

Abstract
Apoptosis and necrosis are considered conceptually and morphologically distinct forms of cell death. Here, we report that demise of human T cells caused by two classic apoptotic triggers (staurosporin and CD95 stimulation) changed from apoptosis to necrosis, when cells were preemptied of adenosine triphosphate (ATP). Nuclear condensation and DNA fragmentation did not occur in cells predepleted of ATP and treated with either of the two inducers, although the kinetics of cell death were unchanged. Selective and graded repletion of the extramitochondrial ATP/pool with glucose prevented necrosis and restored the ability of the cells to undergo apoptosis. Pulsed ATP/depletion/repletion experiments also showed that ATP generation either by glycolysis or by mitochondria was required for the active execution of the final phase of apoptosis, which involves nuclear condensation and DNA degradation.
Apoptosis and necrosis are two forms of cell death, with clearly distinguishing morphological and biochemical features (1). However, these two types of demise can occur simultaneously in tissues or cell cultures exposed to the same stimulus (2–5). Often, the intensity of the same initial insult decides the prevalence of either apoptosis or necrosis (6, 7). This suggests that while some early events may be common to both types of cell death, a downstream controller may be required to direct cells towards the organized execution of apoptosis. Previously, we have shown that intracellular energy levels are dissipated in necrosis, but not in apoptosis of neuronal cells (3). To investigate the role of ATP in apoptosis and necrosis here, we used Jurkat cells, a lymphoid cell line, and two well-established inducers of apoptosis: (a) antiCD95 antibodies (αCD95) that elicit apoptosis by activating cell surface CD95 receptors (8, 9); (b) the protein kinase inhibitor staurosporin (STS) that, at high concentrations, triggers apoptosis in a wide variety of mammalian cells (10). Neither of these stimuli requires a functional respiratory chain for the induction of apoptosis (11, 12).
We examined the mode of cell death elicited by these typical apoptosis triggers under conditions of intracellular ATP depletion, which was obtained by blocking mitochondrial and/or glycolytic ATP generation. After the initial findings that ATP is required for the progression of apoptosis, we defined the threshold ATP concentration needed to execute fully the apoptotic program, and the time window during which the intracellular ATP level decides the mode of cell death in this system.
Materials and Methods
Cell Cultures.
Jurkat cells were grown in RPMI-1640 medium supplemented with 10% FCS. Before the experiments, cells were washed and resuspended in serum-free medium without glucose containing 2 mM pyruvate. After adaptation to this medium, cells were exposed to 0 or 2.5 μM oligomycin for 45 min, and then challenged with STS or an agonist mAb (clone CH-11) against CD95.
Viability Assays and Membrane Alteration.
Cultures were stained with a mixture of the membrane permeant dye H-33342 (500 ng/ml) and the membrane impermeant dye SYTOX (500 nM) (Molecular Probes, Eugene, OR). Necrotic (damaged plasma membrane; noncondensed nuclei) and apoptotic cells (intact plasma membrane; characteristically condensed or fragmented nuclei) were scored. Alternatively, cells were stained with a combination of 0.5 μM calcein-AM, 1 μM ethidium homodimer (EH-1), and 500 ng/ml H-33342. Stained cells (immobilized on poly-l-lysine–coated cover slips at the bottom of a 35-mm chamber) were examined with a Leica TCS-4D confocal microscope simultaneously for membrane integrity (calcein retention and EH-1 exclusion) and morphological changes of the nucleus and the cell (polarization contrast and H-33342 stain). Results obtained with both methods were similar. Digital images of SYTO-13/EH-1–stained cells were acquired on a Leica DM-IRB microscope (40× lens) coupled to an imaging system (Imaging Corporation, St. Catherins, Canada). Apoptotic nuclei appeared hyperfluorescent and condensed. In necrotic nuclei, the fainter green SYTO-13 stain was displaced by the red EH-1 stain (3). Surface phosphatidylserine expression was analyzed by annexin V–staining and FACS® analysis as described (13).
ATP Determination.
ATP was determined luminometrically (14). Addition of glucose (⩽10 mM) to cells in the absence of oligomycin resulted in increases of the ATP concentration ⩽20% of the initial value (data not shown) and did not modify apoptosis. With ATP concentrations between 30% and 50% of the resting control level intermediate forms of cell death (e.g., partially condensed nuclei in ruptured cells), and a slight delay in the kinetics of necrosis were occasionally observed after stimulation with either αCD95 or STS. Addition of oligomycin decreased the intracellular ATP concentration (as compared with initial control levels: 60% within 10 min, 75% within 20 min, and ⩾90% within 30 min) irrespective of the presence of other medium supplements or stimuli. Conversely, after treatment with glucose, ATP rose to ⩾70% within 5 min and reached a new steady state after 15 min.
DNA Fragmentation.
Field-inverted gel electrophoresis (FIGE) was performed as described previously (3) after embedding 5 × 105 cells into 40-μl agarose blocks. Yeast chromosomes and λDNA concatemers were used as molecular weight markers. Conventional agarose gel electrophoresis (CAGE) was performed on 1.0% agarose gels (15). Gels were stained with SYBR green (Molecular Probes, Eugene, OR). Alternatively, histone-containing DNA fragments were determined quantitatively by ELISA (Boehringer Mannheim, Germany) (16). In this assay we used a lysate obtained from ∼500 cells.
Lamin Proteolysis.
Western blots were performed using an anti-human lamin B antibody (clone 101 B7, Oncogene Research Products, Cambridge, MA). All lanes of 10% polyacrylamide gels were loaded with 10 μg protein, and homogeneous transfer to nitrocellulose membranes was controlled by Ponceau red staining. For immunocytochemistry, cells were fixed with 80% MeOH. Incubation with anti-lamin B antibody (101 B7) was followed by staining with a secondary BODIPY-coupled anti-mouse antibody and 1 μg/ml H-33342. Cells were visualized by threechannel confocal microscopy with a 100× UV lens (polarization, lamin, chromatin).
Results and Discussion
Initially, we examined the characteristics of cell death caused by the two inducers under conditions of intracellular ATP depletion. Jurkat cells were maintained in a glucose-free RPMI-1640 medium supplemented with 2 mM pyruvate to allow only mitochondrial ATP production. Exposure to either STS or αCD95-induced apoptosis. Intracellular ATP levels were maintained during the first 90 min, but eventually declined after the appearance of the first apoptotic cells (Fig. 1, a and b). When cells were preemptied of ATP using oligomycin, a blocker of mitochondrial ATP synthesis, death occurred by necrosis (Fig. 1, a and c).
Figure 1.
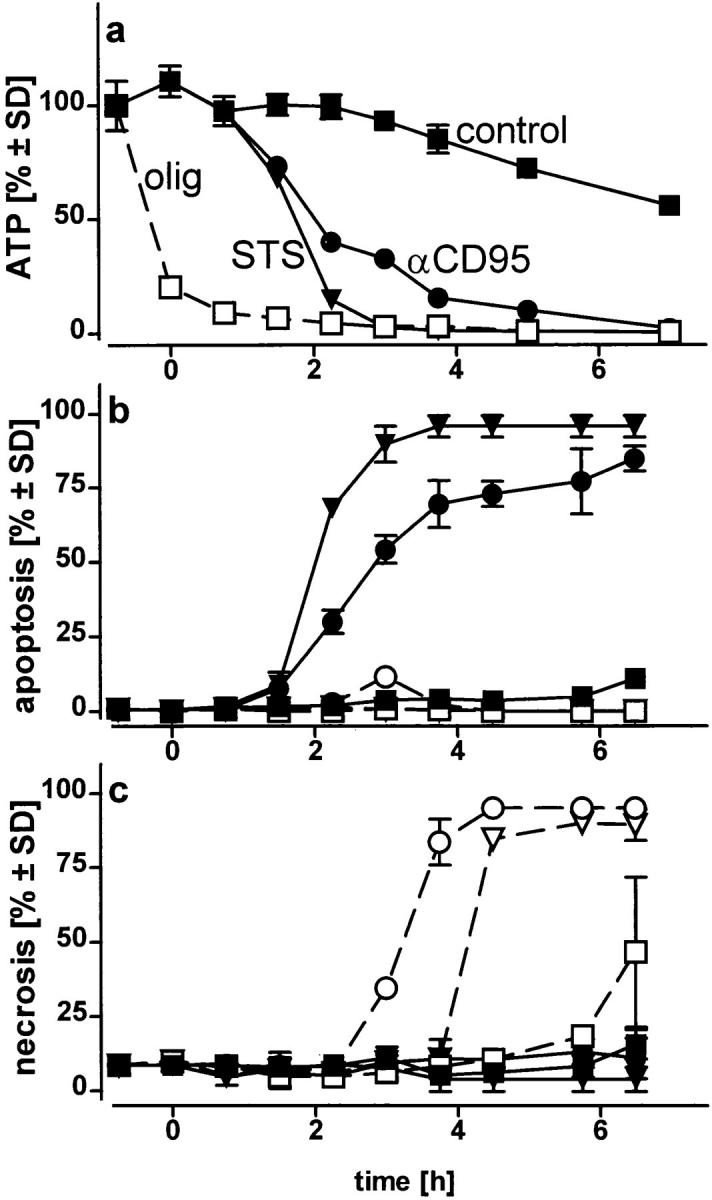

ATP-depletion and the shape of cell death: apoptosis or necrosis. Cells, in a glucose-free medium supplemented with 2 mM pyruvate, were incubated with solvent (control) (squares), 1.2 μM STS (triangles), or 100 ng/ml αCD95 (circles). Pretreatment with 2.5 μM oligomycin (olig) is shown by the open symbols; (a) intracellular ATP concentration was measured at the time indicated and expressed as percentage of untreated controls (1.6 nmol/mg protein; 4.5 nmol/106 cells). ATP concentrations in cells treated with oligomycin plus STS or αCD95 did not differ from those of cells treated with oligomycin alone (open squares); (b and c) the percentage of apoptotic and necrotic cells was determined after staining of cultures with H-33342 plus SYTOX; in d, Jurkat cell were incubated with 1.2 μM STS (left), STS plus oligomycin (middle), or STS plus oligomycin plus 5 mM glucose (right) for 4 h, before staining with SYTO-13 (green) and EH-1 (red). Data are means ± SD of triplicate determinations and representative of at least four experiments.
We asked ourselves whether maintenance of a certain ATP level was needed to prevent an early breakdown of the plasma membrane. In this case, the development of apoptosis, which would not necessarily require ATP, could have been precluded by a premature demise with apparent necrotic features. However, it became clear that necrosis elicited by either of the two inducers in ATP-depleted cells occurred with a similar or rather longer time course than apoptosis. In this case, cell demise had entirely different morphological and biochemical features (Fig. 1 d ). Nuclei of cells pretreated with oligomycin and then exposed to STS/ αCD95 had normal size and showed no apoptotic chromatin condensation, while the cell membrane was clearly permeable to fluorescent dyes. The typical apoptotic DNA cleavage into 700/300 kbp, 50 kbp, and oligonucleosomal fragments did not occur in Jurkat cells preemptied of ATP (Fig. 2). In addition, exposure of phosphatydylserines on the outer surface of the plasma membrane, a process leading to recognition of apoptotic cells (13), did not occur in ATP-depleted cells, which later underwent necrosis (annexin binding cells after 210 min: STS, 85 ± 9%; oligomycin, 19 ± 4%; STS + oligomycin, 13 ± 7%; control, 11 ± 5%). This suggests that ATP was required for the execution of several distinct processes in the apoptotic program.
Figure 2.
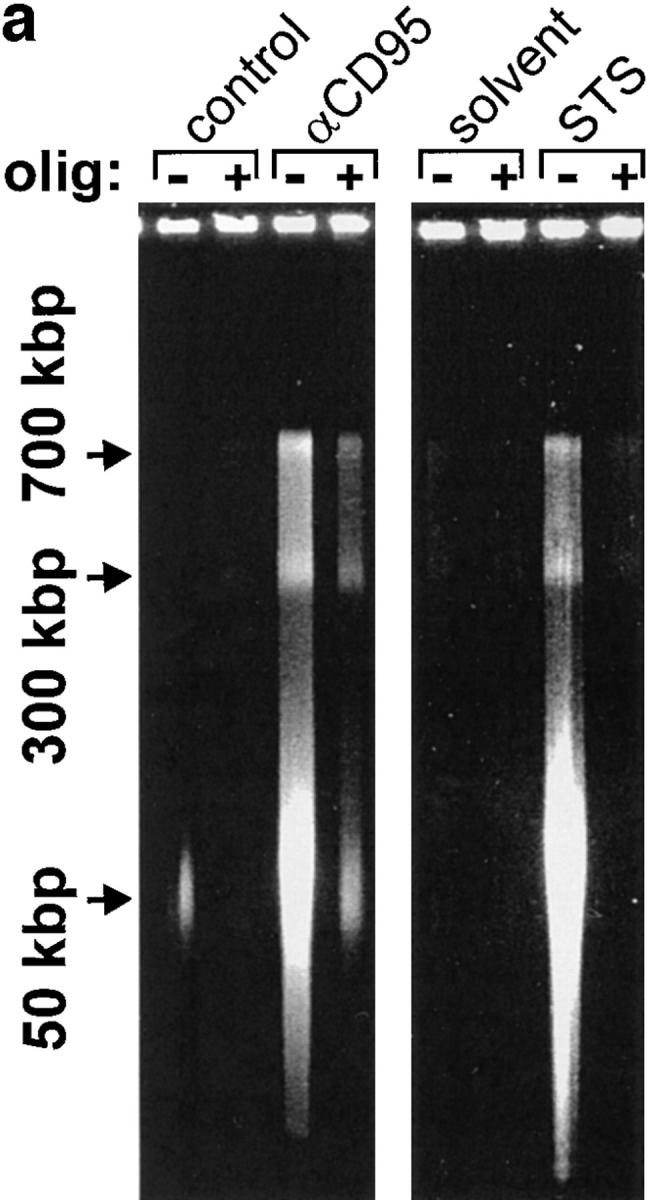

Lack of apoptotic DNA cleavage in ATP-depleted cells. Jurkat cells were incubated with αCD95 or STS in the presence or absence of oligomycin (olig). (a) After a 90 min incubation, high molecular weight DNA fragmentation was determined by FIGE; (b) after 3 h, low molecular weight oligonucleosomal fragments were determined by CAGE. At the selected time points, the two types of DNA cleavage had reached the maximum as determined by pilot experiments.
To estimate the ATP concentration sufficient to allow the progression of apoptosis, we clamped the ATP concentration of oligomycin-treated cells at different levels by supplementing the medium with glucose (Fig. 3 a). Cells were then treated with the inducers. In STS-treated cells, an ATP depletion >50% (i.e., a residual ATP concentration <0.8 nmol × mg prot−1 45 min after the addition of the stimulus, when the apoptotic features were not yet apparent) was sufficient to change the mode of demise from apoptotic to necrotic. Higher ATP concentrations instead favored the ordered continuation of the apoptotic program. With αCD95, an ATP loss ⩾70% (i.e., a residual ATP concentration of ⩽0.5 nmol × mg prot−1) was invariably followed by necrosis (Fig. 3, b and c). Similar results were obtained in experiments with fructose replenishment. In cells pretreated with oligomycin and then challenged with αCD95, a fructose concentration (40 mM) that limited the ATP loss (i.e., residual ATP above 50%) changed the mode of cell death from necrosis to apoptosis. Addition of similar metabolic substrates, e.g., glycerol (50 mM), dihydroxyacetone (50 mM), or lower fructose concentrations (10 mM), which were not able to restore intracellular ATP levels, did not modify the shape of cell death. Thus, it appears that the availability of the ATP generated by glycolysis is sufficient to allow the execution of cell death by apoptosis. In the absence of glucose and oligomycin, mitochondrial ATP generation stimulated by the addition of pyruvate would naturally replenish also the extramitochondrial pool.
Figure 3.
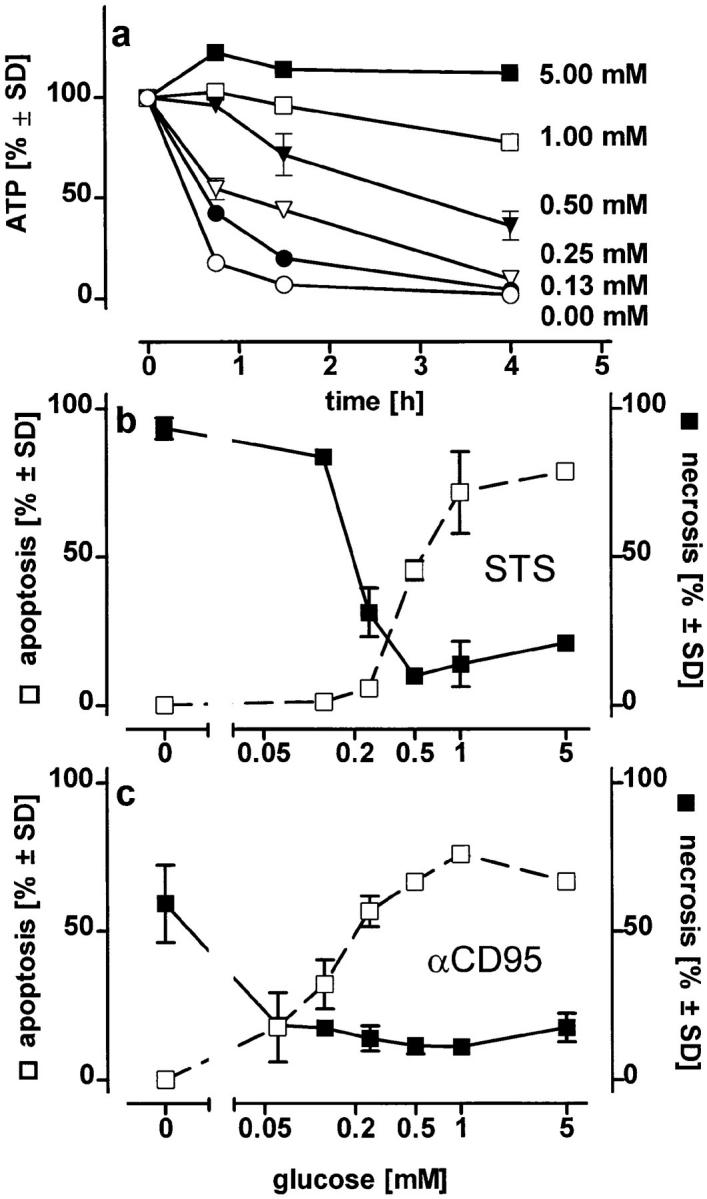

Changes in the mode of cell death after clamping the intracellular ATP concentration at defined levels or at different times. (a) Jurkat cells in glucose-free medium containing pyruvate (2 mM) were exposed to 2.5 μM oligomycin plus the indicated concentrations of glucose. Intracellular ATP concentrations were determined at the times indicated; (b and c) Jurkat cells were preincubated for 45 min in medium containing 2.5 μM oligomycin plus the concentration of glucose indicated. STS (b) or αCD95 (c) were then added and the mode of cell death was determined after further 3.5 h; (d ) intracellular ATP levels were manipulated during incubation of Jurkat cells with either STS or αCD95. Left, cells were incubated in pyruvate-supplemented medium without glucose and challenged (at t = 0) with 100 ng/ml αCD95 or 1.2 μM STS. At the times indicated oligomycin was added to deplete ATP, and the mode of cell death was determined 4 h after the challenge. Right, cells were first depleted of ATP by preincubation with oligomycin (at t = −1 h). At t = 0, STS or αCD95 were added. At the times indicated, intracellular ATP was replenished by adding 10 mM glucose to the incubation medium. The dashed bold line indicates the cellular ATP concentration (percentage of untreated control cultures in standard pyruvate medium), which was reached 15 min after each glucose supplementation. Data are means ± SD of triplicate determinations.
To determine at which phase of the death program ATP was required, we added oligomycin at different times after triggering cell death with STS or αCD95. When ATP depletion was inflicted up to 90 min after the challenge with either of the two stimuli, cell death still shifted from apoptosis to necrosis (Fig. 3 d ). In a complementary experimental setup, we found that glucose addition to replenish ATP in cells treated with either STS or αCD95 plus oligomycin directed the death process towards apoptosis up to 2 h after the challenge (Fig. 3 d ). Thus, it seemed that ATP was required immediately before the characteristic nuclear changes, perhaps to ensure that nuclear collapse, the ordered packaging of chromatin, and DNA degradation occur before cell lysis. Accordingly, DNA fragmentation was strongly reduced in necrotic cells (i.e., challenged with STS or αCD95 in the presence of oligomycin) as compared with those undergoing apoptosis, although the total percentage of cell death was approximately the same (Fig. 4, a and b). Differences in the fate of the nucleus became evident when we examined lamin degradation, a process pivotal for the nuclear collapse that is effected by the activation of certain caspases during apoptosis (17, 18). Cleavage of lamin B was significantly reduced under conditions of ATP depletion leading to necrosis (Fig. 4, c and d ), which suggests that activation of the proteolytic system responsible for lamin cleavage is specific for apoptosis.
Figure 4.



Activation of caspases in apoptotic or necrotic cell death. (a and b) Cells were preincubated for 45 min with oligomycin and/or 75 μM zVAD-fmk as indicated and then challenged with αCD95. After 4 h, (a) the mode of cell death (closed bars, apoptotic; open bars, necrotic) or (b) DNA fragmentation were determined by dual fluorescent staining or ELISA, respectively; (c) cells were incubated with αCD95 or STS for 200 min in the presence or absence of 2.5 μM oligomycin (olig). Lamin B cleavage was determined by Western blot analysis. The original lamin band and the proteolytic fragment, typically found in apoptotic cells are indicated by arrowheads; (d ) Lamin disintegration was also studied by immunostaining. Cells untreated or treated with oligomycin were then challenged by STS. Lamins were stained with an anti-lamin antibody, whereas nuclei were counterstained with H-33342. Confocal images show that breaks/dissolution of the lamin structure (middle-left) as well as chromatin condensation (middle-right) did not occur in cells undergoing necrosis (i.e., treated with oligomycin) (bottom).
Because activation of caspases is also an integral component of the CD95 signaling system that triggers apoptosis (19, 20) we used caspase inhibitors to test whether the same signaling system would be active in necrosis and apoptosis. In this case, caspase inhibitors prevented αCD95-induced cell death regardless whether apoptotic or necrotic (Fig 4, a and b), in agreement with other findings that both types of cell death share similar signals and initial mechanisms (21–23).
A cytoplasmic controller (24) seems to direct downstream events resulting in the typical apoptotic features including proteolysis of nuclear elements and DNA degradation. Furthermore, according to a series of elegant experiments with reconstituted cell-free systems, an apoptogenic protease (25) released from mitochondria upon permeability transition (26) can promote the nuclear changes. In cells, mitochondrial permeability transition can be triggered by a variety of stimuli acting on the mitochondria directly, or by a yet unknown upstream controller (25, 26). Thus, either the upstream controller or the execution system(s) may be modulated by the availability of ATP. The latter may be provided by gycolysis or, in tissues with high metabolic demand, primarily by the mitochondria (3, 27). In the model described here, restoration of glycolytic ATP generation was sufficient to allow the ordered execution of apoptosis. Thus, apoptosis and necrosis would be two extremes of a continuum of possible types of cell demise, whose shape and implications for the neighboring tissue would be decided by the availability of ATP, and likely by additional factors in the dying cell and in scavenger cells. This would explain the frequent coexistence of both types of cell death in pathological situations, e.g., cerebral ischemia (28) or inflammatory liver failure (5, 6), where individual cell death within the tissue would be decided by the energy supply.
Acknowledgments
We are grateful to Ms. H. Naumann and to Mr. T. Schmitz for the excellent technical assistance and to Dr. E. May for stimulating discussions.
This work has been supported by grants from the European Community (EV5V CT94 0508) (ENV4CT96-0169), by the European Science Foundation to A.F. Castoldi.
References
- 1.Wyllie AH, Kerr JF, Currie AR. Cell death: the significance of apoptosis. Int Rev Cytol. 1980;68:251–306. doi: 10.1016/s0074-7696(08)62312-8. [DOI] [PubMed] [Google Scholar]
- 2.Shimizu S, Eguchi Y, Kamiike W, Itoh Y, Hasegawa J, Yamabe K, Otsuki Y, Matsuda H, Tsujimoto Y. Induction of apoptosis as well as necrosis by hypoxia and predominant prevention of apoptosis by Bcl-2 and Bcl-xL. Cancer Res. 1996;56:2161–2166. [PubMed] [Google Scholar]
- 3.Ankarcrona M, Dypbukt JM, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton SA, Nicotera P. Glutamateinduced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15:961–973. doi: 10.1016/0896-6273(95)90186-8. [DOI] [PubMed] [Google Scholar]
- 4.Leist M, Gantner F, Künstle G, Bohlinger I, Tiegs G, Bluethmann H, Wendel A. The 55 kD tumor necrosis factor receptor and CD95 independently signal murine hepatocyte apoptosis and subsequent liver failure. Mol Med. 1996;2:109–124. [PMC free article] [PubMed] [Google Scholar]
- 5.Leist M, Gantner F, Bohlinger I, Tiegs G, Germann PG, Wendel A. Tumor necrosis factor–induced hepatocyte apoptosis precedes liver failure in experimental murine shock models. Am J Pathol. 1995;146:1220–1234. [PMC free article] [PubMed] [Google Scholar]
- 6.Bonfoco E, Krainc D, Ankarcrona M, Nicotera P, Lipton SA. Apoptosis and necrosis: two distinct events induced respectively by mild and intense insults with NMDA or nitric oxide/superoxide in cortical cell cultures. Proc Natl Acad Sci USA. 1995;92:7162–7166. doi: 10.1073/pnas.92.16.7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dypbukt JM, Ankarcrona M, Burkitt M, Sjöholm A, Ström K, Orrenius S, Nicotera P. Different prooxidant levels stimulate growth, trigger apoptosis or produce necrosis of insulin-secreting RINm5F cells. The role of intracellular polyamines. J Biol Chem. 1994;269:30533–30560. [PubMed] [Google Scholar]
- 8.Trauth BC, Clas C, Peters AMJ, Matzku S, Möller P, Falk W, Debatin K, Krammer PH. Monoclonal antibody-mediated tumor regression by induction of apoptosis. Science (Wash DC) 1989;245:301–305. doi: 10.1126/science.2787530. [DOI] [PubMed] [Google Scholar]
- 9.Yonehara S, Ishii A, Yonehara M. A cell-killing monoclonal antibody (anti-Fas) to a cell surface antigen codownregulated with the receptor of tumor necrosis factor. J Exp Med. 1989;169:1747–1756. doi: 10.1084/jem.169.5.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weil M, Jacobson MD, Coles HSR, Davies TJ, Gardner RL, Raff KD, Raff MC. Constitutive expression of the machinery for programmed cell death. J Cell Biol. 1996;133:1053–1059. doi: 10.1083/jcb.133.5.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jacobson MD, Raff MC. Bcl-2 blocks apoptosis in cells lacking mitochondrial DNA. Nature (Lond) 1995;374:814–816. doi: 10.1038/361365a0. [DOI] [PubMed] [Google Scholar]
- 12.Anel A, Gamen S, Alava MA, Schmitt-Verhulst A-M, Pineiro A, Naval J. Role of oxidative damage and IL-1beta-converting enzyme-like proteases in Fas-based cytotoxicity exerted by effector T cells. Int Immunol. 1996;8:1173–1183. doi: 10.1093/intimm/8.7.1173. [DOI] [PubMed] [Google Scholar]
- 13.Martin, S.J., C.P.M. Reutlingsperger, and D.R. Green. 1996. Annexin V: a specific probe for apoptotic cells. In Techniques in Apoptosis. T.G. Cotter and S.J. Martin, editors. Portland Press, Ltd., London. 107–121.
- 14.Bergmeyer, H.U. 1984. Methods of Enzymatic Analysis, Vol. 82, 3rd edition. Verlag Chemie, Weinheim, Germany. 607–612.
- 15.Wyllie AH. Glucocorticoid-induced thymocyte apoptosis is associated with endogenous endonuclease activation. Nature (Lond) 1980;284:555–556. doi: 10.1038/284555a0. [DOI] [PubMed] [Google Scholar]
- 16.Leist M, Gantner F, Bohlinger I, German PG, Tiegs G, Wendel A. Murine hepatocyte apoptosis induced in vitro and in vivo by TNF-α requires transcriptional arrest. J Immunol. 1994;154:1307–1316. [PubMed] [Google Scholar]
- 17.Lazebnik YA, Takahashi A, Moir RD, Goldman RD, Poirier GG, Kaufmann SH, Earnshaw WC. Studies of the lamin proteinase reveal multiple parallel biochemical pathways during apoptotic execution. Proc Natl Acad Sci USA. 1995;92:9042–9046. doi: 10.1073/pnas.92.20.9042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oberhammer FA, Hochegger K, Fröschl G, Tiefenbacher R, Pavelka M. Chromatin condensation during apoptosis is accompanied by degradation of lamin A+B, without enhanced activation of cdc2 kinase. J Cell Biol. 1994;126:827–837. doi: 10.1083/jcb.126.4.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muzio M, Chinnaiyan AM, Kischkel FC, O'Rourke K, Shevchenko A, Ni J, Scaffidi C, Bretz JD, Zhang M, Gentz R, et al. FLICE, a novel FADD-homologous ICE/ CED-3–like protease, is recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell. 1996;85:817–827. doi: 10.1016/s0092-8674(00)81266-0. [DOI] [PubMed] [Google Scholar]
- 20.Boldin MP, Goncharov TM, Goltsev YV, Wallach D. Involvement of MACH, a novel MORT1/ FADD-interacting protease, in Fas/APO-1– and TNF receptor–induced cell death. Cell. 1996;85:803–815. doi: 10.1016/s0092-8674(00)81265-9. [DOI] [PubMed] [Google Scholar]
- 21.Shimizu S, Eguchi Y, Kamiike W, Waguri S, Uchiyama Y, Matsuda H, Tsujimoto Y. Bcl-2 blocks loss of mitochondrial membrane potential while ICE inhibitors act at a different step during inhibition of death induced by respiratory chain inhibitors. Oncogene. 1996;13:21–29. [PubMed] [Google Scholar]
- 22.Shimizu S, Eguchi Y, Kamiike W, Waguri S, Uchiyama Y, Matsuda H, Tsujimoto Y. Retardation of chemical hypoxia-induced necrotic cell death by Bcl-2 and ICE inhibitors: possible involvement of common mediators in apoptotic and necrotic signal transductions. Oncogene. 1996;12:2045–2050. [PubMed] [Google Scholar]
- 23.Zhong L-T, Sarafian T, Kane DJ, Charles AC, Mah SP, Edwards RH, Bredesen DE. bcl-2 inhibits death of central neural cells induced by multiple agents. Proc Natl Acad Sci USA. 1993;90:4533–4537. doi: 10.1073/pnas.90.10.4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jacobson MD, Burne JF, Raff MC. Programmed cell death and Bcl-2 protection in the absence of a nucleus. EMBO (Eur Mol Biol Organ) J. 1994;13:1899–1910. doi: 10.1002/j.1460-2075.1994.tb06459.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Susin SA, Zamzami N, Castedo M, Hirsch T, Marchetti P, Macho A, Daugas E, Geuskens M, Kroemer G. Bcl-2 inhibits the mitochondrial release of an apoptogenic protease. J Exp Med. 1996;184:1331–1341. doi: 10.1084/jem.184.4.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zamzami N, Susin SA, Marchetti P, Hirsch T, GómezMonterrey I, Castedo M, Kroemer G. Mitochondrial control of nuclear apoptosis. J Exp Med. 1996;183:1533–1544. doi: 10.1084/jem.183.4.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Richter C, Schweizer M, Cossarizza A, Franceschi C. Control of apoptosis by the cellular ATP level. FEBS Lett. 1996;378:107–110. doi: 10.1016/0014-5793(95)01431-4. [DOI] [PubMed] [Google Scholar]
- 28.Li Y, Sharov VG, Jiang N, Zaloga C, Sabbah HN, Chopp M. Ultrastructural and light microscopic evidence of apoptosis after middle cerebral artery occlusion in the rat. Am J Pathol. 1995;146:1045–1051. [PMC free article] [PubMed] [Google Scholar]