (Fig. 3) is consistent with studies examining recombinant or purified factors in vivo. Administrations of TNF, IL-1, and IL-6 increase circulating ACTH and corticosterone levels (5, 7) with IL-1β being the most potent and rapid inducer (7). However, IL-1α, IL-1β, and TNFα can all stimulate IL-6 production, with IL-1α being a more potent inducer than either IL-1β or TNF (5, 23). In addition, IL-1α and IL-6 have been shown to synergize for release of ACTH (23, 24). This ability of IL-1α to induce and synergize with IL-6 can explain the observation that corticosterone responses after IL-6 administration alone are not as great as those after IL-1 administration (23). As a downstream or cooperative role of IL-6 has not been previously distinguished from direct stimulation, the reported IL-1 requirement for corticosterone responses to pro-inflammatory stimuli may be due to this factor's ability to induce and synergize with IL-6 (23, 24). In addition, given the different potencies of IL-1α and IL-1β for IL-6 induction (5), yet similar activity on the HPA axis (24), IL-1β alone may induce glucocorticoids, but optimal glucocorticoid responses to IL-1α may require IL-6. It is likely that serum IL-1α values reported here are actually an underestimate of overall induced levels, as IL-1α is generally expressed in a membrane-bound form (19).
Although the glucocorticoid response dependency on IL-1α-induced IL-6 is consistent with results from administering cytokines, the IL-6 requirement in response to challenge with particular agents appears to be specific to viral infections. Two additional systems have not found IL-6 essential for glucocorticoid induction; these are turpentine and LPS challenges (13). IL-1β is readily detectable after injection of LPS (6), however, it is undetectable in serum from MCMV-infected mice. Therefore, during viral infection, the cytokines leading to glucocorticoid production may be more dependent upon an IL-1α to IL-6 cascade, whereas LPS and/or turpentine may induce other cytokines, including IL-1β, that either alone or in combination induce glucocorticoids independently of IL-6 (3). The poly I:C–induced responses also appear to occur under conditions of minimal IL-1β expression. Thus, the ability to define the IL-1α to IL-6 pathway for glucocorticoid induction, in response to MCMV and/or poly I:C, is most likely possible because of the absence of parallel or overlapping pathways for induction. A model for pathways of glucocorticoid induction, after viral as compared to bacterial stimulation, is presented in Fig. 5.
Figure 5.

Model for microbial stimulation of glucocorticoid induction.
Our results can be contrasted to others examining virus activation of the HPA axis after exposure to Newcastle disease virus (NDV), influenza virus, and herpes simples type 1 (HSV-1) (25–27). NDV stimulates IL-1–dependent ACTH and corticosterone release (25). However, NDV does not productively infect mice and, similar to poly I:C responses, increases in serum corticosterone are observed within hours of injection (12, 25). Therefore, NDV does not reflect actual events occurring during infection of a permissive host. Influenza virus infections and ocular infections of herpes simplex virus type 1 (HSV-1) also induce increased corticosterone levels, but, in comparison to MCMV infection, these responses peak much later (7 d after infection) and are prolonged (26, 27). As both HSV-1 and influenza virus infections can cause considerable additional physical distress which may itself stimulate the HPA axis, it is likely that other pathways are contributing to induction of glucocorticoid responses at later times after these infections. Interestingly, several other viruses have been shown to stimulate IL-6 mRNA expression in vitro (28). Although a thorough comparison of IL-6 production and corticosterone responses has not been performed during each of these infections, our results suggest that induction of circulating IL-6 may determine whether or not glucocorticoid responses and their consequences are elicited early during viral infections. Taken together, these observations suggest that the kinetics and magnitude of corticosterone production induced by systemic viral infection may be specific to the virus, the extent of virus-induced pathology, and/or the levels of virus-induced IL-6.
In summary, these results demonstrate rapid and dramatic increases in serum cytokines and glucocorticoids early during MCMV infection. An essential role for IL-6 in the induction of peak endogenous glucocorticoid responses is defined, with IL-1 playing an accessory role for production of, and/ or synergism, with IL-6. Taken together with work in other systems, these studies show a stringent regulation of systemic cytokines responses during early MCMV infection, and define a previously uncharacterized cytokine pathway for glucocorticoid induction during a natural infection.
Acknowledgments
The authors thank Kimberly D'Eramo, Theodore Johnson, Khuong (Ken) Nguyen, Gary Pien, Michael Primiano, Stephen Taubenfeld, and Leslie Cousens for their help in harvesting low stress samples, as well as Tracy Pisell for technical assistance with the corticosterone assays. The authors also thank Drs. R. Coffman, P. Scott, G. Trinchieri, J. Stein-Streilein, and A. Campbell as well as Genetics Institute, Synergen, and Celltech for reagents.
This work was supported by National Institutes of Health grants RO1-MH47674, KO2-MH00680, T32ES07272, and RO1-CA41268.
Footnotes
1 Abbreviations used in this paper: ACTH, adrenocorticotropin hormone; CRH, corticotropin-releasing hormone; HPA, hypothalamic-pituitaryadrenal; MCMV, murine cytomegalovirus; NDV, Newcastle disease virus; poly I:C, polyinosinic-polycytidylic acid.
References
- 1.Ozmen L, Pericin M, Hakimi J, Chizzonite RA, Wysocka M, Trinchieri G, Gately M, Garotta G. Interleukin 12, interferon γ, and tumor necrosis factor α are the key cytokines of the generalized Shwartzman reaction. J Exp Med. 1994;180:907–915. doi: 10.1084/jem.180.3.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zanetti G, Heumann D, Gerain J, Kohler J, Abbet P, Barras C, Lucas R, Glauser M-P, Baumgartner J-D. Cytokine production after intravenous or peritoneal gram-negative bacterial challenge in mice. Comparative protective efficacy of antibodies to tumor necrosis factor-α and to lipopolysaccharide. J Immunol. 1992;148:1890–1897. [PubMed] [Google Scholar]
- 3.Fantuzzi G, Dinarello CA. The inflammatory response in interleukin-1β-deficient mice: comparison with other cytokine-related knock-out mice. J Leuk Biol. 1996;59:489–493. doi: 10.1002/jlb.59.4.489. [DOI] [PubMed] [Google Scholar]
- 4.Imura H, Fukata J. Endocrine-paracrine interaction in communication between the immune system and endocrine systems. Activation of the hypothalamic-pituitaryadrenal axis in inflammation. Eur J Endocrinol. 1994;130:32–37. doi: 10.1530/eje.0.1300032. [DOI] [PubMed] [Google Scholar]
- 5.Libert C, Brouckaert P, Shaw A, Fiers W. Induction of interleukin 6 by human and murine recombinant interleukin 1 in mice. Eur J Immunol. 1990;20:691–694. doi: 10.1002/eji.1830200333. [DOI] [PubMed] [Google Scholar]
- 6.Fong Y, Tracey KJ, Moldawer LL, Hesse DG, Manogue KB, Kenney JS, Lee AT, Kuo GC, Allison AC, Lowry SF, Cerami A. Antibodies to cachectin/ tumor necrosis factor reduce interleukin 1β and interleukin 6 appearance during lethal bacteremia. J Exp Med. 1989;170:1627–1633. doi: 10.1084/jem.170.5.1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Besedovsky HO, Del Rey A, Klusman I, Furukawa H, Monge G, Arditi, Kabiersch A. Cytokines as modulators of the hypothalamus-pituitary-adrenal axis. J Steroid Biochem Molec Biol. 1991;40:613–618. doi: 10.1016/0960-0760(91)90284-c. [DOI] [PubMed] [Google Scholar]
- 8.Kunicka JE, Talle MA, Denhardt GH, Brown M, Prince CA, Goldstein G. Immunosuppression by glucocorticoids: inhibition of production of multiple lymphokines by in vivoadministration of dexamethasone. Cell Immunol. 1993;149:39–49. doi: 10.1006/cimm.1993.1134. [DOI] [PubMed] [Google Scholar]
- 9.Schleimer RP, Jacques A, Shin HS, Lichtenstein LM, Plaut M. Inhibition of T cell-mediated cytotoxicity by anti-inflammatory steroids. J Immunol. 1984;132:266–271. [PubMed] [Google Scholar]
- 10.Dhabhar FS, Miller AH, Stein M, McEwen BS, Spencer RL. Diurnal and stress-induced changes in distribution of peripheral blood leukocyte populations. Brain Behav Immun. 1994;8:66–73. doi: 10.1006/brbi.1994.1006. [DOI] [PubMed] [Google Scholar]
- 11.Khansari DN, Murgo AJ, Faith RE. Effects of stress on the immune system. Immunol Today. 1990;11:170–175. doi: 10.1016/0167-5699(90)90069-l. [DOI] [PubMed] [Google Scholar]
- 12.Miller, A.H., R.L. Spencer, B.D. Pearce, T.L. Pisell, J.J. Leung, F.S. Dhabar, B.S. McEwen, and C.A. Biron. 1997. Effects of viral infection on corticosterone secretion and glucocorticoid receptor binding in immune tissues. Psychoneuroendocrinology. In press. [DOI] [PubMed]
- 13.Fattori E, Cappelletti M, Costa P, Sellitto C, Cantoni L, Carelli M, Faggioni R, Fantuzzi G, Ghezzi P, Poli V. Defective inflammatory response in interleukin 6–deficient mice. J Exp Med. 1994;180:1243–1250. doi: 10.1084/jem.180.4.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Biron CA, Orange JS. IL12 in acute viral infectious disease. Res Immunol. 1995;146:590–600. doi: 10.1016/0923-2494(96)83036-7. [DOI] [PubMed] [Google Scholar]
- 15.Orange JS, Biron CA. An absolute and restricted requirement for IL-12 in natural killer cell IFN-γ production and antiviral defense. J Immunol. 1996;156:1138–1142. [PubMed] [Google Scholar]
- 16.Orange JS, Biron CA. Characterization of early IL-12, IFN-αβ and TNF effects on antiviral state and NK cell responses during murine cytomegalovirus infection. J Immunol. 1996;156:4746–4756. [PubMed] [Google Scholar]
- 17.Shanley JD, Biczak L, Forman SJ. Acute murine cytomegalovirus infection induces lethal hepatitis. J Infect Dis. 1993;167:264–269. doi: 10.1093/infdis/167.2.264. [DOI] [PubMed] [Google Scholar]
- 18.Price P, Olver SD, Gibbons AE, Teo HK, Shellam GR. Characterization of thymic involution induced by murine cytomegalovirus infection. Immunol Cell Biol. 1993;71:155–165. doi: 10.1038/icb.1993.18. [DOI] [PubMed] [Google Scholar]
- 19.Dinarello CA. Biological basis for interleukin-1 in disease. Blood. 1996;87:2095–2147. [PubMed] [Google Scholar]
- 20.Dhabhar FS, Miller AH, McEwen BS, Spencer RL. Effects of stress on immune cell distribution. Dynamics and hormonal mechanisms. J Immunol. 1995;154:5511–5527. [PubMed] [Google Scholar]
- 21.Oldenburg HSA, Rogy MA, Lazarus DD, Van Zee KJ, Keeler BP, Chizzonite RA, Lowry SF, Moldawer LL. Cachexia and the acute-phase protein response in inflammation are regulated by interleukin-6. Eur J Immunol. 1993;23:1889–1894. doi: 10.1002/eji.1830230824. [DOI] [PubMed] [Google Scholar]
- 22.Kapcala LP, Chautard T, Eskay RL. The protective role of the hypothalmic-pituitary-adrenal axis against lethality produced by immune, infectious and inflammatory stress. Ann NY Acad Sci. 1995;771:419–437. doi: 10.1111/j.1749-6632.1995.tb44699.x. [DOI] [PubMed] [Google Scholar]
- 23.Neta R, Perlstein R, Vogel SN, Ledney GD, Abrams J. Role of interleukin 6 (IL-6) in protection from lethal irradiation and in endocrine responses to IL-1 and tumor necrosis factor. J Exp Med. 1992;175:689–694. doi: 10.1084/jem.175.3.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perlstein RS, Mougey EH, Jackson WE, Neta R. Interleukin-1 and interleukin-6 act synergistically to stimulate the release of adrenocorticotropic hormone in vivo . Lymphokine Cytokine Res. 1991;10:141–146. [PubMed] [Google Scholar]
- 25.Besedovsky H, Del Rey A. Mechanism of virusinduced stimulation of the hypothalamus-pituitary-adrenal axis. J Exp Med. 1989;54:235–239. doi: 10.1016/0022-4731(89)90087-3. [DOI] [PubMed] [Google Scholar]
- 26.Hermann G, Tovar CA, Beck FM, Sheridan JF. Kinetics of glucocorticoid response to restraint stress and/or experimental influenza viral infection in two inbred strains of mice. J Neuroimmunol. 1994;49:25–33. doi: 10.1016/0165-5728(94)90177-5. [DOI] [PubMed] [Google Scholar]
- 27.Ben-Hur T, Conforti N, Itzik A, Weidenfeld J. Effects of HSV-1, a neurotropic virus, on the hypothalamicpituitary-adrenocortical axis in rats. Brain Res. 1995;702:17–22. doi: 10.1016/0006-8993(95)00806-7. [DOI] [PubMed] [Google Scholar]
- 28.Sehgal PB, Helfgott DC, Santhanam U, Tatter SB, Clarick RH, Ghrayeb J, May LT. Regulation of the acute phase and immune responses in viral disease. Enhanced expression of the β2-interferon/hepatocyte-stimulating factor/interleukin 6 gene in virus-infected human fibroblasts. J Exp Med. 1988;167:1951–1956. doi: 10.1084/jem.167.6.1951. [DOI] [PMC free article] [PubMed] [Google Scholar]